

Abstract
Patients with inflammatory bowel disease are at increased risk for colon cancer due to augmented oxidative stress. These patients also have compromised antioxidant defenses as the result of nutritional deficiencies. The micronutrient selenium is essential for selenoprotein production and is transported from the liver to target tissues via selenoprotein P (SEPP1). Target tissues also produce SEPP1, which is thought to possess an endogenous antioxidant function. Here, we have shown that mice with Sepp1 haploinsufficiency or mutations that disrupt either the selenium transport or the enzymatic domain of SEPP1 exhibit increased colitis-associated carcinogenesis as the result of increased genomic instability and promotion of a protumorigenic microenvironment. Reduced SEPP1 function markedly increased M2-polarized macrophages, indicating a role for SEPP1 in macrophage polarization and immune function. Furthermore, compared with partial loss, complete loss of SEPP1 substantially reduced tumor burden, in part due to increased apoptosis. Using intestinal organoid cultures, we found that, compared with those from WT animals, Sepp1-null cultures display increased stem cell characteristics that are coupled with increased ROS production, DNA damage, proliferation, decreased cell survival, and modulation of WNT signaling in response to H2O2-mediated oxidative stress. Together, these data demonstrate that SEPP1 influences inflammatory tumorigenesis by affecting genomic stability, the inflammatory microenvironment, and epithelial stem cell functions.
Introduction
Inflammatory bowel disease (IBD) primarily comprises two types of chronic inflammatory disorders of the intestine: Crohn’s disease and ulcerative colitis. IBD affects approximately 1.4 million Americans, with typical onset occurring at between 15 and 30 years of age (1). While the etiology of IBD is incompletely understood, prevailing thought presents a model in which the interplay of (a) intestinal microbiota or other undefined environmental exposures, (b) genetic susceptibility, and (c) inappropriately sustained and severe autoimmune inflammatory responses leads to repetitive injury of the GI tract, resulting in a protumorigenic microenvironment rich in ROS and reactive nitrogen species that predisposes to colon cancer (2, 3). Colitis-associated cancer (CAC) risk in IBD is influenced by several factors, including age of diagnosis, disease extent, and severity of inflammation (4–8). Importantly, as increased disease activity is associated with increased cancer risk, understanding modifiers of IBD severity and CAC risk factors is of paramount importance.
Selenium is a trace element that is specifically incorporated into selenoproteins as selenocysteine through a specialized UGA codon encoded in the mRNA. Selenium deficiency results in stimulation of the WNT and NRF2 pathways (9–11), which are both heavily implicated in the pathogenesis of colon cancer (12–14). Selenoproteins are enzymes that participate in oxidative defense and are thought to protect against colon cancer, especially in settings of high oxidant stress, such as chronic inflammatory states. Several epidemiological studies have inversely correlated nutritional selenium status and cancer risk, particularly in colon cancer (15). Recently, our laboratory has demonstrated that selenium deficiency in mice exacerbates intestinal injury in response to chemical models of colitis and increases tumorigenesis in CAC modeling (16).
Selenoprotein P (SEPP1) is a major selenoprotein and is thought to serve two main roles: supplying tissues with selenium and acting in an antioxidant defense capacity. The transport role is carried out by the C-terminal domain of SEPP1, which contains 9 selenocysteine residues (17, 18). The antioxidant role is carried out by a single selenocysteine within a UXXC redox motif at amino terminal residues 40–43 (19), a domain that has recently been shown to have peroxidase activity (20). As selenoproteins are produced in a hierarchical manner based on selenium availability, the importance of SEPP1 is underscored by the fact that, even under conditions of severe selenium deficiency, its production perseveres (19). Previous studies have shown that SEPP1 is downregulated in colorectal cancers (CRCs) (21) and SNPs within the gene are associated with advanced adenoma risk (22). Because SEPP1 serves as the major selenium transport protein, the known effects of selenium on WNT and NRF2 signaling in target tissues may partially depend on SEPP1. The antioxidant function of SEPP1 also suggests that it could play a role in cancer prevention, particularly in the context of inflammatory cancers characterized by increased oxidative stress, such as CAC (23). Indeed, the gastrointestinal tract is susceptible to oxidative damage resulting from direct contact of the colonic epithelium with microbial and food-derived ROS, and the loss of SEPP1 might result in increased oxidative damage–induced tumor initiation in the colonic mucosa. This potential damage is augmented in the context of IBD, during which oxidative stress plays a major role (23). Thus, SEPP1 may regulate stem cell pathways and protect from CAC.
In the present study, we investigated the role of SEPP1 in colonic epithelial biology and how its loss affects colitis-associated carcinoma. We demonstrate that SEPP1 loss contributes to increases in colonic epithelial stem cell properties and modulation of WNT tone and, in response to oxidative stress, decreases cell survival and increases oxidative DNA damage in intestinal organoids. These SEPP1-dependent phenotypes ultimately contribute to an increase in inflammatory tumorigenesis with Sepp1 heterozygosity or upon loss of either the redox-active site or the selenium transport domain of SEPP1. SEPP1 manipulation was found to alter the tumor microenvironment and genomic integrity. Our findings establish a critical role for SEPP1 in intestinal biology, homeostasis, injury response, preservation of genomic integrity, and inflammatory carcinogenesis.
Results
Loss of SEPP1 enhances both tumor promotion and initiation.
Because SEPP1 mRNA expression is decreased as early as the adenoma stage in CRC (ref. 24 and Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI76099DS1) and in tumors of mice subjected to an inflammatory carcinogenesis protocol (Supplemental Figure 1B), we predicted that SEPP1 loss would enhance tumorigenesis. In order to test the impact of SEPP1 loss on tumor initiation and promotion, respectively, we used both a 6-month azoxymethane (AOM) protocol (Figure 1A) and a chronic dextran sodium sulfate (DSS) administration protocol (Figure 1B). AOM is a procarcinogen that is metabolically activated to a potent alkylating agent forming O6-methyl-guanine (25). DSS serves as a tumor-promoting agent, and repeated DSS administration produces chronic colonic inflammation (26). To determine whether SEPP1 deletion influences colonic tumorigenesis, we used Sepp1–/– mice. These mice are globally null for SEPP1, and a lack of expression could be seen in both the Sepp1–/– colon and small intestine as compared with WT mice (Figure 1C). Methylene blue staining of colons 6 months after AOM administration revealed an increase in aberrant crypt foci (ACF) in Sepp1–/– mice compared with that in WT mice (6.0 ± 0.6 vs. 2.5 ± 0.3 ACF per colon, P < 0.001; Figure 1D), indicating a role for SEPP1 loss in tumor initiation. Endoscopic injury analysis of mice subjected to chronic DSS administration using the murine endoscopic index of colitis severity (MEICS) scoring system (27) revealed increased injury in response to DSS after 3 cycles (8.1 ± 0.4 vs. 4.1 ± 0.5 MEICS score, P < 0.0001; Figure 1E). Furthermore, chronic DSS treatment led to increased ACF formation in Sepp1–/– mice compared with that in both WT and Sepp1+/– mice (Sepp1–/–, 3.2 ± 0.4 in tumors per colon vs. Sepp1+/–, 2.1 ± 0.3, and WT, 1.9 ± 0.3 tumors per colon, P < 0.05; Figure 1E), suggesting that complete SEPP1 loss exacerbates injury and tumorigenesis in response to DSS. In addition, Sepp1–/– mice demonstrated decreased proliferation after chronic DSS treatment in comparison with that of Sepp1+/– and WT mice (Figure 1E), with no differences in apoptosis (data not shown) among the cohorts.
Figure 1. Absence of SEPP1 exacerbates tumorigenesis in response to AOM and injury after chronic DSS treatment.

(A) Schematic of the AOM protocol used. Mice were injected with AOM and aged for 6 months before being sacrificed on day 180 after AOM treatment. (B) Schematic of the chronic DSS protocol used. Mice were subjected to three 5-day cycles of 3% DSS ad libitum. There were 16 days of recovery between each DSS administration, and mice were monitored for injury by endoscopy (black circles) 4 days after each DSS cycle (gray rectangles). (C) Immunofluorescent staining of SEPP1 (red) within the colons and small intestines of WT and Sepp1–/– mice (original magnification, ×100). (D) ACF counts per mouse in WT (n = 4), Sepp1+/– (n = 4), and Sepp1–/– (n = 3) mice subjected to the AOM protocol. (E) Endoscopic colitis score, ACF counts, and Ki67 staining in mice subjected to the chronic DSS protocol (7, WT; 10, Sepp1+/–; 10, Sepp1–/–). *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test.
SEPP1 is a haploinsufficient tumor suppressor in inflammatory carcinogenesis.
Based on the impact of SEPP1 loss on initiation in response to AOM and DSS as single modalities, we predicted that SEPP1 would play a critical role in ensuring tumor cell survival after inflammatory injury. We used a CAC model in which WT, Sepp1+/–, and Sepp1–/– mice received AOM and repeated-dose DSS (AOM/DSS). As expected, complete loss of SEPP1 decreased colonic selenium content (Figure 2A), though we noted that this decrease was modest, indicating that SEPP1 is not the only mechanism of selenium supply in the colon. Complete loss of SEPP1 led to a decrease in tumor multiplicity (2.7 ± 0.7 in Sepp1–/– vs. 5.2 ± 0.8 tumors per mouse in WT, P = 0.02; Figure 2, B and C) and alteration in tumor size in Sepp1–/– mice compared with that in Sepp1+/– mice but not WT mice (2.5 ± 0.4 in Sepp1–/– vs. 4.2 ± 0.5 mm2 in Sepp1+/–, P = 0.01; Figure 2, B and D), suggesting that complete loss of SEPP1 impaired survival or proliferation of initiated epithelial cells. In contrast, partial loss of SEPP1 led to increased tumor multiplicity (8.3 ± 1.2 in Sepp1+/– vs. 5.2 ± 0.8 tumors per mouse in WT, P = 0.03; Figure 2, B and C) but no change in tumor size. However, histopathological examination suggested more advanced dysplasia in the Sepp1+/– mice (Figure 2E).
Figure 2. Sepp1 haploinsufficiency augments inflammatory carcinogenesis.

(A) Quantification of colonic selenium in WT (n = 8), Sepp1+/– (n = 5), and Sepp1–/– (n = 8) mice fed a selenium-supplemented (1.0 PPM) diet. (B) Representative gross colon images. (C) Tumor number (tumors per mouse) and (D) average tumor size (mm2) per mouse (17, WT; 15, Sepp1+/–; 20, Sepp1–/–). (E) Representative images of Swiss-rolled colons from each genotype (original magnification, ×25). Scale bar: 500 μm. *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test.
The decrease in tumorigenesis in the Sepp1–/– mice was concomitant with increased apoptosis, as measured by TUNEL staining (74.4 ± 10.9 vs. 34.3 ± 3.1 TUNEL+ cells per tumor high-power field [HPF] in WT, P < 0.01; Figure 3A, left, and Supplemental Figure 2, A and B, top) and Western blotting for markers within the intrinsic apoptosis pathway (Figure 3A and Supplemental Figure 2B, bottom). Therefore, we hypothesized that decreased tumor multiplicity might be caused in part by clearance of initiated cells due to oxidative injury beyond that which total SEPP1 loss can compensate. This phenomenon has been hypothesized to play a role in the initiation process of many cancers and was recently demonstrated in the process of ferroptosis, a mechanism of cell death triggered via the production of iron-dependent ROS (28, 29). Moreover, proliferation was increased in Sepp1+/– tumors (85.9 ± 2.9 vs. 55.6 ± 2.8 Ki67+ cells per tumor HPF in WT, P < 0.0001; Figure 3B and Supplemental Figure 2D) and decreased in Sepp1–/– tumors (19.0 ± 6.9 vs. 55.6 ± 2.8 Ki67+ cells per tumor HPF in WT, P < 0.0001; Figure 3B and Supplemental Figure 2D) and crypts (1.6 ± 0.2 vs. 4.6 ± 0.2 Ki67+ cells per crypt in WT, P < 0.0001; Supplemental Figure 2C). These data support a role for clearance of initiated cells in Sepp1–/– tumors.
Figure 3. Intratumoral apoptosis and DNA damage are increased in response to complete Sepp1 knockout, and proliferation is increased in Sepp1+/– tumors.

(A) Quantification of intratumoral apoptosis, as determined by TUNEL+ cells per tumor HPF (original magnification, ×40; 6, WT; 10, Sepp1+/–; 4 Sepp1–/–), and quantification of cleaved caspase-3 protein normalized to caspase-3 and cleaved PARP protein normalized to β-actin. Quantification is shown as fold change relative to WT (n = 6 per group). (B) Quantification of intratumoral proliferation, as determined by Ki67+ cells per tumor HPF (6, WT; 10, Sepp1+/–; 4, Sepp1–/–). (C) Quantification of intratumoral DNA damage, as measured by 8-hydroxyguanine+ cells per tumor HPF (6, WT; 10, Sepp1+/–; 4 Sepp1–/–). *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test.
Intratumoral DNA damage increases in a dose-dependent manner with decreased SEPP1 levels.
Selenium has been identified as a potential contributor to the maintenance of genomic stability (30, 31). However, the role of SEPP1 in DNA damage repair is unknown. We wanted to test the impact of SEPP1 deficiency on crypt and intratumoral DNA damage. 8-hydroxyguanine (8-OHdG) staining, a measure of oxidative DNA damage, demonstrated that intratumoral DNA damage increases with decreased Sepp1 expression (WT: 8.1 ± 0.8 8-hydroxyguanine+ cells per tumor HPF, Sepp1+/–: 15.3 ± 1.5, 8-hydroxyguanine+ cells per tumor HPF, P = 0.002; Sepp1–/–: 23.3 ± 3.4 8-hydroxyguanine+ cells per tumor HPF, P = 0.0007; Figure 3C and Supplemental Figure 2F), though crypt DNA damage was not altered when compared with that of WT mice in response to SEPP1 deficiency (Supplemental Figure 2E). These data suggest that, within the tumor microenvironment, decreasing Sepp1 expression leads to an increase in oxidative DNA damage.
Protumorigenic M2 macrophage polarization is increased in Sepp1+/– tumors.
The most significantly induced gene after polarization of macrophages to the M2 phenotype is SEPP1 (32–34). Moreover, proinflammatory cytokines induce iNOS, which results in downregulation of SEPP1 (35). To determine whether SEPP1 influences macrophage phenotypes and inflammation, we analyzed tumors from WT, Sepp1+/–, and Sepp1–/– mice after AOM/DSS treatment for macrophage polarization. Immunohistochemistry of tumors for the pan-macrophage marker F4/80 and either the M1 macrophage marker IL-1β or the M2 macrophage marker arginase I (Arg1) revealed an increase of total macrophages in Sepp1+/– tumors (WT: 13.1 ± 1.3, Sepp1+/–: 23.2 ± 3.0, P < 0.01, Sepp1–/–: 13.1 ± 0.8 F4/80+ cells per tumor HPF; Figure 4A), apparently resulting from an increased M2 macrophage presence (WT: 8.4 ± 0.8, Sepp1+/–: 18.0 ± 3.9, Sepp1–/–: 8.3 ± 1.1 F4/80+/ARG1+ cells per tumor HPF; Figure 4C), as M1 macrophage numbers remain unaltered (Figure 4B). Interestingly, Sepp1–/– tumors did not demonstrate increased M2 polarized macrophage infiltration. In order to determine whether this effect was due to recruitment or polarization, we isolated bone marrow macrophages from WT, Sepp1+/–, and Sepp1–/– mice and treated them with IFN-γ and LPS to polarize to the M1 phenotype and IL-13 to polarize to the M2 phenotype. Analysis of M1 markers Inos and Il1b demonstrated decreased M1 polarization in Sepp1+/– macrophages (Inos: 19,494 ± 593.8 vs. 14,961 ± 782.3 fold change, P < 0.001; Il1b: 337.6 ± 0.78 vs. 225.1 ± 20.66 fold change, P < 0.001; Figure 4D), while the M2 marker Ym1 indicated increased polarization to M2 (13.1 ± 3.3 vs. 26.6 ± 0.3, P < 0.001; Figure 4E). These data indicate that Sepp1 haploinsufficiency leads to altered macrophage polarization upon directed stimulation, a phenotype observed in vivo in the inflammatory infiltrates in Sepp1+/– tumors. Therefore, it is possible that altered immune responses may contribute to a protumorigenic microenvironment in these mice.
Figure 4. SEPP1 regulates protumorigenic M2 macrophage polarization.

Quantification of (A) intratumoral total macrophage staining, as determined by F4/80+ cells per tumor HPF; (B) M1 macrophage staining, as determined by F4/80+/IL-1β+ cells per tumor HPF; and (C) M2 macrophage staining, as determined by F4/80+/ARG1+ cells per tumor HPF (8, WT; 12, Sepp1+/–; 8, Sepp1–/–). (D) Inos (n = 5 per genotype) and Il1b (n = 6 per genotype) and (E) Ym1 mRNA expression (n = 6 per group) in WT, Sepp1+/–, and Sepp1–/– in vitro–activated bone marrow macrophages. Graphs demonstrate fold change in expression relative to unstimulated (US) WT macrophages normalized to Gapdh. *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test.
Loss of the selenium-rich C-terminal domain of SEPP1 promotes inflammatory tumorigenesis.
SEPP1 is considered to be a selenium transport protein, and deletion of the selenium-rich C-terminal domain (Sepp1Δ240–361/Δ240–361) results in severe selenium deficiency in the brain and testes of mice (36). Colonic selenium levels were also decreased in Sepp1Δ240–361/Δ240–361 mice (394.5 ± 19.6 vs. 260.2 ± 10.6 ng selenium/g colon, P = 0.0001; Figure 5A), yet the presence of selenium suggests that, while SEPP1 contributes to selenium delivery to the colon, it is not the sole source. Because selenium has been implicated in DNA damage repair via regulation of p53 (37), WNT and NRF2 signaling, and modification of several cancer types, including colorectal, lung, laryngeal, hepatic, and prostate (38–41), we determined whether the loss of the selenium-rich domain of SEPP1 would contribute to tumorigenesis. We subjected WT and Sepp1Δ240–361/Δ240–361 mice to the AOM/DSS protocol and analyzed them for tumor burden. We found that SEPP1 truncation resulted in an increase in tumor number (7.9 ± 1.4 vs. 3.9 ± 0.7 tumors per mouse, P = 0.02; Figure 5B). We have previously determined that selenium deficiency results in increased dysplasia grade in this model (16). Similarly, tumor grade was more advanced in Sepp1Δ240–361/Δ240–361 mice (P < 0.05; Figure 5C).
Figure 5. Both the selenium-rich region and the putative antioxidant domain of SEPP1 protect from inflammatory tumorigenesis.

(A) Quantification of colonic selenium in WT (n = 6) and Sepp1Δ240–361/Δ240–361 (n = 6) mice and (B) number of tumors per mouse (16, WT; 15, Sepp1Δ240–361/Δ240–361). *P < 0.05, ***P < 0.001, 2-tailed unpaired t test. (C) Percentage of total tumors per genotype with either high-grade dysplasia (HGD) or low-grade dysplasia (LGD) (n = 15 per group, ***P < 0.001, χ2 contingency analysis) and representative images from tumors of the given genotypes (original magnification, ×40). Scale bar: 200 μm. (D) Quantification of intratumoral proliferation, as measured by Ki67+ cells per tumor HPF (12, WT; 17, Sepp1Δ240–361/Δ240–361). (E) Crypt DNA damage, as measured by 8-hydroxyguanine+ cells per crypt averaged from 20 crypts within each mouse (8, WT; 9, Sepp1Δ240–361/Δ240–361). (F) Intratumoral DNA damage, as measured by 8-hydroxyguanine+ cells per tumor HPF (8, WT; 17, Sepp1Δ240–361/Δ240–361). (G) Number of tumors and (H) average tumor size within either WT (n = 12) or Sepp1U40S/U40S (n = 16) mice. (I) Quantification of crypt proliferation, as measured by Ki67+ cells per crypt averaged from 20 crypts within each mouse (8, WT; 8, Sepp1U40S/U40S); (J) intratumoral proliferation, as measured by Ki67+ cells per tumor HPF (11, WT; 19, Sepp1U40S/U40S); (K) crypt DNA damage, as measured by 8-hydroxyguanine+ cells per crypt averaged from 20 crypts within each mouse (8, WT; 8, Sepp1U40S/U40S); and (L) intratumoral DNA damage, as measured by 8-hydroxyguanine+ cells per tumor HPF (9, WT; 8, Sepp1U40S/U40S). *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed unpaired t test.
Along with an increase in tumor number, we also observed an increase in intratumoral proliferation (20.3 ± 2.2 vs. 13.4 ± 1.7 Ki67+ cells per tumor HPF, P = 0.04; Figure 5D and Supplemental Figure 3A) but no change in crypt proliferation (Supplemental Figure 5A) or crypt or tumor apoptosis (Supplemental Figure 4, B and C). Consistent with a role of selenium in DNA damage, there was an increase in crypt (1.0 ± 0.08 vs. 0.7 ± 0.04 8-OHdG+ cells per crypt, P = 0.02; Figure 5E and Supplemental Figure 3B) and intratumoral (29.8 ± 2.8 vs. 17.1 ± 2.7 8-OHdG+ cells per tumor HPF, P = 0.01; Figure 5F and Supplemental Figure 3C) DNA damage index scores in mice lacking the selenium-rich C-terminal domain compared with that in WT mice. These data suggest that the selenium-rich domain of SEPP1 may protect from tumor initiation by preserving genomic integrity.
Mutation of the redox-active selenocysteine in SEPP1 promotes inflammatory tumorigenesis.
Residues 40–43 of the N-terminal domain of SEPP1 comprise a UXXC redox motif that acts as a peroxidase (19, 20). As such, SEPP1 may contribute to antioxidant activity independent of its roles in selenium transport. In order to test whether loss of this antioxidant domain would modify tumorigenesis, we subjected mice in which the selenocysteine (U) at amino acid 40 was mutated to an enzymatically dead serine (S) (Sepp1U40S/U40S) to the AOM/DSS protocol. Compared with those in WT mice, SEPP1-mutated mice displayed increased tumor number (7.3 ± 1.1 vs. 3.8 ± 0.7 tumors per mouse, P = 0.02; Figure 5G) and tumor size (3.1 ± 0.3 vs. 1.6 ± 0.2 mm2, P = 0.002; Figure 5H). This increase in tumorigenesis was likely at least partially dependent upon an increase in crypt proliferation (0.5 ± 0.04 vs. 0.3 ± 0.02 Ki67+ cells per crypt, P = 0.0009; Figure 5I and Supplemental Figure 5A) and intratumoral proliferation (33.5 ± 2.6 vs. 15.2 ± 1.6 Ki67+ cells per tumor HPF, P < 0.0001; Figure 5J and Supplemental Figure 5B), compared with those in WT mice, despite a lack of change in apoptosis (Supplemental Figure 6, A and B). We did note an increase in crypt DNA damage (0.6 ± 0.02 vs. 0.3 ± 0.03 8-OHdG+ cells per crypt, P < 0.0001; Figure 5K and Supplemental Figure 5C) and intratumoral DNA damage (28.7 ± 2.6 vs. 10.8 ± 1.2 8-OHdG+ cells per tumor HPF, P < 0.0001; Figure 5L and Supplemental Figure 5D) in the SEPP1-mutated mice compared with those in WT mice, suggesting that the redox site of SEPP1 contributes to protection from DNA damage.
Sepp1 haploinsufficiency-driven CAC persists in mice fed normal selenium diets.
Sepp1–/– mice require selenium supplementation to survive (42); therefore, all of the above experiments were conducted in the setting of supranutritional selenium levels. Our results indicate that Sepp1 haploinsufficiency augments tumorigenesis. We next determined whether Sepp1 heterozygosity would also augment tumorigenesis if mice were maintained on a normal selenium diet (0.25 PPM selenium). Compared with WT mice, Sepp1+/– mice demonstrated an increase in tumor number, as determined by both endoscopy (9.8 ± 1.2 vs. 4.6 ± 0.8 tumors per mouse, P < 0.01; Figure 6, A and B) and analysis of tumor burden at sacrifice (11.6 ± 1.0 vs. 5.5 ± 0.9 tumors per mouse, P < 0.001; Figure 6, C and D), and, in general, these tumors were also larger than those observed in WT colons (P < 0.05; Figure 6E). When comparing overall survival of cohorts maintained on a normal selenium diet (0.25 PPM selenium) with that of those on a selenium-supplemented diet (1.0 PPM selenium), cohorts maintained on the selenium-supplemented diet demonstrated a significant survival benefit (P < 0.001; Figure 6F). These results may have potential translational implications, as the 0.25 PPM diet more closely approximates selenium levels in the typical Western diet.
Figure 6. Sepp1 haploinsufficiency-driven CAC persists in mice maintained on normal selenium diets.

(A) Endoscopic images of WT and Sepp1+/– colons after the second cycle of DSS administration. (B) Quantitative assessment of endoscopic tumor burden (11, WT; 13, Sepp1+/–). **P < 0.01, 2-tailed unpaired t test. (C) Gross representative images of tumors (D) and tumor counts at necropsy (11, WT; 13, Sepp1+/–). ***P < 0.001, 2-tailed unpaired t test. (E) Tumor size distribution. *P < 0.05, unpaired t test with Welch’s correction. (F) Survival analysis of AOM/DSS-treated mice maintained on the indicated diets. ***P < 0.001, unpaired t test with Welch’s correction.
Absence of SEPP1 augments stem cell properties and reduces the ability of enteroids to survive in response to hydrogen peroxide.
SEPP1 regulates oxidative stress and toxicity (43, 44) and, consistent with a function in WNT signaling, has been shown to play a role in differentiation and stem cell properties (45, 46). Recently, a model for epithelial crypt growth has been established based on the isolation of stem cells from the small intestinal crypt. From single crypts, complex structures can be grown in vitro that closely resemble their ex vivo counterparts (enteroids) (47). Using this model system, we sought to determine how SEPP1 influences intestinal epithelial biology. Importantly, Sepp1 expression in intestinal organoids was similar to expression in both the colon and the brain, a tissue known to produce high levels of SEPP1 (48), indicating these cultures would accurately recapitulate an in vivo setting (Figure 7A).
Figure 7. Sepp1–/– enteroids display increased stem cell characteristics as well as ROS production, proliferation, and decreased survival after oxidative stress.

(A) Sepp1 mRNA expression in tissue isolated from WT mice (n = 3 per group). Fold change expression normalized to Gapdh and relative to colonic Sepp1 expression. ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test. (B) Percentage of surviving enteroids 1 day after plating, percentage of branching enteroids and stem spheroids counted 3 days after plating, and average number of branches per branching enteroid at 4 days after plating (n = 4 per group per experiment). *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed unpaired t test. (C) ROS quantification by carboxy-H2DCFDA staining intensity, as measured 2 hours after treatment with H2O2. Fold change in intensity relative to WT enteroids treated with 0 μM H2O2 (n = 4 per group). Representative images of the single carboxy-H2DCFDA channel and merged carboxy-H2DCFDA and TO-PRO3 channels in WT and Sepp1–/– enteroids treated with either 0 μM or 800 μM H2O2 (original magnification, ×100). *P < 0.05, 1-way ANOVA, Newman-Keuls multiple comparison test. (D) Proliferation, as determined by EdU+ cells per crypt area within WT and Sepp1–/– enteroids after 2 hours of treatment with either 0 μM or 800 μM H2O2 (n = 4 per group with analysis of 10 enteroids per genotype) and representative images of EdU staining in WT and Sepp1–/– enteroids after treatment with either 0 μM or 800 μM H2O2 (original magnification, ×100). *P < 0.05, **P < 0.01, 1-way ANOVA, Newman-Keuls multiple comparison test. (E) Survival curves for WT, Sepp1+/–, and Sepp1–/– enteroids after daily treatment with 400 μM H2O2.
Enteroids can be grown from a single stem cell or complete crypts (49), and the “stemness” of enteroids has been measured by several growth parameters (50–52). For instance, augmented stem cell survival can be measured by an increase in the number of crypts that survive plating versus the total number of crypts plated (plating efficiency). Furthermore, the percentage of branching enteroids or stem spheroids present at certain time points can establish the differentiation capacity of the stem cells and the extent of stem cell signaling, respectively. Finally, the number of branches within each enteroid indicates the ability of the stem cells to self-propagate and establish new crypts. To determine whether Sepp1 contributes to epithelial cell growth and stem cell function at baseline, we analyzed Sepp1–/– enteroids for each of these properties (plating efficiency: 96.5% ± 2.4% enteroids of total crypts plated; 61.5% ± 1.8% branching enteroids; 14.5% ± 0.2% stem spheroids; 3.2 ± 0.2 number of branches per branching enteroid; Figure 7B) and observed increased stem cell fitness and growth abilities compared with those of WT enteroids (plating efficiency: 65.5% ± 2.0% enteroids of total crypts plated, P < 0.0001; 42.1% ± 2.4% branching enteroids, P = 0.02; 7.6% ± 0.9% stem spheroids, P = 0.002; number of branches per branching enteroid: 1.9 ± 0.1, P = 0.003; Figure 7B).
Because SEPP1 serves as a peroxidase in vitro (20), we next determined the response of Sepp1–/– enteroids to hydrogen peroxide treatment. Analysis of ROS produced after treatment with hydrogen peroxide revealed a decreased ability of Sepp1–/– small intestine to respond to oxidative stress, as demonstrated by increased 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) staining intensity (800 μM H2O2: 1.6 ± 0.4 vs. 3.3 ± 0.4 fold change intensity relative to 0 μM H2O2 WT, P = 0.04; Figure 7C); however, there was no change in staining intensity of the oxidative-insensitive analog carboxy-DCFDA. Moreover, baseline proliferation was increased in Sepp1–/– enteroids (1.0 ± 0.06 vs. 1.4 ± 0.1 fold change EdU+ cells per crypt area relative to 0 μM H2O2 WT, P < 0.05; Figure 7D) and, while treatment with hydrogen peroxide resulted in decreased Sepp1–/– proliferation compared with that of untreated null enteroids (1.4 ± 0.1 vs. 0.9 ± 0.09 fold change EdU+ cells per crypt area relative to 0 μM H2O2 Sepp1–/–, P < 0.01; Figure 7D), Sepp1–/– enteroids retained increased proliferation compared with that of WT enteroids (0.4 ± 0.09 vs. 0.9 ± 0.09 fold change EdU+ cells per crypt area relative to 0 μM H2O2 WT, P < 0.05; Figure 7D). Finally, Sepp1–/– enteroids demonstrated decreased survival in response to multiple daily administrations of hydrogen peroxide (P = 0.005; Figure 7E). These data indicate that absence of SEPP1 increases baseline stem phenotypes and proliferation and compromises the antioxidant potential of enteroids. These data are particularly compelling, because they indicate an epithelial cell–autonomous role for SEPP1 in processes that would likely contribute to tumorigenesis in CAC, a cancer characterized by increased microenvironmental oxidative stress.
WNT signaling and oxidative stress gene signatures are altered in Sepp1–/– epithelium.
Enteroids rely on the addition of growth factors EGF, noggin, and R-spondin for growth and differentiation (47). As Sepp1–/– enteroids demonstrate increased stem cell properties and fitness, we next determined growth factor dependency of Sepp1–/– enteroids. Depletion of EGF, noggin, and R-spondin resulted in similar decreases in survival in all 3 genotypes, indicating that all 3 pathways are important for enteroid persistence (Figure 8A).
Figure 8. WNT signaling plays a pivotal role in the Sepp1–/– phenotype.

(A) Survival curves in response to growth factor depletion in WT, Sepp1+/–, and Sepp1–/– enteroids. (B) Quantification of WNT proteins LEF-1, cyclin D1, and MMP-7 normalized to β-actin. Quantification is shown as fold change relative to WT and representative images of blots from 3 individual tumors from each genotype. *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA, Newman-Keuls multiple comparison test.
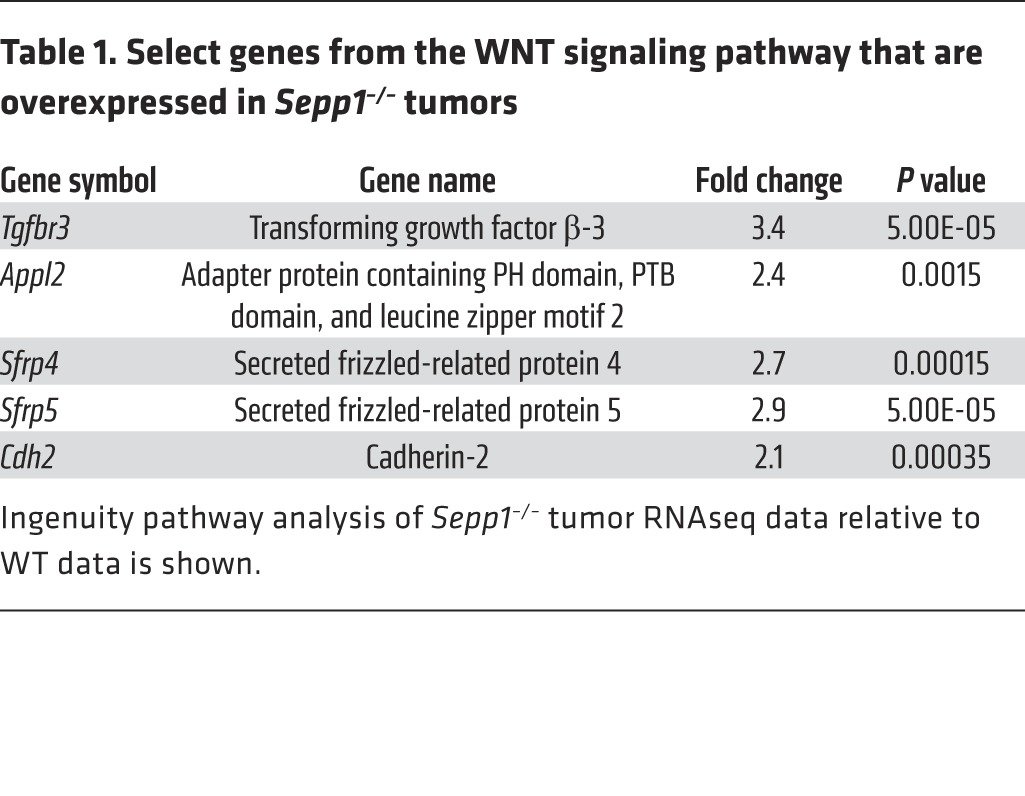
In order to acquire an unbiased understanding of transcriptome changes within tumors of Sepp1–/– mice, we performed RNA sequencing (RNAseq) analysis of tumors isolated from WT and Sepp1–/– mice subjected to the AOM/DSS protocol. Ingenuity pathway analysis indicated that the WNT pathway was among the most significantly altered canonical pathways (Supplemental Figure 8), as Sepp1–/– tumors demonstrated increases in Tgfbr3, Appl2, Sfrp4, Sfrp5, and Cdh2 expression, among other abnormalities (Table 1 and Supplemental Figure 9). As further validation of altered WNT signaling in these tumors, protein analysis demonstrated increased expression of the WNT targets LEF-1, cyclin D1, and MMP-7 upon loss of Sepp1 expression (Figure 8B). We next determined whether SEPP1 loss influenced production of other oxidative defense genes. First, we tested for compensation by other selenoproteins at the RNA level and did not observe differences in their message between WT and Sepp1–/– tumors. We did, however, note significant alterations in several cytochrome P450 gene family members as well as β-hydroxysteroid dehydrogenase genes (Supplemental Figure 7), suggesting that other antioxidant genes are upregulated in an attempt to compensate for SEPP1 loss.
Table 1. Select genes from the WNT signaling pathway that are overexpressed in Sepp1–/– tumors.
Discussion
The efficacy of antioxidants as chemopreventive agents of malignancy in the general population has been disappointing. However, most studies have been conducted in the setting of relatively normal selenium levels, which ensures sufficient production of the majority of selenoproteins. Patients with IBD are reported to be selenium deficient, potentially skewing selenoprotein production. Our findings suggest that modifying production of selenoproteins (i.e., SEPP1) in patients with IBD may impact cancer risk. Here, we show that reduced levels of a major selenoprotein, SEPP1, result in increased colonic epithelial stem cell properties, such as increased branching and increased presence of stem spheroids as well as increased WNT signaling. Reduced expression of SEPP1 also results in increased DNA damage in response to oxidative stress. These SEPP1-dependent phenotypes ultimately contribute to an increase in inflammatory tumorigenesis upon loss of either the redox-active site or the selenium transport domain of SEPP1 via its effects on the tumor microenvironment and preservation of genomic integrity. These data establish a critical role for SEPP1 in intestinal biology, homeostasis, injury response, preservation of genomic integrity, and inflammatory carcinogenesis.
Induction of several signaling pathways, including NRF2 (11) and WNT (10), has been linked to selenium depletion, and there is little known about the contribution of SEPP1 to the observed alterations in these pathways. We therefore performed an unbiased RNAseq analysis of tumors from WT and Sepp1–/– mice after subjecting them to the AOM/DSS protocol. Among the most severely altered pathways in response to SEPP1 loss was the WNT pathway. While genes were both upregulated and downregulated in this pathway, some of particular importance included modulators of WNT signaling, Sfrp4 and Sfrp5; the transcriptional effector Lef (also known as Tcf); and the WNT target Mmp7, which we also validated at the protein level. Genes that demonstrated increased expression included those coding for the TGF-β receptor, N-cadherin, and the liver receptor homolog-1 (LRH-1), which is important for maintaining stem cell pluripotence during development (53). Importantly, ingenuity pathway analysis identified the WNT/β-catenin signaling pathway as being stimulated in Sepp1–/– tumors. This is of particular importance because SEPP1 is a selenium transporter and selenium deficiency leads to an increase in WNT signaling, suggesting that the selenium transport function of SEPP1 may contribute to WNT modulation.
The literature has extensively implicated selenium in the modulation of immune processes, particularly in relation to macrophage activity (54–56). We noted an increase in M2 polarized macrophages in tumors of AOM/DSS-treated Sepp1+/– mice and hypothesized that this was due to an effect of SEPP1 on macrophage polarization. Isolation of bone marrow macrophages from Sepp1+/– mice and subsequent polarization with either IFN-γ and LPS (M1) or IL-13 (M2) resulted in deficits in expression of the M1 markers Inos and Il1b and increased expression of the M2 marker Ym1. This altered polarization may contribute to tumorigenesis by altering the tumor microenvironment in Sepp1+/– mice. As such, SEPP1 may reduce inflammatory tumorigenesis by attenuating proinflammatory immune cell activation. This is especially important to note when considering the potential positive impact of selenium supplementation on patients with nutritional deficiencies due to diseases, such as IBD.
The influence of plasma SEPP1 on delivery of selenium to target tissues has been extensively studied (36, 57, 58). Our enteroid studies demonstrate a role for tissue-produced SEPP1 in intestinal biology, as loss of intestinal epithelial cell–autonomous Sepp1 affects cellular growth, differentiation, and response to ROS. Because it is a secreted protein, we hypothesize that SEPP1 provides localized antioxidant benefit to the proximate epithelial microenvironment. It was recently shown that SEPP1 antioxidant capacity is due, at least in part, to its ability to act as a substrate for thioredoxin reductase-1, which catalyzes NADPH oxidation (59). From these studies, we postulate that extracellular SEPP1 within the near-cell environment may be cooperating with intracellular selenoproteins, such as thioredoxin reductase-1, Gpx2, and Gpx4, to protect the epithelium from oxidative damage.
Whether selenium supplementation is of potential value in reducing cancer risk has been studied. The US case-control SELECT study did not demonstrate any clear association between serum selenium levels and the risk of recurrent CRC (60), and a meta-analysis of intervention trials concluded that oral administration of antioxidants, including selenium, was not effective in preventing colorectal neoplasia in the general population (61). A major limitation of these studies, however, was patient selection, as patients in both studies had near-normal selenium levels. Our data suggest that targeted supplementation in populations known to be selenium deficient might be an effective prevention strategy. Patients with IBD are selenium deficient, as measured by both plasma selenium and SEPP1 levels (62). Furthermore, the plasma concentration of SEPP1 is reduced 53% in patients with Crohn’s disease compared with that in healthy controls (63). This suggests that patients with IBD are selenium deficient to such an extent that selenium supplementation might increase SEPP1 levels and subsequently reduce risk of CAC arising from IBD. Indeed, in line with the idea of a protective role for selenium supplementation, our study demonstrated a significant survival benefit in cohorts of mice that were on high selenium diet in comparison to those on normal selenium diet when subjected to an AOM/DSS inflammatory carcinogenesis protocol.
Importantly, several SNPs have been identified in SEPP1 that might contribute to decreased expression in tumors. For instance, SNPs in SEPP1 are associated with decreased plasma SEPP1 in men and increased prostate cancer risk (40, 64). Furthermore, 4 SEPP1 variants have been significantly associated with advanced colorectal adenoma risk, including the variant SEPP1 -4166G, which exists within the promoter region, 2 loci in the 3′ region of SEPP1, and a third locus in the 3′ region of SEPP1, which is inversely associated with risk of colorectal adenoma (22). Additionally, genetic instability has been observed within the (T)17 repeat motif within the SEPP1 promoter in CRC to the mutator phenotype. Thus, this SEPP1 repeat structure may be of functional relevance to SEPP1 gene expression and thus modify tumorigenesis (65). A mechanism by which some of these polymorphisms may alter SEPP1 status is by either altering expression or proportions of SEPP1 isoforms. Several functional polymorphisms in the SEPP1 gene have been shown to influence isoform expression, and it is thought that increased expression of the 60-kDa isoform of SEPP1 may increase selenoprotein synthesis and decrease CRC risk (66). Thus, genotyping of SEPP1 in patients with CAC may indicate increased responsiveness to selenium supplementation.
In conclusion, we demonstrate a cell-autonomous role for SEPP1 in the intestinal epithelium and that its reduction, in vivo, augments inflammatory carcinogenesis. These in vivo effects occur via independent enzymatic and transport activities. Our studies establish a critical role for SEPP1 in intestinal biology, homeostasis, injury response, preservation of genomic integrity, and inflammatory carcinogenesis and suggest that SEPP1 could serve as a therapeutic target in the prevention of CAC.
Methods
Murine carcinogenesis protocols.
Sepp1–/– mice exhibit decreased survival on standard chow diet (42) and require selenium supplementation. Therefore, these mice were fed Torula yeast-based diets supplemented with 1.0 mg selenium (sodium selenite was used) per kg. Accordingly, to properly control the experiment, WT and Sepp1+/– mice were also maintained on an identical diet. In a separate experiment, WT and Sepp1+/– mice were maintained on a normal selenium Torula yeast-based diet (0.25 mg selenium [sodium selenite was used] per kg). These diets were prepared and pelleted to our specifications (42) by Harlan-Teklad. For AOM only experiments (Figure 1A), 8- to 12-week-old C57BL/6 WT (n = 4), Sepp1+/– (n = 4), or Sepp1–/– (n = 4) mice (17, 18) were injected with 12.5 mg/kg AOM (Sigma-Aldrich) intraperitoneally. Mice were monitored for tumor burden by endoscopy once per month for 6 months and sacrificed 180 days after AOM injection. For chronic DSS experiments, 8- to 12-week-old C57BL/6 WT (n = 7), Sepp1+/– (n = 10), or Sepp1–/– (n = 10) mice (17, 18) were subjected to three 5-day cycles of 3% DSS ad libitum, with each cycle being followed by a 16-day recovery period (Figure 1B). Mice were monitored for colonic injury by endoscopy 4 days into the recovery period after each DSS administration. Injury was evaluated based on the MEICS scoring system, which grades mucosal thickening, vasculature pattern, granularity, exudate, and stool consistency (67). For AOM/DSS protocols, 8- to 12-week-old C57BL/6 WT (n = 13), Sepp1+/– (n = 9), or Sepp1–/– (n = 11) mice (17, 18) (constitutional knockout experiment with mice fed 1.0 mg selenium per kg diet); WT (n = 16) or Sepp1+/– (n = 17) mice (constitutional knockout experiment with mice fed 0.25 mg selenium per kg diet); WT (n = 16) or Sepp1Δ240–361/Δ240–361 (n = 14) mice (36) (truncation experiments); and WT (n = 12) or Sepp1U40S/U40S (n = 15) (20) mice (redox motif mutant experiments) (Table 2) were injected with 12.5 mg/kg AOM (Sigma-Aldrich) intraperitoneally. Three days after injection, the animals were started on the first of 3 cycles of 3% DSS ad libitum. Each cycle lasted 5 days and was followed by a 16-day recovery period. During each cycle of recovery, colonoscopy was performed to assess injury, tumor multiplicity, and grade (67). All mice were sacrificed on day 70, with the exception of cohorts maintained on 0.25 mg selenium per kg diet, which were sacrificed on day 52 due to increased mortality following the third cycle of DSS. All tumor counts and measurements were performed in a blinded fashion under stereo-dissecting microscopy. Histologic analysis was performed in a blinded fashion for severity of inflammation (68) and dysplasia on H&E-stained “Swiss-rolled” colons by a gastrointestinal pathologist in house.
Table 2. The genotypes of mice subjected to the AOM/DSS protocol and the symbols used for each genotype.

Immunohistochemistry and immunofluorescence staining.
For SEPP1 and phalloidin staining, colons and small intestines were embedded in OCT media and flash frozen in liquid nitrogen. Five-micrometer sections were cut and SEPP1 staining was performed as described previously (69). For all other staining, standardized protocols were followed to ensure the integrity of the samples for immunohistochemistry/immunofluorescence applications. In brief, immediately upon sacrifice, the colons were harvested, flushed with ice-cold PBS, Swiss rolled without delay, and fixed in fresh neutral buffered 10% formalin, which was subsequently exchanged for 70% EtOH. Processing was performed at the Vanderbilt University Translational Pathology Shared Resource. Five-micrometer sections of paraffin-embedded colons were cut, and H&E staining was performed. For immunohistochemistry, cut sections were dewaxed, hydrated, and quenched of endogenous peroxidase activity with 0.03% hydrogen peroxide in methanol. Antigen retrieval was conducted using Antigen Unmasking Reagent (Vector Laboratories Inc.) according to the manufacturer’s instructions. After blocking, primary antibody was added (α-Ki67 [NeoMarkers], 1:1,000; α-ARG1 [Santa Cruz], 1:500; α–IL-1β [R&D Systems], 1:40; α-F4/80 [ABd Serotec], 1:1,000; α-8-OHdG [Santa Cruz], 1:500) and incubated overnight at 4°C. Isotype-matched antibodies were included as negative controls. Identification of intratumoral apoptotic cells was conducted using the ApopTag Plus Peroxidase In Situ Apoptosis Kit (Chemicon) according to the manufacturer’s protocol. Control slides were obtained by omitting the terminal transferase enzyme. For immunofluorescence staining of proliferation, macrophages, and DNA damage, slides were counterstained and mounted with ProLong Gold antifade, including DAPI (Invitrogen). Apoptosis, proliferation, DNA damage, and M1 and M2 macrophage indices were generated by counting either the number of positive cells per HPF (×40 objective) within each tumor or the number of positive cells per crypt in 20 crypts per mouse by a blinded observer. The average score was then calculated for each Swiss-rolled colon.
Colonic selenium measurements.
The determination of colonic selenium was carried out using a modification of the fluorometric assay of Koh and Benson (70) developed by Sheehan and Gao (71). Briefly, tissue was digested in nitric and perchloric acids and, after adjustment of pH, selenium (as selenite) was complexed with diaminonaphthalene. Selenium-diaminonaphthalene was extracted into cyclohexane, and fluorescence was measured in a Perkin-Elmer LS 55 fluorometer as described previously (58).
Western blot analysis of apoptosis and WNT pathway proteins.
Flash-frozen sections of colon (n = 6 for each genotype) were lysed in RIPA buffer, including 1× protease inhibitor cocktail (Sigma-Aldrich), using a rotor homogenizer (Janke & Kunkel IKA Labortechnik Ultra-Turrax T25). Protein quantification was performed using a Pierce BCA Protein Assay Kit (Thermo) according to the manufacturer’s instructions. Samples were suspended in loading buffer, boiled, and 40 μg of protein was run on an SDS-PAGE gel for immunoblotting. The Apoptosis Antibody Sampler (Mouse Preferred) Kit (9930, Cell Signaling Technology) and the WNT Signaling Antibody Sampler Kit (2915, Cell Signaling Technology) were used according to the manufacturer’s protocol. Quantification was performed using Odyssey Imaging Software and normalized either to total caspase or β-actin.
Bone marrow macrophage polarization and analysis.
Bone marrow macrophages were isolated and activated as has been previously published (72, 73). For the production of classically activated M1 macrophages, bone marrow macrophages were primed with 150 U/ml IFN-γ for 6 hours and subsequently stimulated with 10 ng/ml LPS. For the production of alternatively activated M2 macrophages, bone marrow macrophages were treated with 20 U/ml IL-13 for 12 hours. RNA was isolated using the RNEasy Mini Kit (Qiagen). 1 μg RNA was used to perform reverse transcription PCR using the iScript cDNA Synthesis Kit (Bio-Rad). 1 μl cDNA was used for qRT-PCR analysis of Inos (Bio-Rad 100-31225), Il1b (RealTimePrimers.com VMPS-3072), and Ym1 (Bio-Rad 100-31225).
Plasma SEPP1 protein determination.
Plasma SEPP1 was measured by ELISA (36). Plasma samples were diluted in PBS with 0.05% Tween-20 containing 2% rat plasma cleared of SEPP1 by monoclonal antibody 8F11 as described previously (74).
Small intestinal organoid culturing.
Six centimeters of the distal small intestine were dissected, flushed with ice-cold PBS, dissected into 1-cm pieces, suspended in 5 ml ice-cold PBS, and vortexed for 3 seconds. PBS was removed with a pipettor, and the wash was repeated. Tissue was transferred to 5 ml chelation buffer (1 mM EDTA, made fresh in DPBS) and rocked for 10 minutes at 4°C prior to washing twice with 10 ml PBS. 5 ml PBS was added and shaken gently for 2 minutes. The supernatant from the first shake was poured off. 5 ml PBS was added, and the shake was repeated for 2 minutes before the supernatant was poured off. 5 ml fresh chelation buffer was added, and chelation was performed for 10 minutes at 4°C with rocking. Crypts were filtered through a 70-μm filter into a prechilled 50-ml tube. The filter was rinsed with 5 ml cold shaking buffer (PBS with 43.3 mM sucrose and 54.9 mM sorbitol). Complete crypts were counted and enough volume for 1,200 crypts was transferred to a prechilled 5-ml round-bottomed tube. Crypts were centrifuged at 150 g for 10 minutes at 4°C. Shaking buffer was aspirated, and crypts were resuspended in 50 μl Matrigel (BD Bioscience 356237) per well, containing 50 ng/ml EGF (2028-EG-200, R&D Systems), 100 ng/ml Noggin (1967-NG-025/CF, R&D Systems), 500 ng/ml R-Spondin (3474-RS-050, R&D Systems), and 200 ng/ml WNT3a (GF-160, Millipore). Matrigel was overlaid with 500 μl Minigut culture media (Advanced DMEM/F12 [12634-010, Invitrogen], l-Glutamine [25030, Invitrogen], Pen-Strep [15140-148, Invitrogen], HEPES [25-060-CI, Mediatech], N2 Supplement [390155, R&D Systems], B27 Supplement [17504044, Invitrogen]). Every 4 days, media were replaced with fresh Minigut media plus growth factors.
Whole-mount carboxy-H2DCFDA enteroid staining.
Enteroids were grown for 4 days after plating. The morning of hydrogen peroxide treatment, enteroid media were replaced with fresh Minigut media. 0 μM, 400 μM, or 800 μM hydrogen peroxide was added directly to media. Two hours after hydrogen peroxide treatment, growth media were removed from cells. Cells were treated with prewarmed DPBS containing either the probe carboxy-H2DCFDA (mp-36103, Invitrogen) or, as a negative control, carboxy-DCFDA to provide a final working concentration of 5 mM. Enteroids were incubated at 37°C in a cell culture incubator for 1 hour. Loading buffer was removed, and cells were returned to prewarmed Minigut media. Cells were incubated at 37°C for 15 minutes. Media were removed, and enteroids were fixed overnight at 4°C in 2% formaldehyde with gentle rocking. Fixative was removed, and enteroids were washed twice with DPBS. Enteroids were imaged with a LSM 510 confocal microscope using the same specifications for imaging of all enteroids. ROS were quantified as staining intensity using ImageJ Image Analysis software. The graph represents the fold change intensity relative to WT enteroids treated with 0 μM hydrogen peroxide (Figure 7C). Error bars represent the SEM for 3 separate experiments performed in duplicate.
Whole-mount enteroid proliferation staining.
Proliferation was determined using the Click-iT EdU cell proliferation assay (C-10337, Invitrogen) according to the manufacturer’s instructions. Enteroids were grown to 4 days after plating. Enteroid media were replaced with fresh Minigut media, and either 0 μM or 800 μM hydrogen peroxide was added directly to media. Two hours after hydrogen peroxide treatment, media were replaced with fresh Minigut media and a 10 μM working solution of EdU was added to each well of the plate. Enteroids were incubated for 15 minutes under normal growth conditions. After incubation, media were removed and 1 ml of 2% formaldehyde in PBS was added to each well and incubated overnight at 4°C. The fixative was removed and enteroids were washed twice with 1 ml of 3% BSA in PBS. Wash buffer was removed, and 1 ml of 0.5% Triton X-100 in PBS was added to each well and incubated at room temperature for 20 minutes. Click-iT reaction cocktail was prepared, and 0.5 ml was added to each well. The wash solution was removed, and enteroids were stained with TO-PRO-3 (1:500 in PBS for 15 minutes at room temperature). Enteroids were imaged with a LSM 510 confocal microscope using the same specifications for imaging of all enteroids. Proliferation was quantified as EdU+ cells per crypt area where crypt area was determined using ImageJ Image Analysis software. The graph represents the fold change intensity relative to WT enteroids treated with 0 μM hydrogen peroxide (Figure 7D). Error bars represent the SEM for 3 separate experiments performed in duplicate.
RNAseq assay and analysis.
Tumors were dissected from WT and Sepp1–/– mice, and RNA was isolated using the RNEasy Mini Kit (Qiagen). RNA integrity was determined using the Experion RNA StdSens Analysis Kit (Bio-Rad). All RNA samples had RNA integrity numbers >8 and were deemed suitable for analysis. Initial raw sequencing data were aligned to reference mouse genome (mm10) using TopHat (version 2.0.8) software with default parameters (75). The Cuffdiff software (version 2.1.1) was used to estimate read count for expression of each gene and to detect differentially expressed genes. For count-based gene expression data, Cuffdiff uses the beta negative binomial model for each gene in each condition in order to estimate the null distribution of its log fold change under the null hypothesis (76). The P values from Cuffdiff were adjusted by Benjamini and Hochberg’s method to control false discovery rate (77).
Statistics.
Analyses comparing 2 groups were analyzed using the 2-tailed Student’s t test. One-way ANOVA and Newman-Keuls post-test were used to compare multiple groups. Data are presented as the mean ± the SEM in bar graphs, and a line identifying the mean is shown when all data points are plotted. Percentages of mice displaying altered dysplasia grade were determined using χ2 contingency analysis for each grade. All of these analyses were performed using GraphPad Prism 5.0c.
Study approval.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (8th ed. The National Academies Press. 2011.). The study protocol was approved by the Institutional Animal Care and Use Committee at Vanderbilt University (protocol no. M/10-355).
Supplementary Material
Acknowledgments
The authors thank Teri Stevenson and Michelle Chatterton for animal husbandry and members of the Williams and Burk laboratories for thoughtful discussions about this research project. This work was supported by NIH grants DK080221 (to C.S. Williams), R37 ES002497 (to R.F. Burk), P50CA095103 (to M.K. Washington), R01AT004821 (to K.T. Wilson), R01AT004821-S1 (to K.T. Wilson), R01DK053620 (to K.T. Wilson), P01CA028842 (to K.T. Wilson), P01CA116087 (to K.T. Wilson), 1F31CA167920 (to C.W. Barrett), 1F30DK103498 (to V.K. Reddy), T32GM07347 (to V.K. Reddy), 1F30DK096718 (to B. Parang), T32 GM07347 (to B. Parang), and 1F30CA165726 (to T. Freeman) and Merit Review Grants from the Office of Medical Research, Department of Veterans Affairs, 1I01BX001426 (to C.S. Williams), 1I01BX001453 (to K.T. Wilson), ACS-RSG 116552 (to C.S. Williams), and T32CA009592-26 (to C.W. Barrett). This publication was also supported in part by the National Cancer Institute Cancer Center support grant P30CA068485, utilizing the Translational Pathology shared resource, which provided tissue processing and staining services. Core Services performed through Vanderbilt University Medical Center’s Digestive Disease Research Center were supported by NIH grant P30DK058404 Core Scholarship. Support was also supplied by the National Center for Research Resources, grant UL1 RR024975-01, which is now the National Center for Advancing Translational Sciences, grant 2 UL1 TR000445-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(7):2646–2660. doi:10.1172/JCI76099.
References
- 1.Loftus EV, Jr, Sandborn WJ. Epidemiology of inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31(1):1–20. doi: 10.1016/S0889-8553(01)00002-4. [DOI] [PubMed] [Google Scholar]
- 2.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361(21):2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204(7):511–524. doi: 10.1016/j.prp.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Jess T, Simonsen J, Jorgensen KT, Pedersen BV, Nielsen NM, Frisch M. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology. 2012;143(2):375–381. doi: 10.1053/j.gastro.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Herrinton LJ, Liu L, Levin TR, Allison JE, Lewis JD, Velayos F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology. 2012;143(2):382–389. doi: 10.1053/j.gastro.2012.04.054. [DOI] [PubMed] [Google Scholar]
- 6.Neumann H, Vieth M, Langner C, Neurath MF, Mudter J. Cancer risk in IBD: how to diagnose and how to manage DALM and ALM. World J Gastroenterol. 2011;17(27):3184–3191. doi: 10.3748/wjg.v17.i27.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen GC, Bressler B. A tale of two cohorts: are we overestimating the risk of colorectal cancer in inflammatory bowel disease? Gastroenterology. 2012;143(2):288–290. doi: 10.1053/j.gastro.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 8.Rubin DT, et al. Colorectal cancer prevention in inflammatory bowel disease and the role of 5-aminosalicylic acid: a clinical review and update. Inflamm Bowel Dis. 2008;14(2):265–274. doi: 10.1002/ibd.20297. [DOI] [PubMed] [Google Scholar]
- 9.Kipp A, et al. Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to limited selenium intake in mouse colon. Mol Nutr Food Res. 2009;53(12):1561–1572. doi: 10.1002/mnfr.200900105. [DOI] [PubMed] [Google Scholar]
- 10.Brigelius-Flohe R, Kipp AP. Selenium in the redox regulation of the nrf2 and the wnt pathway. Methods Enzymol. 2013;527:65–86. doi: 10.1016/B978-0-12-405882-8.00004-0. [DOI] [PubMed] [Google Scholar]
- 11.Burk RF, et al. Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic Biol Med. 2008;44(8):1617–1623. doi: 10.1016/j.freeradbiomed.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim TH, et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011;71(6):2260–2275. doi: 10.1158/0008-5472.CAN-10-3007. [DOI] [PubMed] [Google Scholar]
- 13.Chang LC, et al. Immunohistochemical study of the Nrf2 pathway in colorectal cancer: Nrf2 expression is closely correlated to Keap1 in the tumor and Bach1 in the normal tissue. Appl Immunohistochem Mol Morphol. 2013;21(6):511–517. doi: 10.1097/PAI.0b013e318282ac20. [DOI] [PubMed] [Google Scholar]
- 14.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 15.Shamberger RJ, Willis CE. Selenium distribution and human cancer mortality. CRC Crit Rev Clin Lab Sci. 1971;2(2):211–221. doi: 10.3109/10408367109151308. [DOI] [PubMed] [Google Scholar]
- 16.Barrett CW, et al. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One. 2013;8(7):e67845. doi: 10.1371/journal.pone.0067845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schomburg L, Schweizer U, Holtmann B, Flohe L, Sendtner M, Kohrle J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J. 2003;370(pt 2):397–402. doi: 10.1042/BJ20021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill KE, et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem. 2003;278(16):13640–13646. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- 19.Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 20.Kurokawa S, et al. Sepp1 forms are N-terminal selenoprotein P truncations that have peroxidase activity when coupled with thioredoxin reductase-1. Free Radic Biol Med. 2014;69:67–76. doi: 10.1016/j.freeradbiomed.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Taie OH, et al. Expression profiling and genetic alterations of the selenoproteins GI-GPx and SePP in colorectal carcinogenesis. Nutr Cancer. 2004;48(1):6–14. doi: 10.1207/s15327914nc4801_2. [DOI] [PubMed] [Google Scholar]
- 22.Peters U, et al. Variation in the selenoenzyme genes and risk of advanced distal colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2008;17(5):1144–1154. doi: 10.1158/1055-9965.EPI-07-2947. [DOI] [PubMed] [Google Scholar]
- 23.Dincer Y, Erzin Y, Himmetoglu S, Gunes KN, Bal K, Akcay T. Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Dig Dis Sci. 2007;52(7):1636–1641. doi: 10.1007/s10620-006-9386-8. [DOI] [PubMed] [Google Scholar]
- 24.Smith JJ, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138(3):958–968. doi: 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pegg AE. Methylation of the O6 position of guanine in DNA is the most likely initiating event in carcinogenesis by methylating agents. Cancer Invest. 1984;2(3):223–231. doi: 10.3109/07357908409104376. [DOI] [PubMed] [Google Scholar]
- 26.Okayasu I, Ohkusa T, Kajiura K, Kanno J, Sakamoto S. Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut. 1996;39(1):87–92. doi: 10.1136/gut.39.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker C, et al. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut. 2005;54(7):950–954. doi: 10.1136/gut.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]
- 29.Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferguson LR, Karunasinghe N, Zhu S, Wang AH. Selenium and its’ role in the maintenance of genomic stability. Mutat Res. 2012;733(1):100–110. doi: 10.1016/j.mrfmmm.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Bera S, De Rosa V, Rachidi W, Diamond AM. Does a role for selenium in DNA damage repair explain apparent controversies in its use in chemoprevention? Mutagenesis. 2013;28(2):127–134. doi: 10.1093/mutage/ges064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solinas G, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. 2010;185(1):642–652. doi: 10.4049/jimmunol.1000413. [DOI] [PubMed] [Google Scholar]
- 33.Ghassabeh GH, et al. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood. 2006;108(2):575–583. doi: 10.1182/blood-2005-04-1485. [DOI] [PubMed] [Google Scholar]
- 34.Bosschaerts T, et al. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol. 2008;180(9):6168–6175. doi: 10.4049/jimmunol.180.9.6168. [DOI] [PubMed] [Google Scholar]
- 35.Speckmann B, Pinto A, Winter M, Förster I, Sies H, Steinbrenner H. Proinflammatory cytokines down-regulate intestinal selenoprotein P biosynthesis via NOS2 induction. Free Radic Biol Med. 2010;49(5):777–785. doi: 10.1016/j.freeradbiomed.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 36.Hill KE, et al. The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for the supply of selenium to brain and testis but not for the maintenance of whole body selenium. J Biol Chem. 2007;282(15):10972–10980. doi: 10.1074/jbc.M700436200. [DOI] [PubMed] [Google Scholar]
- 37.Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci U S A. 2002;99(22):14548–14553. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaworska K, et al. A low selenium level is associated with lung and laryngeal cancers. PLoS One. 2013;8(3):e59051. doi: 10.1371/journal.pone.0059051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kasaikina MV, et al. Contrasting roles of dietary selenium and selenoproteins in chemically induced hepatocarcinogenesis. Carcinogenesis. 2013;34(5):1089–1095. doi: 10.1093/carcin/bgt011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinbrecher A, et al. Effects of selenium status and polymorphisms in selenoprotein genes on prostate cancer risk in a prospective study of European men. Cancer Epidemiol Biomarkers Prev. 2010;19(11):2958–2968. doi: 10.1158/1055-9965.EPI-10-0364. [DOI] [PubMed] [Google Scholar]
- 41.Connelly-Frost A, Poole C, Satia JA, Kupper LL, Millikan RC, Sandler RS. Selenium, folate, and colon cancer. Nutr Cancer. 2009;61(2):165–178. doi: 10.1080/01635580802404188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J Nutr. 2004;134(1):157–161. doi: 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- 43.Xiao W, et al. Selenoprotein P regulates 1-(4-Chlorophenyl)-benzo-2,5-quinone-induced oxidative stress and toxicity in human keratinocytes. Free Radic Biol Med. 2013;65:70–77. doi: 10.1016/j.freeradbiomed.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arteel GE, Mostert V, Oubrahim H, Briviba K, Abel J, Sies H. Protection by selenoprotein P in human plasma against peroxynitrite-mediated oxidation and nitration. Biol Chem. 1998;379(8):1201–1205. [PubMed] [Google Scholar]
- 45.Sampson N, et al. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol Endocrinol. 2011;25(3):503–515. doi: 10.1210/me.2010-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Chen X. Reducing selenoprotein P expression suppresses adipocyte differentiation as a result of increased preadipocyte inflammation. Am J Physiol Endocrinol Metab. 2011;300(1):E77–E85. doi: 10.1152/ajpendo.00380.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 48.Steinbrenner H, Sies H. Selenium homeostasis and antioxidant selenoproteins in brain: implications for disorders in the central nervous system. Arch Biochem Biophys. 2013;536(2):152–157. doi: 10.1016/j.abb.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 49.Fuller MK, Faulk DM, Sundaram N, Shroyer NF, Henning SJ, Helmrath MA. Intestinal crypts reproducibly expand in culture. J Surg Res. 2012;178(1):48–54. doi: 10.1016/j.jss.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato T, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durand A, et al. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc Natl Acad Sci U S A. 2012;109(23):8965–8970. doi: 10.1073/pnas.1201652109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farin HF, Van Es JH, Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology. 2012;143(6):1518–1529 e1517. doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 53.Gu P, et al. Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol Cell Biol. 2005;25(9):3492–3505. doi: 10.1128/MCB.25.9.3492-3505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vunta H, Belda BJ, Arner RJ, Channa Reddy C, Vanden Heuvel JP, Sandeep Prabhu K. Selenium attenuates pro-inflammatory gene expression in macrophages. Mol Nutr Food Res. 2008;52(11):1316–1323. doi: 10.1002/mnfr.200700346. [DOI] [PubMed] [Google Scholar]
- 55.Carlson BA, et al. Role of selenium-containing proteins in T-cell and macrophage function. Proc Nutr Soc. 2010;69(3):300–310. doi: 10.1017/S002966511000176X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carlson BA, et al. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 2009;10:57. doi: 10.1186/1471-2172-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burk RF, Hill KE. Selenoprotein P. A selenium-rich extracellular glycoprotein. J Nutr. 1994;124(10):1891–1897. doi: 10.1093/jn/124.10.1891. [DOI] [PubMed] [Google Scholar]
- 58.Burk RF, Hill KE, Motley AK, Austin LM, Norsworthy BK. Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole-body selenium content. Biochim Biophys Acta. 2006;1760(12):1789–1793. doi: 10.1016/j.bbagen.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurokawa S, et al. Sepp1 forms are N-terminal selenoprotein P truncations that have peroxidase activity when coupled with thioredoxin reductase-1. Free Radic Biol Med. 2014;69:67–76. doi: 10.1016/j.freeradbiomed.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lippman SM, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papaioannou D, et al. Antioxidants in the chemoprevention of colorectal cancer and colorectal adenomas in the general population: a systematic review and meta-analysis. Colorectal Dis. 2011;13(10):1085–1099. doi: 10.1111/j.1463-1318.2010.02289.x. [DOI] [PubMed] [Google Scholar]
- 62.Ojuawo A, Keith L. The serum concentrations of zinc, copper and selenium in children with inflammatory bowel disease. Cent Afr J Med. 2002;48(9):116–119. [PubMed] [Google Scholar]
- 63.Andoh A, et al. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition. 2005;21(5):574–579. doi: 10.1016/j.nut.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 64.Cooper ML, Adami HO, Gronberg H, Wiklund F, Green FR, Rayman MP. Interaction between single nucleotide polymorphisms in selenoprotein P and mitochondrial superoxide dismutase determines prostate cancer risk. Cancer Res. 2008;68(24):10171–10177. doi: 10.1158/0008-5472.CAN-08-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Al-Taie OH, et al. A complex DNA-repeat structure within the selenoprotein P promoter contains a functionally relevant polymorphism and is genetically unstable under conditions of mismatch repair deficiency. Eur J Hum Genet. 2002;10(9):499–504. doi: 10.1038/sj.ejhg.5200811. [DOI] [PubMed] [Google Scholar]
- 66.Meplan C, et al. Relative abundance of selenoprotein P isoforms in human plasma depends on genotype, se intake, and cancer status. Antioxid Redox Signal. 2009;11(11):2631–2640. doi: 10.1089/ARS.2009.2533. [DOI] [PubMed] [Google Scholar]
- 67.Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nat Protoc. 2006;1(6):2900–2904. doi: 10.1038/nprot.2006.446. [DOI] [PubMed] [Google Scholar]
- 68.Dieleman LA, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114(3):385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olson GE, Winfrey VP, Hill KE, Burk RF. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J Biol Chem. 2008;283(11):6854–6860. doi: 10.1074/jbc.M709945200. [DOI] [PubMed] [Google Scholar]
- 70.Koh TS, Benson TH. Critical re-appraisal of fluorometric method for determination of selenium in biological materials. J Assoc Off Anal Chem. 1983;66(4):918–926. [PubMed] [Google Scholar]
- 71.Sheehan TM, Gao M. Simplified fluorometric assay of total selenium in plasma and urine. Clin Chem. 1990;36(12):2124–2126. [PubMed] [Google Scholar]
- 72.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14:Unit 14.1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mosser DM, Zhang X. Activation of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14:Unit 14.2. doi: 10.1002/0471142735.im1402s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Read R, Bellew T, Yang JG, Hill KE, Palmer IS, Burk RF. Selenium and amino acid composition of selenoprotein P, the major selenoprotein in rat serum. J Biol Chem. 1990;265(29):17899–17905. [PubMed] [Google Scholar]
- 75.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1):279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.