

Abstract
The primary afferent nociceptor was used as a model system to study mechanisms of pain induced by chronic opioid administration. Repeated intradermal injection of the selective mu-opioid receptor (MOR) agonist DAMGO induced mechanical hyperalgesia and marked prolongation of prostaglandin E2 (PGE2) hyperalgesia, a key feature of hyperalgesic priming. However, in contrast to prior studies of priming induced by receptor-mediated (i.e., TNFα, NGF, or IL-6 receptor) or direct activation of protein kinase Cε (PKCε), the pronociceptive effects of PGE2 in DAMGO-treated rats demonstrated the following: (1) rapid induction (4 h compared with 3 d); (2) protein kinase A (PKA), rather than PKCε, dependence; (3) prolongation of hyperalgesia induced by an activator of PKA, 8-bromo cAMP; (4) failure to be reversed by a protein translation inhibitor; (5) priming in females as well as in males; and (6) lack of dependence on the isolectin B4-positive nociceptor. These studies demonstrate a novel form of hyperalgesic priming induced by repeated administration of an agonist at the Gi-protein-coupled MOR to the peripheral terminal of the nociceptor.
SIGNIFICANCE STATEMENT The current study demonstrates the molecular mechanisms involved in the sensitization of nociceptors produced by repeated activation of mu-opioid receptors and contributes to our understanding of the painful condition observed in patients submitted to chronic use of opioids.
Keywords: β/γ subunit, chronic pain, hyperalgesia, hyperalgesic priming, mu-opioid receptor
Introduction
Repeated exposure to mu-opioid receptor (MOR) agonists can induce, in addition to the craving associated with opioid tolerance and dependence (Aley et al., 1995; Kolesnikov et al., 1996; Aley and Levine, 1997a, 1997b), long-lasting mechanical hyperalgesia, a phenomenon referred to as opioid-induced hyperalgesia (OIH) (Mao, 2002; Angst and Clark, 2006; Lee et al., 2011). Despite its potential clinical importance, the mechanism underlying OIH is still not well understood. Others (Vanderah et al., 2001; Mao, 2002; Ossipov et al., 2005; Chu et al., 2008; Chen et al., 2010) and we (Aley et al., 1995; Aley and Levine, 1997a, 1997b; Joseph et al., 2010) have used the MOR-containing primary afferent nociceptor as a model system to study the mechanism underlying the tolerance, dependence, and pronociceptive effect of chronic opioid administration (Aley et al., 1995; Aley and Levine, 1997a, 1997b; Ossipov et al., 2005; Chu et al., 2008; Chen et al., 2010; Joseph et al., 2010).
In the present experiments, we evaluated the hypothesis that the mechanical hyperalgesia induced by repeated exposure to a MOR-selective agonist reflects a form of hyperalgesic priming (Aley et al., 2000; Joseph et al., 2010), a model of the transition to chronic pain. Classical hyperalgesic priming is induced by activation of receptors for pronociceptive mediators that signal via protein kinase Cε (PKCε) or by direct activation of PKCε in the peripheral terminal of the nociceptor (Aley et al., 2000; Parada et al., 2003b). In the PKCε-primed nociceptor, exposure to prostaglandin E2 (PGE2) and other pronociceptive mediators produces markedly prolonged, PKCε-dependent mechanical hyperalgesia mediated by isolectin B4-positive (IB4+) nociceptors (Joseph and Levine, 2010a), which can be reversed by the administration of inhibitors of protein translation to the peripheral terminal of the primed nociceptor (Ferrari et al., 2013b) in male but not female rats (Joseph et al., 2003). We report that repeated administration of DAMGO induces PKA-dependent, rather than PKCε-dependent, prolongation of PGE2 hyperalgesia independently of protein translation in the peripheral terminal of the nociceptor occurring in IB4− nociceptors and, in female as well as male rats. Therefore, whereas a similar phenotype (increased response to PGE2) is observed in this model of change in nociceptor function, because it involves distinct mechanisms, we designate it as type II hyperalgesic priming.
Materials and Methods
Animals.
Experiments were performed on 230–280 g adult male and female Sprague Dawley rats (Charles River Laboratories). Animals were housed in a controlled environment at the animal care facility of the University of California–San Francisco (UCSF), under a 12 h light/dark cycle. Food and water were available ad libitum. Experiments were approved by the Institutional Animal Care and Use Committee at UCSF and adhered to guidelines of the American Association of Laboratory Animal Care, the National Institutes of Health, and the Committee for Research and Ethical Issues of the International Association for the Study of Pain. In the design of the experiments, effort was made to minimize the number of animals used and their suffering.
Mechanical nociceptive threshold testing.
Mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter (Randall-Selitto paw-withdrawal test; Stoelting), which applies a linearly increasing mechanical force to the dorsum of the rat's hindpaw, as described previously (Taiwo and Levine, 1989; Taiwo et al., 1989; Ferrari and Levine, 2015). The nociceptive threshold was defined as the force in grams at which the rat withdrew its paw; baseline paw-pressure threshold was defined as the mean of the three readings taken just before a test agent was injected. Each paw was treated as an independent measure and each experiment was performed on a separate group of rats. Data are presented as the mean change from baseline mechanical nociceptive threshold.
Drugs and their administration.
The chemicals used in this study were as follows: cordycepin 5′-triphosphate sodium salt (protein translation inhibitor), Quin 2 potassium salt hydrate (a calcium chelator), PGE2 (a hyperalgesic agent that sensitizes nociceptors directly), [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt (DAMGO, a MOR agonist); mastoparan (a G-protein activator via Gαi and Gαo stimulation), SU6656 (an Src inhibitor), U-73122 (an inhibitor of phospholipase Cγ), wortmannin (a PI3K inhibitor), Z-VAD-FMK (a caspase inhibitor), and pertussis toxin (a Gi-protein inhibitor) (all from Sigma-Aldrich); H-89 dihydrochloride (an inhibitor of PKA) and gallein (an inhibitor of G-protein β/γ) (both from Santa Cruz Biotechnology); G-Protein βγ Binding Peptide (a G-protein β/γ activator) and PKCεV1-2 (PKCε-I, a PKCε-specific translocation inhibitor peptide (Johnson et al., 1996; Khasar et al., 1999) (both from Calbiochem); and NG-monomethyl-l-arginine acetate (L-NMMA, an inhibitor of nitric oxide synthase) and the potent cell-permeable cAMP analog 8-bromo cAMP (both from Tocris Bioscience).
The selection of the doses used was based on previous studies showing their effectiveness when injected intradermally on the dorsum of the rat hindpaw (Taiwo et al., 1989; Taiwo and Levine, 1991; Aley and Levine, 1997b; Aley et al., 2001; Dina et al., 2009; Joseph and Levine, 2010b; Ferrari et al., 2012). The stock solution of PGE2 (1 μg/μl) was prepared in 10% ethanol and additional dilutions made with physiological saline (0.9% NaCl), yielding a final ethanol concentration of <1%. DAMGO, cordycepin, 8-bromo cAMP, and pertussis toxin were dissolved in saline. All other drugs were dissolved in 100% DMSO (Sigma-Aldrich) and further diluted in saline containing 2% Tween 80 (Sigma-Aldrich). The final concentration of DMSO and Tween 80 was ∼2%. All drugs were administered intradermally on the dorsum of the hindpaw in a volume of 5 μl using a 30-gauge hypodermic needle adapted to a 50 μl Hamilton syringe. The administration of all drugs, except PGE2, DAMGO, and 8-bromo cAMP, was preceded by a hypotonic shock to facilitate cell permeability to these agents (2 μl of distilled water separated by a bubble of air to avoid mixing in the same syringe) to facilitate the entry of compounds into the nerve terminal (Borle and Snowdowne, 1982; Burch and Axelrod, 1987).
Intrathecal administration of IB4-saporin.
IB4-saporin, a neurotoxin for the IB4+ nociceptor (Advanced Targeting Systems), was diluted with saline and a dose of 3.2 μg in a volume of 20 μl was administered intrathecally 10 d before priming experiments. This dose of IB4-saporin and the time point chosen for evaluation of its effect was based in previous reports from us and others (Vulchanova et al., 2001; Joseph et al., 2008; Joseph and Levine, 2010a; Nishiguchi et al., 2004). Rats were briefly anesthetized with 2.5% isoflurane (Phoenix Pharmaceuticals) in 97.5% O2. Then, a 30-gauge hypodermic needle was inserted on the midline into the subarachnoid space between the L4 and L5 vertebrae. The control treatment consisted of intrathecal injection of saline (20 μl). Animals regained consciousness ∼1 min after removal from the anesthetic chamber. There was no effect of IB4-saporin on the mechanical nociceptive threshold per se.
Oligodeoxynucleotide antisense to PLC-β3.
The oligodeoxynucleotide (ODN) antisense (AS) sequence for PLC-β3 was directed against a unique region of the rat PLC-β3 mRNA sequence (GenBank accession number NM_033350). The AS-ODN sequence was 5′-CCTTCAAGACCTCACCCT AC-3′ and the ODN mismatch sequence (MM-ODN) was designed by mismatching 8 bases (denoted by bold face) of the PLC-β3 AS-ODN sequence, 5′-GGTTGAAGAGGTCACGCAAG-3′ (Joseph et al., 2007). We have shown previously that spinal intrathecal administration of AS-ODN with this sequence decreases PLC-β3 protein in dorsal root ganglia (Joseph et al., 2007).
Before use, ODNs were lyophilized and reconstituted in 0.9% NaCl to a concentration of 2 μg/μl. During each injection, rats were briefly anesthetized with 2.5% isoflurane in 95% O2. A 30-gauge hypodermic needle was then inserted on the midline into the subarachnoid space between the L4 and L5 vertebrae. A total of 40 μg of ODN, in a volume of 20 μl, was slowly injected. The intrathecal site of injection was confirmed by a sudden tail flick, a reflex that is evoked by subarachnoid space access and bolus injection (Mestre et al., 1994). Animals regained consciousness ∼1 min after the injection. The use of antisense to manipulate the expression of proteins in nociceptors, important for their role in nociceptor sensitization, is well supported by previous studies by others (Song et al., 2009; Su et al., 2011; Quanhong et al., 2012; Sun et al., 2013), as well as our group (Parada et al., 2003a; Ferrari et al., 2010; Bogen et al., 2012; Ferrari et al., 2012).
DAMGO-induced changes in nociceptor function.
We have shown previously that, whereas a single injection of the selective MOR agonist DAMGO alone has no effect on nociceptive threshold and attenuates the mechanical hyperalgesia induced by PGE2 (Levine and Taiwo, 1989; Taiwo and Levine, 1990), when injected repeatedly, it produces changes in nociceptor function such as tolerance, no longer producing an antihyperalgesic effect and producing mechanical hyperalgesia by itself (Aley et al., 1995; Aley and Levine, 1997a). The repeated injection of DAMGO also induces a latent state of hyperresponsiveness to subsequent injection of proalgesic mediators (Joseph et al., 2010), here referred to as type II hyperalgesic priming. Similar to the classical, type I, hyperalgesic priming (Aley et al., 2000; Reichling and Levine, 2009; Ferrari et al., 2014), this model of neuroplasticity is expressed as prolongation of the mechanical hyperalgesia produced by PGE2, lasting at least 4 h, as opposed to the injection of PGE2 in naive paws, in which hyperalgesia dissipated by 2 h (Aley and Levine, 1999). To study the mechanism involved in the hyperalgesia produced by the repeated activation of the MOR, 3 hourly intradermal injections of DAMGO (1 μg) were performed on the dorsum of the hindpaw; mechanical hyperalgesia was observed starting after a fourth injection of DAMGO (see Fig. 1). To investigate the changes in nociceptor function produced by previous repeated injection of DAMGO—measured as prolonged response to a hyperalgesic mediator at a point in time when the mechanical nociceptive threshold was not different from pre-DAMGO baseline levels—PGE2 (100 ng) was injected at the same site and hyperalgesia was evaluated after 30 min and again at 4 h. The presence of hyperalgesia at the fourth hour is characteristic of priming (Aley et al., 2000; Ferrari et al., 2014). To differentiate the intracellular signaling pathways that play a role in type I and type II priming and to investigate the mechanisms that play a role in the induction of the changes in nociceptor function produced by the repeated activation of the MOR, pharmacological agents were injected before DAMGO (prevention protocol). To investigate the second messengers involved in the expression of the neuroplasticity, the inhibitors were administered before the injection of PGE2 in the primed paw (inhibition protocol). To evaluate the role of messengers in the maintenance of the neuroplasticity, PGE2 was injected again at a time point when the inhibitors were no longer present (reversal protocol).
Figure 1.

Repeated exposure to DAMGO induces acute mechanical hyperalgesia and prolongation of PGE2 hyperalgesia in male rats. A, Male rats received repeated (hourly, ×4) intradermal injections of vehicle (control, black bars) or DAMGO (1 μg, white bars) on the dorsum of the hindpaw and the mechanical nociceptive threshold was evaluated at the injection site 30 min after the first, third, and fourth administration by the Randall–Sellitto paw-withdrawal test. Significant hyperalgesia was observed after a fourth injection of DAMGO, but not to DAMGO in the vehicle-treated paws (F(1,20) = 18.72; ***p < 0.001, when both groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); B, A separate group of rats received repeated (hourly, ×3) intradermal injection of DAMGO (1 μg) on the dorsum of the hindpaw. A fourth injection 1 h after the third dose of DAMGO, vehicle (gray bars), or PGE2 (100 ng, white bars) was performed at the same site and the mechanical nociceptive threshold was evaluated 30 min and 4 h later. We observed that, whereas injection of vehicle did not induce significant change in the mechanical threshold, the injection of PGE2 induced hyperalgesia that was still present at the fourth hour (F(1,20) = 280.56; ***p < 0.0001, when both groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating the presence of priming; C, Rats that were treated with 4 injections of vehicle (black bars) or DAMGO (white bars) 1 week before received PGE2 (100 ng) injected at the same site. Mechanical nociceptive threshold was then evaluated 30 min and 4 h later. By the time that PGE2 was injected, the mechanical nociceptive thresholds were not different from preinjection control baseline (t(5) = 0.6956; p = 0.5177, for the vehicle group; t(5) = 1.320; p = 0.2441, for the DAMGO group; paired Student's t test). In both groups, PGE2 induced significant hyperalgesia. However, whereas in the vehicle-treated group, the effect of PGE2 was no longer present at 4 h, in the group previously treated with DAMGO (hourly, ×3), the hyperalgesia induced by PGE2 was still present, indicating the presence of priming (***p < 0.0001 when both groups are compared at the fourth hour, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). D, Thirty-eight days after the vehicle (×3) or DAMGO (×3) treatments (no difference in the average mechanical nociceptive thresholds from pretreatments levels was observed: p = 0.0706 for the vehicle, t(5) = 2.291; p = 0.1613 for the DAMGO group, t(5) = 1.643; paired Student's t test), PGE2 was injected again at the same site. We observed that it produced prolonged hyperalgesia in the group previously treated with DAMGO (×3), but not in the vehicle-treated control group, and this was significant 4 h after injection (***p < 0.0001, when both groups are compared, two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the repeated injection of DAMGO produced long-term plastic changes in nociceptors. n = 6 paws per group.
Statistics.
In all experiments, the dependent variable was paw-withdrawal threshold, expressed as percentage change from baseline. The average paw-withdrawal threshold before the three injections of DAMGO (initial baseline mechanical nociceptive threshold) and before the subsequent injection of DAMGO (a fourth injection or injection 1 week later; see Fig. 2B), PGE2 (varying from 1 h to 38 d after 3 hourly injections of DAMGO, depending on the experiment), or 8-bromo cAMP (1 h after 3 injections of DAMGO; see Fig. 12) were 120.0 ± 0.5 g and 118.8 ± 0.8 g, respectively; paired Student's t test showed no significant difference between these values (t(317) = 1.156, p = 0.2505), indicating that the induction of type II priming by DAMGO does not affect the mechanical threshold. The total number of rats used in this study was 159 (318 paws). To compare the percentage change in the hyperalgesia induced by repeated injections of the neuroplasticity inducer (DAMGO, or activators of G-protein subunits, e.g., mastoparan and G-Protein βγ Binding Peptide), a one-way or two-way repeated-measures ANOVA, followed by Bonferroni post test, was performed to compare the effect of PGE2 in different groups in the presence or absence of inhibitors. GraphPad Prism 5.0 software was used to plot graphs and to perform the statistical analyses. p < 0.05 was considered statistically significant. Data are presented as mean ± SEM.
Figure 2.
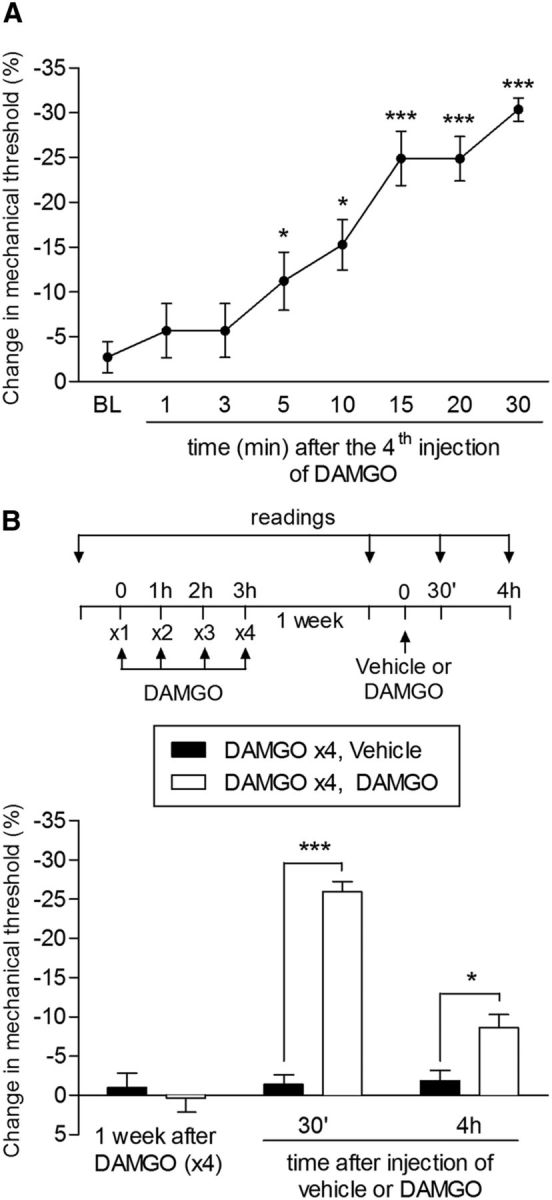
Rapid onset of DAMGO hyperalgesia and long-term persistence of type II priming. A, Mechanical nociceptive threshold was evaluated 1, 3, 5, 10, 15, 20, and 30 min after a fourth injection of DAMGO (1 μg) on the dorsum of the hindpaw. Significant hyperalgesia was already observed 5 min after the fourth injection (*p < 0.05 and ***p < 0.005, compared with the baseline (BL), paired Student's t test). B, Rats that were treated with repeated injection of DAMGO (1 μg/h ×4) 1 week before received, at the same site, an injection of DAMGO (1 μg, white bars) or vehicle (black bars). Mechanical hyperalgesia, evaluated 30 min and 4 h after injection, was observed only in the paws previously treated with repeated injection of DAMGO, showing the persistence of the changes in the nociceptor produced by the repeated stimulation of the MOR (F(1,20) = 88.69; ***p < 0.001 and *p < 0.005, two-way repeated-measures ANOVA followed by Bonferroni post hoc test; n = 6 paws per group).
Figure 12.
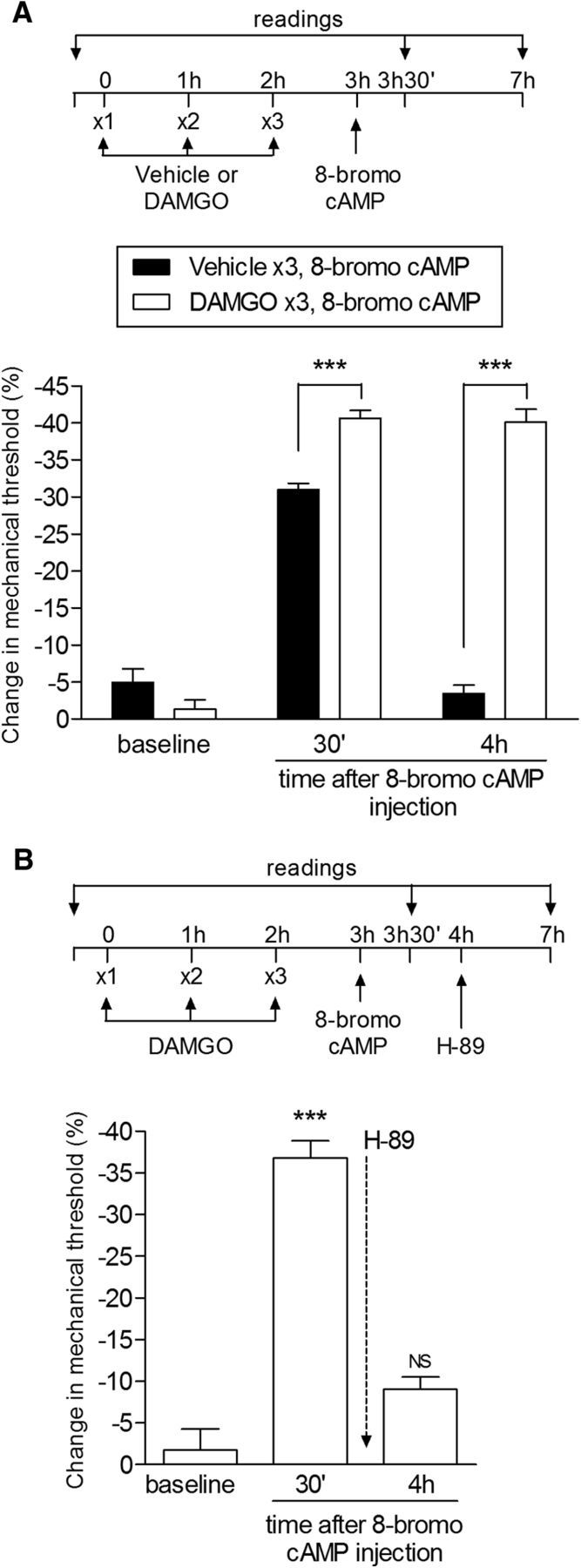
Changes in PKA signaling contribute to the prolongation of PGE2-induced hyperalgesia in type II priming. A, Rats received 3 hourly intradermal injections of vehicle or DAMGO (1 μg) on the dorsum of the hindpaw. 8-bromo cAMP (1 μg) was injected at the same site 1 h later and mechanical hyperalgesia was evaluated after 30 min and 4 h. 8-bromo cAMP produced mechanical hyperalgesia in both groups at 30 min. However, in the group previously treated with DAMGO, it was significantly enhanced and prolonged and still present after 4 h, as opposed to the vehicle-treated group (F(1,20) = 96.82; ***p < 0.0001, when comparing both groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), suggesting an increased activation of PKA signaling by repeated MOR activation; B, Rats received 3 hourly intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw and a fourth injection of 8-bromo cAMP (1 μg) at the same site. Significant mechanical hyperalgesia was observed 30 min later. Injection of H-89 (1 μg) 1 h after 8-bromo cAMP inhibited the ongoing hyperalgesia evaluated 3 h later that was produced by the previous treatment with repeated injections of DAMGO (NS, p > 0.05 vs baseline; one-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating a role of PKA in the increased activation of the cAMP signaling pathway produced by repeated MOR agonist administration. n = 6 paws per group.
Results
Distinguishing a novel type of hyperalgesic priming
Although 3 hourly intradermal injections of the highly selective MOR agonist DAMGO (1 μg) had no effect on nociceptive threshold (Aley et al., 1995; Aley and Levine, 1997a), injection of a fourth dose 1 h later induced mechanical hyperalgesia (Fig. 1A). In addition, injection of the pronociceptive inflammatory mediator PGE2 (100 ng) 1 h after rats had been treated with 3 injections of DAMGO induced markedly prolonged mechanical hyperalgesia compared with PGE2-induced hyperalgesia in naive rats (Joseph et al., 2010), still being present 4 h after administration (Fig. 1B). These results indicate the presence of a plastic change in the nociceptor produced by the repeated injection of DAMGO, an effect that persisted for at least 38 d. When PGE2 was injected at the same site 7 d (Fig. 1C) and 38 d (Fig. 1D) after the injection of 4 doses of DAMGO, it produced prolonged hyperalgesia. This marked prolongation of PGE2-induced mechanical hyperalgesia resembles that observed in the setting of hyperalgesic priming induced by activation of PKCε (Aley et al., 2000; Parada et al., 2005; Reichling and Levine, 2009; Joseph and Levine, 2010a; Ferrari and Levine, 2015). However, in contrast to classical hyperalgesic priming, which takes at least 72 h to develop (Bogen et al., 2012; Ferrari et al., 2015), this neuroplasticity developed much more rapidly, with the onset of the hyperalgesia produced by the fourth injection of DAMGO already significant by 5 min (Fig. 2A). In addition, the persistence of the neuroplasticity produced by the initial injections of DAMGO was confirmed by production of hyperalgesia by injection of DAMGO at the same site 1 week later (Fig. 2B).
To investigate whether the mechanisms underlying the neuroplasticity produced by repeated injection of DAMGO was different from the one observed in classical hyperalgesic priming, we evaluated the effect of inhibitors shown previously to attenuate the prolongation of PGE2-induced hyperalgesia in priming induced by activation of PKCε (Aley et al., 2000; Parada et al., 2003a; Ferrari et al., 2013b) in DAMGO-treated animals. We observed that the injection of neither PKCεV1-2 (1 μg/i.d.), a selective PKCε translocation inhibitor (Fig. 3, gray bars), nor cordycepin (1 μg/i.d.), a selective protein translation inhibitor, at the same site as PGE2 on the dorsum of the hindpaw (Fig. 3, white bars) affected the prolongation of PGE2-induced hyperalgesia. Therefore, because PKCε dependence and ongoing local protein translation, two of the key features of classic—here referred to as type I—hyperalgesic priming (Ferrari et al., 2013b; Ferrari et al., 2014; Ferrari et al., 2015), were not observed in the neuroplasticity produced by repeated injection of DAMGO, we referred to this condition as type II hyperalgesic priming.
Figure 3.

Type II priming is not dependent on PKCε or local protein translation. Male rats that were treated with repeated intradermal administration of DAMGO (1 μg/h ×4) on the dorsum of the hindpaw received 1 week later an injection of PGE2 (100 ng) at the same site in the presence of vehicle (control, black bars), PKCε inhibitor (1 μg, gray bars), or the inhibitor of protein translation cordycepin (1 μg, white bars) administered 10 min before. Mechanical nociceptive threshold was evaluated 30 min and 4 h after PGE2. We observed no difference between the groups in the prolongation of the PGE2-induced hyperalgesia (F(4,30) = 0.72; p = 0.5861, two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the neuroplasticity induced by previous repeated injection of DAMGO is not dependent on PKCε or local protein translation. n = 6 paws per group.
Unlike the changes in nociceptor function observed in type I priming (Aley et al., 2000; Parada et al., 2003a; Reichling and Levine, 2009; Ferrari et al., 2014), those induced by repeated administration of DAMGO were inhibited by pretreatment with the selective protein kinase A (Konopka and van Wijhe, 2010) inhibitor H-89 (1 μg)—that is, both DAMGO-induced hyperalgesia (Fig. 4A) and the prolongation of PGE2-induced hyperalgesia (Fig. 4B) were attenuated. Interestingly, although we still observed significant attenuation of the prolongation of PGE2-induced hyperalgesia 6 d after the injection of H-89, when PGE2 was injected 30 d after H-89, the hyperalgesia observed by the fourth hour was similar in magnitude to that in the control group (Fig. 4C). These findings indicate that the effect of H-89 was reversible over time and that PKA is involved only in the expression, not the induction, of type II priming; that is, a PKA-independent mechanism is involved in the induction of the changes in the nociceptor produced by repeated activation of the MOR. Moreover, injection of H-89 in naive (i.e., nonprimed) rats inhibits the hyperalgesia induced by PGE2 injected 10 min later, but not when injected 6 h later (D. Araldi, unpublished observation), indicating that the effect of H-89 lasts no more than a few hours. That PKA has a role in the expression of type II priming was further confirmed when we injected H-89 after the repeated injection of DAMGO; its administration 6 d after DAMGO inhibited PGE2 hyperalgesia at both 30 min and 4 h (inhibition protocol; Fig. 4D), an effect still observed when PGE2 was injected again 6 d later (Fig. 4E, left). However, injection of PGE2 30 d after H-89 produced prolonged hyperalgesia (Fig. 4E, right), demonstrating that the effect of H-89 is reversible and that PKA is involved in the expression, but not the induction or maintenance, of type II priming. We have shown previously that inhibition of PKA did not affect prolongation of PGE2 hyperalgesia associated with type I hyperalgesic priming (Aley et al., 2000). DAMGO-induced, type II priming also differs from type I priming in the following: (1) whereas agonists at cell surface receptors that signal via PKCε only produce priming (type I) in male rats (Joseph et al., 2003), DAMGO induced priming (type II) in female as well as male rats (Fig. 5); and (2) intrathecal administration of IB4-saporin, a neurotoxin for IB4+ nociceptors that eliminates type I priming (Joseph and Levine, 2010a), actually increased the DAMGO-induced hyperalgesia (Fig. 6) that was observed already after the third, instead of the fourth, injection of DAMGO.
Figure 4.

Role of PKA in type II priming. A, Rats were treated with intradermal injection of vehicle (control, black bars) or H-89 (1 μg, white bars) on the dorsum of the hindpaw. Ten minutes later, 4 injections of DAMGO (1 μg, hourly) were performed in both groups of rats. We observed, in the group pretreated with H-89, significant attenuation of the mechanical hyperalgesia induced by the fourth injection of DAMGO compared with the control group (F(1,10) = 18.52; ***p = 0.0016, two-way repeated-measures ANOVA followed by Bonferroni post hoc test); B, Rats were treated with intradermal injection of vehicle or H-89 (1 μg) on the dorsum of the hindpaw. Ten minutes later, 3 injections of DAMGO (1 μg/h ×3) were performed. One hour after the third injection of DAMGO, PGE2 (100 ng) was injected at the same site and the mechanical hyperalgesia was evaluated 30 min and 4 h later. Both at 30 min and 4 h, significant attenuation of the mechanical hyperalgesia induced by PGE2 was observed in the group pretreated with H-89 (F(1,10) = 189.86, ***p < 0.0001, vehicle vs H-89 groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); C, Six days later, a time point when the mechanical nociceptive threshold was not different from the pretreatment levels (t(5) = 2.000; p = 0.1019, for the control group; t(5) = 2.169; p = 0.0822, for the H-89 group; paired Student's t test), PGE2 (100 ng) was injected at the same site and the mechanical hyperalgesia was evaluated 30 min and 4 h later. Although PGE2-induced hyperalgesia was present 30 min after injection, in the group previously treated with H-89, it was significantly reduced at the fourth hour (F(1,20) = 15.67, ***p < 0.001, when both groups are compared at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). To determine whether type II priming was permanently prevented by H-89, PGE2 was injected again at the same site 30 d later. At this time, we observed that the hyperalgesia induced by PGE2 was prolonged in both groups; D, Rats received repeated (hourly, ×4) intradermal injection of DAMGO (1 μg) on the dorsum of the hindpaw. Six days later, when the mechanical thresholds were no longer different from pre-DAMGO levels (t7 = 0.7593; p = 0.4819, paired Student's t test), vehicle (control, black bars) or H-89 (1 μg, white bars) was injected at the same site, followed 10 min later by PGE2 (100 ng). Although PGE2-induced hyperalgesia was still present 4 h later in the group that received vehicle, in the group treated with H-89, it was attenuated at both time points (F(1,28) = 585.86 ***p < 0.0001, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). E, Left, Six days later, when PGE2 was injected again at the same site, the mechanical hyperalgesia in the group that had previously received H-89 (6 d before, white bars) was still significantly attenuated at both 30 min and 4 h (F(2,18) = 133.63, ***p < 0.0001, H-89 vs control groups, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Right, To determine whether the treatment with H-89 had permanently reversed the type II priming, PGE2 was again injected at the same site 30 d later (gray bars). In this case, however, we observed that PGE2 produced significant hyperalgesia that was present 30 min and 4 h after injection in both groups, indicating that type II priming was present. n = 6 paws per group.
Figure 5.

Repeated exposure to DAMGO induces acute mechanical hyperalgesia and prolongation of PGE2 hyperalgesia in female rats. Left, Female rats received repeated hourly (×4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. The mechanical nociceptive threshold was evaluated before and 30 min after the first, third, and fourth administration. We observed significant hyperalgesia after the third and fourth injections of DAMGO (***p = 0.0005 and *p < 0.0001, respectively, third and fourth injections vs baseline threshold; **p = 0.007 third vs fourth injection thresholds, paired Student's t test); Right, One week later, when the mechanical thresholds were no longer different from pre-DAMGO levels (p = 0.3939, paired Student's t test), PGE2 (100 ng) was again injected at the same site and mechanical hyperalgesia was evaluated 30 min and 4 h later. One-way ANOVA followed by Bonferroni post test showed that PGE2 induced significant hyperalgesia that was still present 4 h after its injection (***p < 0.0001, compared with the baseline pre-PGE2 injection). Together, these data demonstrate that, as in males, repeated injection of DAMGO also produces neuroplasticity in female rats, with a more rapid onset in the female. n = 6 paws per group.
Figure 6.

Effect of elimination of IB4+ nociceptors on type II priming. Left, Male rats were treated with vehicle (control, black bars) or IB4-saporin (3.2 μg/20 μl; white bars) by intrathecal injection. Two weeks later, DAMGO (1 μg) was injected (hourly, ×4) on the dorsum of the hindpaw. Although, in the vehicle-treated group, the mechanical hyperalgesia developed after the fourth injection of DAMGO, in the IB4-saporin-treated group, it was already present after the third injection, significantly further increasing after the fourth injection (F(1,20) = 33.11; ***p < 0.001, when both groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Right, One week later, when the mechanical thresholds were not different from the pre-DAMGO baseline (t(5) = 0.9035; p = 0.4077, for the control group; t(5) = 1.419; p = 0.2150, for the IB4-saporin group, paired Student's t test), PGE2 (100 ng) was injected at the same site on the dorsum of the hindpaw and the mechanical hyperalgesia was evaluated 30 min and 4 h later. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test showed PGE2-induced hyperalgesia at 30 min, which was still present at the fourth hour after injection in both groups, with no significant (NS) difference between the groups (p > 0.05, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These data support the suggestion that IB4+ nociceptors do not contribute to the prolonged hyperalgesia induced by PGE2 observed in type II priming. n = 6 paws per group.
Intracellular second messengers involved in the expression of type II priming
We described previously a role of PKA in another type of nociceptor neuroplasticity induced by the transient downregulation of G-protein related kinase 2 (GRK2) induced by intrathecal antisense, which also involves the intracellular second messengers PLC-β3 and Src (Ferrari et al., 2012). Therefore, we tested whether the inhibition of PLC-β3 and Src would affect the expression of priming induced by repeated injection of DAMGO. Pretreatment with AS-ODN against PLC-β3 mRNA inhibited the hyperalgesia induced by the fourth dose of DAMGO and the prolongation of PGE2 hyperalgesia after repeated administration of DAMGO (Fig. 7A). In rats pretreated with DAMGO (×4), PGE2 hyperalgesia was already attenuated 30 min after its injection in the paws of the PLC-β3 AS-ODN-treated rats, suggesting a change in the signaling pathway downstream of the prostaglandin receptor in the hyperalgesia induced by PGE2 in the setting of type II priming; PGE2-induced hyperalgesia in the naive rat is not PLC-β3 dependent (Joseph et al., 2007). In addition, when PGE2 was injected 12 d later, it produced hyperalgesia that was present 4 h after injection, indicating that the knock-down of PLC-β3 inhibited only the expression of type II priming, and did not prevent its induction (Fig. 7A).
Figure 7.

Second messengers involved in type II priming: PLC-β3 and Src. A, Left, Rats were treated with daily spinal intrathecal injections of MM-ODN (black bars) or AS-ODN (white bars) for PLC-β3 mRNA for 3 consecutive days. On the fourth day, repeated (hourly, ×4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw were performed and the mechanical nociceptive threshold was evaluated 30 min after the fourth DAMGO injection. We observed that, in the MM-ODN-treated group, but not in the AS-ODN-treated group, the fourth injection of DAMGO produced significant mechanical hyperalgesia (***p = 0.0009, paired Student's t test, when both groups are compared), suggesting a role for PLC-β3 in DAMGO hyperalgesia. ODN treatment continued for 2 more days and, at a time point when the mechanical thresholds were not different from the pre-DAMGO levels (t(5) = 0.9918; p = 0.3668, for the MM-ODN group; t(5) = 2.490; p = 0.0551, for the AS-ODN group; paired Student's t test), PGE2 (100 ng) was administered in both groups. Evaluation of the mechanical thresholds 30 min and 4 h after injection showed that, whereas PGE2 induced hyperalgesia that was still present 4 h later in the group treated with MM-ODN, in the group treated with AS-ODN against PLC-β3, the hyperalgesia was significantly attenuated at both time points (F(1,20) = 128.33; ***p < 0.0001, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Right, To determine whether the reversal of type II priming by treatment with AS-ODN was permanent, PGE2 was injected again in the same site 12 d after the last ODN injection. We observed that PGE2-induced mechanical hyperalgesia was still present at the fourth hour, indicating the presence of type II priming. B, Rats were treated with intradermal injection of vehicle (control, black bars) or SU6656 (1 μg, white bars). Ten minutes later, repeated injections of DAMGO (1 μg) were performed on the dorsum of the hindpaw. No difference in the mechanical hyperalgesia after the fourth injection of DAMGO was observed when the groups treated with SU6656 or vehicle were compared (nonsignificant, NS; paired Student's t test). Two days later, when the mechanical thresholds were not different from pretreatment levels (t(5) = 2.5690; p = 0.0501, for the control group; t(5) = 0.5423; p = 0.6109, for the SU6656 group; paired Student's t test), PGE2 (100 ng) was injected at the same site and mechanical hyperalgesia was evaluated 30 min and 4 h later. In the group previously treated with SU6656, PGE2-induced hyperalgesia was almost completely inhibited at the fourth hour (F(1,20) = 100.00; ***p < 0.0001, when both groups are compared at the 30 min and fourth hour, respectively; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating a role of Src in type II priming. C, Male rats received repeated (hourly, ×4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. Four days later, when the mechanical thresholds were not different from the pre-DAMGO levels (t(5) = 0.8500; p = 0.4341, for the control group; t(5) = 1.356; p = 0.2332, for the SU6656 group; paired Student's t test), vehicle (control) or the Src inhibitor SU6656 (1 μg) was injected at the same site, followed 10 min later by PGE2 (100 ng). Although PGE2 induced significant hyperalgesia that was still present when evaluated 4 h later in the vehicle-treated group, in the group treated with SU6656, PGE2-induced hyperalgesia was attenuated at the fourth hour (F(1,20) = 184.31; **p < 0.01 and ***p < 0.001, two-way repeated-measures ANOVA followed by Bonferroni post hoc). n = 6 paws per group.
The role of Src in the prolongation of PGE2-induced hyperalgesia induced by previous repeated injection of DAMGO (reversal protocol) was also evaluated. Pretreatment with the Src inhibitor SU6656 did not prevent the induction of hyperalgesia by 4 injections of DAMGO, although, similar to the PLC-β3 AS-ODN, it did prevent the prolongation of the hyperalgesia induced by injection of PGE2 2 d later (Fig. 7B). Moreover, injection of the Src inhibitor SU6656 significantly inhibited the prolongation of PGE2-induced hyperalgesia in DAMGO-induced priming 4 d later (Fig. 7C), indicating a role of Src in the expression of type II priming. Inhibition of other intracellular second messengers shown to play a role in nociceptor sensitization, PLCγ, caspase, PI3K, nitric oxide synthase, and intracellular calcium (Malik-Hall et al., 2005; Bogen et al., 2008; Joseph et al., 2008; Cunha et al., 2010; Alves et al., 2013; Alkhani et al., 2014; Khomula et al., 2014), did not affect the prolongation of PGE2 hyperalgesia observed after repeated injection of DAMGO (Fig. 8), ruling out their involvement in type II priming. The differential effect of the two inhibitors at 30 min for PGE2 hyperalgesia (Fig. 7A for PLC-β3 AS-ODN and Fig. 7B for SU6656) suggests that, at least in part, distinct mechanisms may be involved in the hyperalgesia induced by DAMGO and in the prolongation of PGE2 hyperalgesia in DAMGO-induced type II priming.
Figure 8.

Some second messengers not involved in type II priming. The groups of rats that had been treated with repeated (hourly, ×4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw 6 d before received, in a dose of 1 μg at the same site, injection of vehicle (control, black bars) or of the inhibitors of intracellular messengers wortmannin (PI3K), Z-VAD-FMK (caspase), U-73122 (PLCγ), l-NMMA (nitric oxide synthase), and quin-2 (calcium). Ten minutes later, PGE2 (100 ng) was injected and mechanical hyperalgesia was evaluated 30 min and 4 h after PGE2. In all groups, we observed that PGE2 induced significant hyperalgesia that was still present when evaluated 4 h after injection, ruling out the involvement of these second messengers in type II priming. n = 6 paws per group.
Type II priming involves changes in G-protein subunit signaling
The pronociceptive prostaglandin receptor at which PGE2 acts to produce mechanical hyperalgesia has been described as activating intracellular signaling pathways through a stimulatory G-protein (Gs) (Khasar et al., 2008). However, in type I priming, we observed a contribution of an additional, indirect pronociceptive signaling pathway involving activation of a pertussis toxin (PTX)-sensitive inhibitory G-protein (Gi) coupled to the A1 adenosine receptor (Ferrari et al., 2013a). Because MOR is also a Gi-coupled receptor, to evaluate a possible role of inhibitory G-proteins in the changes in the nociceptor produced by repeated injection of DAMGO, we first tested whether the Gi α subunit inhibitor PTX would prevent the induction of DAMGO hyperalgesia and the prolongation of PGE2 hyperalgesia after repeated DAMGO injection. We found that pretreatment with PTX does not affect the hyperalgesia produced by a fourth injection of DAMGO nor the prolongation of the hyperalgesia (i.e., still present at the fourth hour after injection) induced by injection of PGE2 4 d later (Fig. 9A). Moreover, the repeated activation of the Gi protein α subunit by administration of mastoparan did not induce priming (Fig. 9B), providing further support that Gi does not participate in the induction of type II priming.
Figure 9.
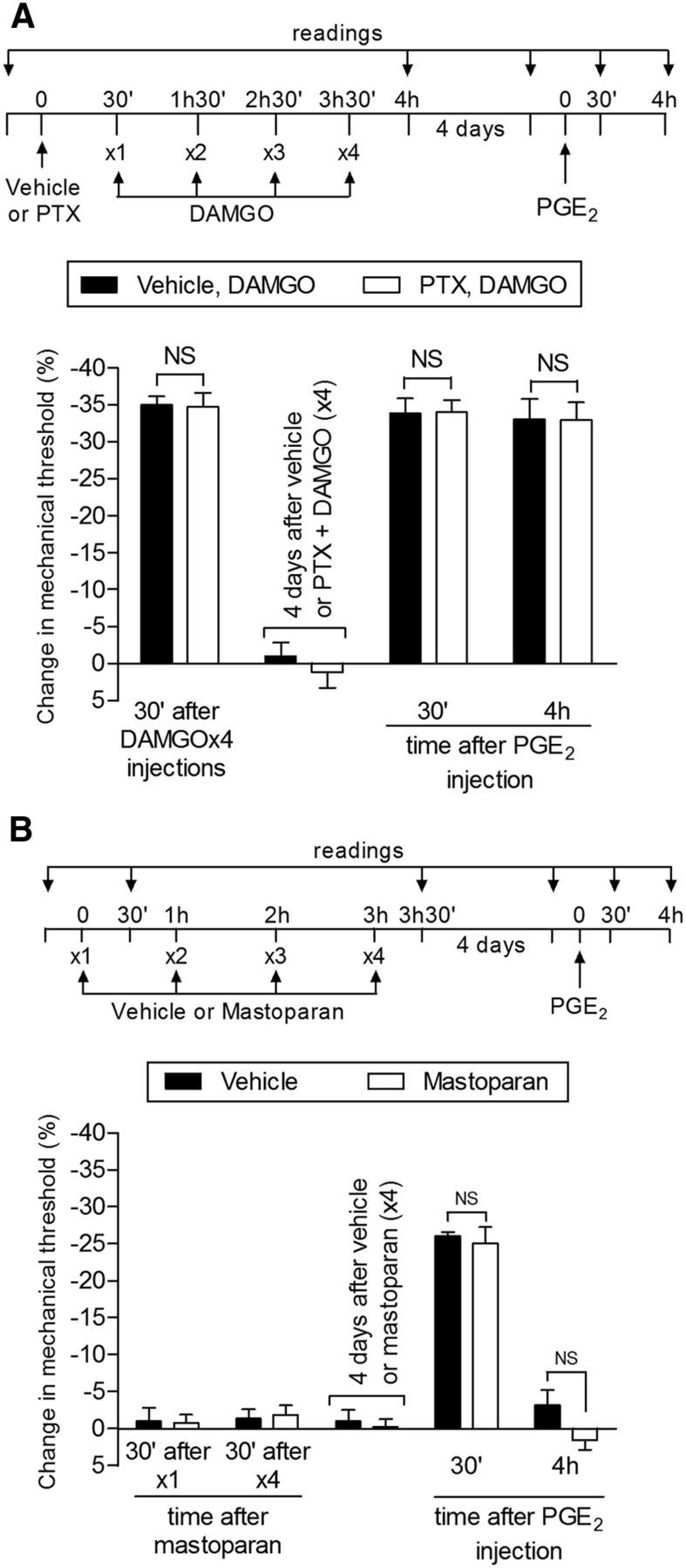
Role of the inhibitory G-protein αi subunit in type II priming. A, Rats received an intradermal injection of vehicle (control, black bars) or PTX (1 μg, white bars) on the dorsum of the hindpaw. Thirty minutes later, DAMGO (1 μg) was injected (hourly, ×4) in the same site. We observed significant mechanical hyperalgesia after the fourth injection of DAMGO, with no significant (NS) difference between groups (t(5) = 0.1075, p = 0.9186, control vs PTX groups, unpaired Student's t test), demonstrating that the αi subunit does not play a role in the hyperalgesia induced by repeated injection of DAMGO. Four days later, when the mechanical thresholds were not different from the prevehicle/PTX injection levels (t(5) = 0.3384; p = 0.7488 and t(5) = 0.5547; p = 0.6030, respectively, paired Student's t test), rats received intradermal injection of PGE2 (100 ng) in the same site and mechanical hyperalgesia was evaluated 30 min and 4 h later. No significant difference was observed in PGE2-induced mechanical hyperalgesia evaluated 30 min and 4 h after injection (F(1,20) = 0.17; p = 0.6859, when comparing both groups; NS, p > 0.05, two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the αi subunit does not play a role in the prolongation of PGE2 hyperalgesia in type II priming; B, Rats received repeated (hourly, ×4) intradermal injections of the G-protein αi subunit peptide activator mastoparan (1 μg) on the dorsum of the hindpaw. The mechanical nociceptive threshold was evaluated before and 30 min after the first and fourth administrations. Unpaired Student's t test showed that the injection of mastoparan did not induce significant change in the mechanical nociceptive threshold (p = 1.0000) even after 4 hourly administrations compared with the control group. Four days later, PGE2 (100 ng) was injected at the same site and mechanical thresholds were evaluated after 30 min and 4 h. In both groups, we observed significant mechanical hyperalgesia induced by PGE2 30 min after injection (NS, p > 0.05) that was no longer present at the fourth hour (NS; F(1,20) = 2.14; p = 0.1747, when comparing both groups at the fourth hour after PGE2 injection; NS, p > 0.05), indicating that an activator of G-protein αi subunit does not induce priming. n = 6 paws per group.
Therefore, we next tested whether the G-protein β/γ subunit plays a role in type II priming. Pretreatment (prevention protocol) with the G-protein β/γ subunit inhibitor gallein reversibly inhibited the expression of type II priming; that is, it attenuated both the hyperalgesia induced by a fourth injection of DAMGO and the prolongation of the hyperalgesia produced by injection of PGE2 4 d later (Fig. 10A, left), but this effect was temporary because, 8 d after gallein, the administration of PGE2 produced prolonged hyperalgesia (Fig. 10A, right). A similar effect of gallein was also observed in the inhibition protocol, in which the hyperalgesia induced by PGE2 in rats that had received repeated injections of DAMGO 4 d prior was markedly attenuated (Fig. 10B). This inhibitory effect of gallein on PGE2 hyperalgesia was already observed 30 min after PGE2 injection, compatible with the suggestion of a change in the signaling pathway activated by PGE2 because gallein in the naive rat does not affect the hyperalgesia induced by PGE2 (Fig. 10C). Repeated injection of the G-protein βγ peptide, an activator of G-protein β/γ, did not induce mechanical hyperalgesia after a fourth injection, nor did it produce prolongation of PGE2 hyperalgesia (Fig. 11), suggesting that activation of the β/γ subunit alone is insufficient to induce type II priming.
Figure 10.

Type II priming is dependent on the G-protein β/γ subunit. A, Rats were treated with intradermal injection of vehicle (control, black bars) or the G-protein β/γ inhibitor gallein (1 μg, white bars) on the dorsum of the hindpaw. Thirty minutes later, repeated injections (hourly, ×4) of DAMGO (1 μg) were performed at the same site and the mechanical nociceptive threshold was evaluated 30 min after the fourth administration. We observed significant mechanical hyperalgesia after the fourth injection of DAMGO in the group pretreated with vehicle, but not in the group pretreated with gallein (***p = 0.0001 when both groups are compared, paired Student's t test). Four days later, a time point when the mechanical thresholds were not different from preinjection levels (t(5) = 2.150; p = 0.0842, for the control group and t(5) = 1.530; p = 0.1852, for the gallein group; paired Student's t test), rats received intradermal injection of PGE2 (100 ng) at the same site and mechanical hyperalgesia was evaluated 30 min and 4 h later. Although in both groups PGE2-induced mechanical hyperalgesia was present 30 min after injection (NS, p > 0.05), in the gallein-treated group, it was markedly attenuated at the fourth hour (F(1,20) = 36.20, ***p < 0.0001, when control and gallein groups are compared at the fourth hour after PGE2 injection; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). However, when PGE2 was injected again 8 d later, it produced mechanical hyperalgesia that was still present 4 h after injection, indicating that the inhibition of the induction of type II priming by gallein was reversible; B, Rats that were treated with repeated hourly (×4) injections of DAMGO on the dorsum of the hindpaw received vehicle (control, black bars) or gallein (1 μg, white bars) 4 d later at the same site followed 10 min later by PGE2 (100 ng). Average mechanical nociceptive thresholds before and 4 d after treatment with DAMGO were 122.7 ± 0.8 g and 120.3 ± 1.4 g, respectively, for the control and the gallein group. Paired Student's t test showed no difference between these values (t(5) = 0.6670; p = 0.5343 for the control group and t(5) = 0.7559; p = 0.4838 for the gallein group). Evaluation of the mechanical nociceptive threshold 30 min and 4 h after PGE2 showed significant attenuation of the hyperalgesia induced by PGE2 at both time points in the group treated with gallein (F(1,12) = 83.65, ***p < 0.0001 when both groups are compared, two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that, in the setting of the neuroplasticity induced by repeated injections of DAMGO, PGE2-induced hyperalgesia depends on the G-protein β/γ subunit; C, Rats treated with intradermal injection of vehicle (5 μl, black bars) or gallein (1 μg, white bars) received PGE2 (100 ng) 30 min later at the same site. We observed significant mechanical hyperalgesia, evaluated 30 min after the injection of PGE2, in both groups (p > 0.05 control- vs gallein-treated groups, two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the β/γ subunit is not involved in the hyperalgesic effect of PGE2 in the naive rat. n = 6 paws per group.
Figure 11.

An activator of the G-protein β/γ subunit does not induce acute mechanical hyperalgesia or type II priming. Rats received repeated (hourly, ×4) intradermal injections of G-protein βγ peptide (1 μg; peptide activator of G-protein β/γ) on the dorsum of the hindpaw. The mechanical nociceptive threshold was evaluated before and 30 min after the first, third, and fourth administration. No significant difference in the mechanical threshold was observed 30 min after the first (p = 0.4838), third (p = 0.3632), or fourth (p = 0.1672) injection of G-protein βγ peptide compared with the preinjection level (paired Student's t test). One week later, PGE2 (100 ng) was injected at the same site and mechanical hyperalgesia was evaluated after 30 min and 4 h. The hyperalgesia induced by PGE2 was present 30 min after injection (***p < 0.0001 vs baseline; one-way repeated-measures ANOVA followed by Bonferroni post hoc test), but had disappeared 4 h later (NS, p > 0.05 vs baseline), indicating that repeated activation of G-protein β/γ does not induce type II priming. n = 6 paws per group.
Changes in PKA signaling in type II priming
It has been reported that chronic administration of MOR agonists lead to supersensitization of cAMP signaling (Sharma et al., 1975b; Brandt et al., 1976; Bie et al., 2005; Al-Hasani and Bruchas, 2011). Therefore, we next addressed which step in the signaling pathway acts to produce this prolongation; its heterotrimeric stimulatory G-protein (Dina et al., 2009) at the level of adenylyl cyclase (Taiwo and Levine, 1991; Parada et al., 2005) or PKA (Aley and Levine, 1999). To explore the role of the components of the adenylyl cyclase-cAMP-PKA second messenger pathway in the expression of type II hyperalgesic priming, we injected 8-bromo cAMP, a potent membrane-permeable cAMP analog that activates PKA, to evaluate whether this produces prolonged hyperalgesia in the setting of type II priming. If so, the administration of activators of more proximal steps in the pathway would add no additional information with respect to mechanism. We found that injection of 8-bromo cAMP produced prolonged mechanical hyperalgesia in MOR agonist-pretreated rats (Fig. 12A).
To determine whether the prolonged hyperalgesia is due to continued activation of PKA as opposed to activity of a target mechanism downstream of PKA, we administered, in rats previously treated with repeated injections of DAMGO the PKA inhibitor H-89 60 min after injection of 8-bromo cAMP, a time point when its hyperalgesic effect would no longer be present in a control animal (Taiwo et al., 1989; Taiwo and Levine, 1991). We observed that, at this time point, H-89 was able to reverse the prolonged hyperalgesia induced by 8-bromo cAMP (Fig. 12B), supporting the suggestion that there is a change in PKA signaling associated with type II priming that is responsible for the prolongation of PGE2-induced mechanical hyperalgesia.
Because we have observed that PKA has a role in DAMGO-induced neuroplasticity and considering reports of long-lasting sensitization of PKA signaling as a result of proteasome-mediated degradation of the regulatory subunit of PKA (Greenberg et al., 1987; Chain et al., 1999; Moss et al., 2002), we investigated whether treatment with a proteasome inhibitor, MG-132, would affect the induction of type II priming by repeated injection of DAMGO. MG-132 did not, however, have any effect on DAMGO-induced priming (Fig. 13).
Figure 13.

Proteasome inhibitor does not affect type II priming. A, Rats were treated with intradermal injection of the proteasome inhibitor MG-132 (1 μg) on the dorsum of the hindpaw. Thirty minutes later, repeated (hourly, ×4) injections of DAMGO (1 μg) were performed at the same site and the mechanical nociceptive threshold was evaluated 30 min after the fourth administration. Significant mechanical hyperalgesia was observed after the fourth injection of DAMGO in the group pretreated with MG-132 compared with baseline (**p = 0.0069, paired Student's t test). B, A different group of rats was treated with intradermal injection of MG-132 (1 μg) and, after 30 min, repeated (hourly, ×3) injections of DAMGO (1 μg) were performed at the same site. One hour after the third injection of DAMGO, PGE2 (100 ng) was injected and mechanical hyperalgesia was evaluated 30 min and 4 h later. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test showed no difference between the groups in PGE2-induced mechanical hyperalgesia (F(1,8) = 1.04; p > 0.05, when comparing both groups; NS, p = 0.3658), indicating that the proteasome system does not play a role in the prolongation of the hyperalgesia induced by PGE2 observed in type II priming. n = 6 paws per group.
Discussion
Repeated administration of DAMGO produces a long-lasting change in nociceptor function such that subsequent doses produce mechanical hyperalgesia (Aley et al., 1995). Further, the administration of PGE2 as soon as 1 h after 3 doses of DAMGO produced markedly prolonged hyperalgesia, similar to PGE2 hyperalgesia in the setting of type I, hyperalgesic priming (Joseph et al., 2010). This effect of a subsequent dose of DAMGO or a dose of PGE2 was still present without attenuation for >1 month. None of the several second messenger inhibitors tested, including those for second messengers implicated in type I priming, prevented the development of the neuroplasticity triggered by the repeated injection of DAMGO.
The involvement of the αi G-protein subunit was considered with respect to the expression of type II priming. Although PTX, an inhibitor of αi, inhibited the prolongation of PGE2 hyperalgesia in the setting of type I hyperalgesic priming (Ferrari et al., 2013a), it failed to inhibit DAMGO hyperalgesia or the prolongation of PGE2 hyperalgesia in type II priming. These findings support the suggestion that, for changes at a very proximal step in signal transduction, the mechanism of DAMGO-induced type II priming can already be distinguished from that for type I hyperalgesic priming. In addition, unlike type I priming, the prolongation of PGE2-induced hyperalgesia in rats pretreated with DAMGO is not attenuated by the PKCε inhibitor PKCεV1-2. To determine which second messengers mediate the induction of type II priming, we evaluated the role of the adenylyl cyclase-cAMP-PKA second messenger pathway, which has been implicated in nociceptor sensitization (Aley and Levine, 1999), in DAMGO-induced hyperalgesia and prolongation of PGE2 hyperalgesia. We found that pretreatment with the PKA inhibitor H-89 attenuated DAMGO hyperalgesia and the prolongation of PGE2 hyperalgesia. However, when PGE2 was injected again 30 d later, it induced prolonged hyperalgesia. These results support a role of the adenylyl cyclase-cAMP-PKA pathway in the expression of type II priming. In addition, because we have shown previously that the Src inhibitor SU6656 and antisense to PLC-β3 inhibited PKA-dependent prolongation of PGE2 hyperalgesia in rats primed by the transient reduction of GRK2 (Ferrari et al., 2012), we evaluated their effect on the hyperalgesia induced by injection of DAMGO or PGE2 in the setting of DAMGO-induced priming. Whereas in rats submitted to repeated injection of DAMGO, SU6656 and antisense to PLC-β3 both also inhibited the prolongation of PGE2-induced hyperalgesia, but only the PLC-β3 antisense inhibited DAMGO hyperalgesia. The interaction of these second messengers in type II priming remains to be determined.
Supersensitization in the adenylyl cyclase-cAMP-PKA signaling pathway has been demonstrated in the setting of opioid dependence (Sharma et al., 1975a; Brandt et al., 1976; Al-Hasani and Bruchas, 2011). To determine whether there is supersensitization in this signaling pathway, we directly activated PKA, in DAMGO-pretreated rats, by intradermal administration of 8-bromo cAMP. In DAMGO-primed animals, 8-bromo cAMP induced prolonged mechanical hyperalgesia, supporting the suggestion that PKA signaling, or a downstream target of PKA, contributes to the prolongation of hyperalgesia. To determine whether a change in function at the level of PKA could alone account for the prolongation of hyperalgesia, we administered the PKA inhibitor during the prolongation phase of 8-bromo cAMP-induced hyperalgesia. That 8-bromo cAMP induces prolonged hyperalgesia and H-89 markedly attenuates the prolongation phase of 8-bromo cAMP-induced hyperalgesia supports the suggestion that a change in signaling by PKA contributes to the prolongation of 8-bromo cAMP in type II priming. Although it is known that the repeated activation of MOR can lead to supersensitization of adenylyl cyclase (Sharma et al., 1975a; Brandt et al., 1976; Al-Hasani and Bruchas, 2011), the findings of the present study are, to the best of our knowledge, the first evidence that it can also induce supersensitization at the level of PKA.
We next addressed possible mechanisms for the supersensitization of PKA. It has been suggested that proteasome-mediated degradation of the regulatory subunit of PKA can produce a long-lasting sensitization of PKA signaling such that even a low concentration of cAMP can induce PKA activation (Yang et al., 2008; Myeku et al., 2012). However, intradermal injection of the proteasome inhibitor MG132 did not prevent DAMGO-induced priming. Alternative mechanisms include autophosphorylation of PKA (Yonemoto et al., 1993; Cauthron et al., 1998; Cheng et al., 1998) or involvement of other elements of its signaling pathway, for example, GRK2 (Penela et al., 1998; Ferrari et al., 2012). We showed recently that a transient decrease in GRK2 also produces a form of hyperalgesic priming that is not inhibited by a selective PKCε inhibitor, which inhibits type I priming (Ferrari et al., 2012), and is not reversed by cordycepin, which permanently reverses type I priming (Ferrari et al., 2013b). Like DAMGO-induced priming, however, this form of priming is inhibited by a selective PKA inhibitor. GRK2 is a β/γ subunit effector (Willets et al., 2003; Smrcka, 2008) and β/γ signaling can be blocked by GRK2 (Smrcka, 2008; Casey et al., 2010). Exactly how signaling between inhibiting β/γ and GRK2 could contribute remains to be established.
In our previous study of the priming induced by the transient attenuation of GRK2 using a large panel of second messenger inhibitors, the only other inhibitors (in addition to that for PKA) that attenuated the prolongation of PGE2 hyperalgesia were an Src inhibitor, SU6656, and PLC-β3 antisense (Ferrari et al., 2012). SU6656 also attenuated the prolongation of PGE2 hyperalgesia in type II priming, without affecting DAMGO hyperalgesia. Although it is known that β/γ subunits signal via Src (Luttrell et al., 1997), the pathway connecting PKA and Src in the expression of type II priming remains to be established. However, in our prior study of priming induced by a transient decrease in GRK2, we did observe that the coadministration of a PKA and Src inhibitors produced no greater reversal of hyperalgesia than either alone (Ferrari et al., 2012), compatible with a shared signaling pathway in which Src is downstream of PKA (Burmeister et al., 2012; Ickowicz et al., 2012).
Although type II and I priming are both dependent on Gi-protein coupled receptor signaling, each uses this G-protein complex differently. In type I priming, receptors that activate PKCε produce a change in adenosine A1 receptor signaling, such that the A1 receptor agonist now induces hyperalgesia (Ferrari et al., 2013a). Although this receptor still activates αi to mediate CPA-induced hyperalgesia, it now signals to activate PKCε rather than inhibit AC-PKA. In contrast, in type II priming, repeated MOR activation produces a state in which the activation of these receptors leads to prolongation of PGE2-induced mechanical hyperalgesia that is β/γ and PKA dependent. Although it is currently unknown to what downstream molecule the MOR signals to induce type II hyperalgesic priming, it is known that the β/γ subunit does signal bidirectionally to GRK2 (Smrcka, 2008) and that transient attenuation of GRK2 produces type II-like priming (Ferrari et al., 2012).
Although type I priming is mediated by IB4+ nociceptors (Joseph and Levine, 2010a), type II priming is mediated by IB4− nociceptors, as indicated by our finding that the prolongation of PGE2-induced hyperalgesia produced by previous repeated injection of DAMGO was not attenuated by IB4-saporin. In fact, DAMGO-induced hyperalgesia was significantly increased in IB4-saporin treated rats, which may suggest an interaction between the different subtypes of nociceptors in the expression of type II priming. In addition, different from type I priming induced by PKCε (Ferrari et al., 2015), type II priming can be induced in both males and females. This is consistent with animal studies (Holtman and Wala, 2007; Elhabazi et al., 2014) and clinical reports (Konopka and van Wijhe, 2010; Schneider and Kirsh, 2010; Akarsu et al., 2012) showing that chronic opioid exposure can produce hyperalgesia as well as tolerance, two conditions that can overlap and be difficult to separate in clinical circumstances (Bekhit, 2010; Chu et al., 2012; Angst, 2015), in both sexes (Konopka and van Wijhe, 2010; Schneider and Kirsh, 2010; Edwards et al., 2011).
Finally, although it has been suggested that opioid-induced hyperalgesia may be due to onset of withdrawal, between opioid doses (Mercadante and Arcuri, 2005; Chu et al., 2008; Chen et al., 2009; Hay et al., 2009; Lee et al., 2011), our finding that the administration of a fourth dose of DAMGO induces a rapid onset mechanical hyperalgesia that was significant by 5 min after injection supports a switch in MOR signaling from an ability to reverse cAMP-dependent hyperalgesia to induction of hyperalgesia. Although the role of type II priming in chronic pain syndromes remains to be established, inflammation can induce a reversible decrease in GRK2 (Kleibeuker et al., 2007; Eijkelkamp et al., 2010), suggesting a possible role in the transition to chronic pain associated with inflammatory diseases (Ferrari et al., 2012). How one might reverse (treat) a chronic pain condition mediated by type II hyperalgesic priming remains a key question.
Footnotes
This work was funded by the National Institutes of Health (Grant NS084545).
The authors declare no competing financial interests.
References
- Akarsu M, Tuncer S, Reisli R, Otelcioğlu S. The role of magnesium in preventing postoperative hyperalgesia [article in Turkish] Agri. 2012;24:15–22. doi: 10.5505/agri.2012.07078. [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD. Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J Neurosci. 1997a;17:3907–3912. doi: 10.1523/JNEUROSCI.17-10-03907.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Levine JD. Multiple receptors involved in peripheral alpha 2, mu, and A1 antinociception, tolerance, and withdrawal. J Neurosci. 1997b;17:735–744. doi: 10.1523/JNEUROSCI.17-02-00735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Green PG, Levine JD. Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J Neurosci. 1995;15:8031–8038. doi: 10.1523/JNEUROSCI.15-12-08031.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhani H, Ase AR, Grant R, O'Donnell D, Groschner K, Seguela P. Contribution of TRPC3 to store-operated calcium entry and inflammatory transductions in primary nociceptors. Mol Pain. 2014;10:43. doi: 10.1186/1744-8069-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves DP, da Motta PG, Romero TR, Klein A, Duarte ID. NO/cGMP production is important for the endogenous peripheral control of hyperalgesia during inflammation. Nitric Oxide. 2013;28:8–13. doi: 10.1016/j.niox.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Angst MS. Intraoperative use of remifentanil for tiva: postoperative pain, acute tolerance, and opioid-induced hyperalgesia. J Cardiothorac Vasc Anesthe. 2015;29:S16–S22. doi: 10.1053/j.jvca.2015.01.026. [DOI] [PubMed] [Google Scholar]
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- Bekhit MH. Opioid-induced hyperalgesia and tolerance. Am J Ther. 2010;17:498–510. doi: 10.1097/MJT.0b013e3181ed83a0. [DOI] [PubMed] [Google Scholar]
- Bie B, Peng Y, Zhang Y, Pan ZZ. cAMP-mediated mechanisms for pain sensitization during opioid withdrawal. J Neurosci. 2005;25:3824–3832. doi: 10.1523/JNEUROSCI.5010-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen O, Joseph EK, Chen X, Levine JD. GDNF hyperalgesia is mediated by PLCgamma, MAPK/ERK, PI3K, CDK5 and Src family kinase signaling and dependent on the IB4-binding protein versican. Eur J Neurosci. 2008;28:12–19. doi: 10.1111/j.1460-9568.2008.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci. 2012;32:2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217:252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- Brandt M, Gullis RJ, Fischer K, Buchen C, Hamprecht B, Moröder L, Wunsch E. Enkephalin regulates the levels of cyclic nucleotides in neuroblastoma x glioma hybrid cells. Nature. 1976;262:311–313. doi: 10.1038/262311a0. [DOI] [PubMed] [Google Scholar]
- Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84:6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister BT, Taglieri DM, Wang L, Carnegie GK. Src homology 2 domain-containing phosphatase 2 (Shp2) is a component of the A-kinase-anchoring protein (AKAP)-Lbc complex and is inhibited by protein kinase A (PKA) under pathological hypertrophic conditions in the heart. J Biol Chem. 2012;287:40535–40546. doi: 10.1074/jbc.M112.385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CM, Smrcka AV, Blaxall BC. Small molecule disruption of G beta gamma signaling inhibits the progression of heart failure. Circ Res. 2010;107:532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauthron RD, Carter KB, Liauw S, Steinberg RA. Physiological phosphorylation of protein kinase A at Thr-197 is by a protein kinase A kinase. Mol Cell Biol. 1998;18:1416–1423. doi: 10.1128/mcb.18.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/S0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- Chen L, Malarick C, Seefeld L, Wang S, Houghton M, Mao J. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain. 2009;143:65–70. doi: 10.1016/j.pain.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ. Ca2+/calmodulin-dependent protein kinase II alpha is required for the initiation and maintenance of opioid-induced hyperalgesia. J Neurosci. 2010;30:38–46. doi: 10.1523/JNEUROSCI.4346-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Ma Y, Moore M, Hemmings BA, Taylor SS. Phosphorylation and activation of cAMP-dependent protein kinase by phosphoinositide-dependent protein kinase. Proc Natl Acad Sci U S A. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24:479–496. doi: 10.1097/AJP.0b013e31816b2f43. [DOI] [PubMed] [Google Scholar]
- Chu LF, D'Arcy N, Brady C, Zamora AK, Young CA, Kim JE, Clemenson AM, Angst MS, Clark JD. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: a double-blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain. 2012;153:1583–1592. doi: 10.1016/j.pain.2012.02.028. [DOI] [PubMed] [Google Scholar]
- Cunha TM, Talbot J, Pinto LG, Vieira SM, Souza GR, Guerrero AT, Sonego F, Verri WA, Jr, Zamboni DS, Ferreira SH, Cunha FQ. Caspase-1 is involved in the genesis of inflammatory hypernociception by contributing to peripheral IL-1beta maturation. Mol Pain. 2010;6:63. doi: 10.1186/1744-8069-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160:501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RR, Wasan AD, Michna E, Greenbaum S, Ross E, Jamison RN. Elevated pain sensitivity in chronic pain patients at risk for opioid misuse. J Pain. 2011;12:953–963. doi: 10.1016/j.jpain.2011.02.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Heijnen CJ, Willemen HL, Deumens R, Joosten EA, Kleibeuker W, den Hartog IJ, van Velthoven CT, Nijboer C, Nassar MA, Dorn GW, 2nd, Wood JN, Kavelaars A. GRK2: a novel cell-specific regulator of severity and duration of inflammatory pain. J Neurosci. 2010;30:2138–2149. doi: 10.1523/JNEUROSCI.5752-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhabazi K, Ayachi S, Ilien B, Simonin F. Assessment of morphine-induced hyperalgesia and analgesic tolerance in mice using thermal and mechanical nociceptive modalities. J Vis Exp. 2014;89:e51264. doi: 10.3791/51264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Levine JD. Plasma membrane mechanisms in a preclinical rat model of chronic pain. J Pain. 2015;16:60–66. doi: 10.1016/j.jpain.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165:896–901. doi: 10.1016/j.neuroscience.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Levine E, Levine JD. Role of a novel nociceptor autocrine mechanism in chronic pain. Eur J Neurosci. 2013a;37:1705–1713. doi: 10.1111/ejn.12145. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013b;14:731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15:312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Reichling DB, Levine JD. Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci. 2015;35:495–507. doi: 10.1523/JNEUROSCI.5147-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Castellucci VF, Bayley H, Schwartz JH. A molecular mechanism for long-term sensitization in Aplysia. Nature. 1987;329:62–65. doi: 10.1038/329062a0. [DOI] [PubMed] [Google Scholar]
- Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. 2009;10:316–322. doi: 10.1016/j.jpain.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Holtman JR, Jr, Wala EP. Characterization of the antinociceptive and pronociceptive effects of methadone in rats. Anesthesiology. 2007;106:563–571. doi: 10.1097/00000542-200703000-00022. [DOI] [PubMed] [Google Scholar]
- Ickowicz D, Finkelstein M, Breitbart H. Mechanism of sperm capacitation and the acrosome reaction: role of protein kinases. Asian J Androl. 2012;14:816–821. doi: 10.1038/aja.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010a;169:431–435. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Multiple PKCepsilon-dependent mechanisms mediating mechanical hyperalgesia. Pain. 2010b;150:17–21. doi: 10.1016/j.pain.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Parada CA, Levine JD. Hyperalgesic priming in the rat demonstrates marked sexual dimorphism. Pain. 2003;105:143–150. doi: 10.1016/S0304-3959(03)00175-1. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Bogen O, Alessandri-Haber N, Levine JD. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain. 2008;9:463–472. doi: 10.1016/j.jpain.2008.01.335. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30:4660–4666. doi: 10.1523/JNEUROSCI.5530-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/S0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–5730. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khomula EV, Borisyuk AL, Viatchenko-Karpinski VY, Briede A, Belan PV, Voitenko NV. Nociceptive neurons differentially express fast and slow T-type Ca(2)(+) currents in different types of diabetic neuropathy. Neural Plast. 2014;2014:938235. doi: 10.1155/2014/938235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleibeuker W, Ledeboer A, Eijkelkamp N, Watkins LR, Maier SF, Zijlstra J, Heijnen CJ, Kavelaars A. A role for G protein-coupled receptor kinase 2 in mechanical allodynia. Eur J Neurosci. 2007;25:1696–1704. doi: 10.1111/j.1460-9568.2007.05423.x. [DOI] [PubMed] [Google Scholar]
- Kolesnikov YA, Jain S, Wilson R, Pasternak GW. Peripheral morphine analgesia: synergy with central sites and a target of morphine tolerance. J Pharmacol Exp Ther. 1996;279:502–506. [PubMed] [Google Scholar]
- Konopka KH, van Wijhe M. Opioid-induced hyperalgesia: pain hurts? Br J Anaesth. 2010;105:555–557. doi: 10.1093/bja/aeq286. [DOI] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14:145–161. [PubMed] [Google Scholar]
- Levine JD, Taiwo YO. Involvement of the mu-opiate receptor in peripheral analgesia. Neuroscience. 1989;32:571–575. doi: 10.1016/0306-4522(89)90279-0. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Della Rocca GJ, van Biesen T, Luttrell DK, Lefkowitz RJ. Gbetagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor: a scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem. 1997;272:4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- Malik-Hall M, Dina OA, Levine JD. Primary afferent nociceptor mechanisms mediating NGF-induced mechanical hyperalgesia. Eur J Neurosci. 2005;21:3387–3394. doi: 10.1111/j.1460-9568.2005.04173.x. [DOI] [PubMed] [Google Scholar]
- Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100:213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Arcuri E. Hyperalgesia and opioid switching. Am J Hosp Palliat Care. 2005;22:291–294. doi: 10.1177/104990910502200411. [DOI] [PubMed] [Google Scholar]
- Mestre C, Pélissier T, Fialip J, Wilcox G, Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods. 1994;32:197–200. doi: 10.1016/1056-8719(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Moss A, Blackburn-Munro G, Garry EM, Blakemore JA, Dickinson T, Rosie R, Mitchell R, Fleetwood-Walker SM. A role of the ubiquitin-proteasome system in neuropathic pain. J Neurosci. 2002;22:1363–1372. doi: 10.1523/JNEUROSCI.22-04-01363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Wang H, Figueiredo-Pereira ME. cAMP stimulates the ubiquitin/proteasome pathway in rat spinal cord neurons. Neurosci Lett. 2012;527:126–131. doi: 10.1016/j.neulet.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi J, Sasaki K, Seki S, Chancellor MB, Erickson KA, de Groat WC, Kumon H, Yoshimura N. Effects of isolectin B4-conjugated saporin, a targeting cytotoxin, on bladder overactivity induced by bladder irritation. Eur J Neurosci. 2004;20:474–482. doi: 10.1111/j.1460-9568.2004.03508.x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003a;120:219–226. doi: 10.1016/S0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003b;17:1847–1852. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Penela P, Ruiz-Gómez A, Castaño JG, Mayor F., Jr Degradation of the G protein-coupled receptor kinase 2 by the proteasome pathway. J Biol Chem. 1998;273:35238–35244. doi: 10.1074/jbc.273.52.35238. [DOI] [PubMed] [Google Scholar]
- Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J. Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res. 2012;1472:38–44. doi: 10.1016/j.brainres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JP, Kirsh KL. Defining clinical issues around tolerance, hyperalgesia, and addiction: a quantitative and qualitative outcome study of long-term opioid dosing in a chronic pain practice. J Opioid Manag. 2010;6:385–395. doi: 10.5055/jom.2010.0036. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci U S A. 1975a;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Nirenberg M, Klee WA. Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci U S A. 1975b;72:590–594. doi: 10.1073/pnas.72.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV. G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Wang YQ, Wu GC. Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull. 2009;78:335–341. doi: 10.1016/j.brainresbull.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011;12:120. doi: 10.1186/1471-2202-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91:545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492:397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience. 1990;38:757–762. doi: 10.1016/0306-4522(90)90068-F. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-M. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R, Honda CN. Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience. 2001;108:143–155. doi: 10.1016/S0306-4522(01)00377-3. [DOI] [PubMed] [Google Scholar]
- Willets JM, Challiss RA, Nahorski SR. Non-visual GRKs: are we seeing the whole picture? Trends Pharmacol Sci. 2003;24:626–633. doi: 10.1016/j.tips.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang S, Sung B, Lim G, Mao J. Morphine induces ubiquitin-proteasome activity and glutamate transporter degradation. J Biol Chem. 2008;283:21703–21713. doi: 10.1074/jbc.M800809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemoto W, Garrod SM, Bell SM, Taylor SS. Identification of phosphorylation sites in the recombinant catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1993;268:18626–18632. [PubMed] [Google Scholar]