

Abstract
μ-Opioid agonists mediate their analgesic effect through GPCRs that are generated via alternate splicing of the Oprm1 transcript. While the majority of μ-opioids interact with receptors comprising the canonical 7 transmembrane (7TM) domain, a recently identified class of μ-opioids appears to require a 6TM domain variant. In this issue of the JCI, Lu and colleagues provide an in vivo proof-of-concept demonstration that a 6TM isoform of the μ-opioid receptor can support functional analgesia in Oprm1-deficent animals. The 6TM isoform was pharmacologically distinct from the canonical 7TM μ-opioid receptor, and 6TM agonists had a reduced side effect profile, which confers a strong therapeutic advantage over standard opioid analgesics. The observations of Lu et al. extend the reach of opioid-receptor neurobiology and pharmacology into a new era of analgesic discovery. This advance emerges from a series of fundamental research analyses in which elements of the endogenous opioid system were frequently in the vanguard.
Opioids in the vanguard
A more complete understanding of the neurobiological, molecular, and cellular systems involved in pain sensation and pain control continues to be a challenging, yet productive, proposition. Opioid drugs are unquestionably the most effective treatment available for moderate to severe pain and can provide additional benefits, such as relieving anxiety, reducing the physical or mental effects of trauma, and acting as a soporific and euphorigenic. Unfortunately, the clinical efficacy of these drugs is accompanied by a panoply of perturbing side effects that include respiratory depression, nausea, vomiting, constipation, and sedation, not to mention the development of tolerance and the potential for addiction. All of these effects can be explained at the neurobiological level by the distribution of μ-opioid receptor–expressing neurons. It is noteworthy that investigations of opioid pharmacology and neurobiology include many early, seminal observations in recombinant DNA technology, molecular pharmacology, and medicinal chemistry.
Opioid receptors were first identified in the brain through evaluation of radioligand binding with a tritiated form of the opioid antagonist naloxone (1), which in turn led to isolation of enkephalin, one of the first neuropeptides identified (2). One of the first mRNAs to be cloned was a partial transcript coding for β-endorphin (3), which was isolated from rodent pituitary tumor cells. Subsequently, the full-length mRNAs coding for the precursor proteins of all three families of endogenous opioids proopiomelanocortin (POMC), preproenkephalin (PENK), and preprodynorphin (PDYN) (4–7) were also cloned. After a concerted research effort, the μ-, δ-, and κ-opioid receptors were cloned and sequenced (8–11). The advent of gene targeting and homologous recombination technologies resulted in the opioid receptors being knocked out one at a time and in combination (12, 13) to evaluate their function and ligands. The KO data clearly showed that morphine analgesia is due to expression of the μ-opioid receptor (14). More recently, the crystal structures of the three receptors have been determined (15–17), and through in vivo expression of receptor-fusion proteins, the circuits and cell types that contain these receptors are being defined (18). Many of these milestones were achieved via long-term research investments, such as the membrane protein structural biology initiative component of the NIH Roadmap for Medical Research that began in 2004 (19). This partial account serves to show how opioid research has frequently been in the vanguard of many research fields, and current opioid pharmacology investigations suggest that new steps are being formed.
Medicinal chemistry
Woven throughout these approximately 40 years has been an intensive and unrelenting search for new opioid analgesics — a search that dates back more than 100 years. Heroin was introduced as an improved opioid analog to avoid the side effects of morphine at the end of the 1800s (20). Heroin not withstanding, medicinal chemistry efforts have yielded a host of new pharmacological agents and inventive preparations to modify receptor-subtype selectivity, duration of action, speed of onset, and routes and methods of administration to treat a wide range of pain problems (21). Manipulation of the two ends of the temporal pharmacodynamic spectrum has led to the development of long-acting analogs and cutaneous patches for sustained pain control and formulations that are ultra-rapidly absorbed for the treatment of cancer breakthrough pain or battlefield wounds (22–24). Nonetheless, attempts to divorce analgesic effects from adverse effects continue to prove difficult. In this issue, the study by Lu et al. on splicing and in vivo analgesia, in conjunction with development of biased agonists, provides new approaches to opioid drug research, with potential for therapeutic translation of GPCR biased agonists (25).
Biased agonism
While cloning, sequencing, and structural determinations of opioid receptors have yielded a deep level of molecular definition, several new levels of functional and pharmacological nuance suggest that the goal of an opioid receptor agonist that retains analgesic properties with fewer deleterious side effects may be achievable. Studies over the past 15 years have indicated that the opioid system has another layer of complexity that resides within differential coupling to signaling pathways downstream of receptor binding. This differential activation allows for compounds to selectively engage the G-protein– or β-arrestin–coupled pathways. Studies of β-arrestin 2–deficient mice showed that β-arrestin 2 is necessary for opiate desensitization but not dependence (26). This observation led to the idea that these biased agonists of opioid receptors could be used to elicit the desired pain-reducing effects of opiates with minimal or no side effects. Indeed, many unwanted side effects are decreased in the absence of β-arrestin 2 (27). Subsequently, endomorphin-2 was identified as an endogenous biased agonist at μ-opioid receptor variant 1 (MOR1), which favors arrestin recruitment (28). Several drugs are currently in development to exploit biased agonism to achieve selective actions at opioid receptors (29).
Results from a phase I study of one of the more advanced candidates were recently published (30). Compared with morphine, the biased agonist was more efficacious in the cold pressor test and had a shorter, more benign effect on respiration and less severe nausea; however, the incidence of other CNS side effects, such as somnolence and dizziness, was similar. These data support the idea that targeting biasing mechanisms confers an advantage in side effect profile without compromising analgesic actions. It seems remarkable that the intracellular conformation of the receptor modifies the extracellular binding pocket to an extent that it is pharmacologically distinguishable. Nonetheless, the use of the biased agonism principle is generating new potential analgesic agents.
Alternative receptor splicing
A number of recent studies have identified alternative mRNA splicing as an additional process that generates a receptor capable of discriminating between desired and undesired opiate effects. Early work examined the apparent heterogeneity of μ-receptor splicing and showed that some 5′ splicing generated an N-terminally truncated protein that lacks the extracellular domain and the first transmembrane loop, yielding a 6TM protein. In animal models, the opiate action observed in Oprm1–exon 11 KO animals was different that of Oprm1–exon 1 KO animals (31). Recently, the 6TM proteins that result from the Oprm1–exon 11 splice variant have been proposed as novel analgesic targets of the splice-specific agonist 3-iodobenzoyl-6β-naltrexamide (IBNtxA), which acts on the exon 11–containing variants of MOR1. IBNtxA has analgesic actions, even in animals in which exon 1 variants of MOR1 are removed by gene deletion and in animals that also lack the κ- and δ-opioid receptor paralogs (32).
Lu and colleagues generated mice harboring a disruption of exons 11 (the 5′ most exon) and 1 from the Oprm1 locus, resulting in no possibility of generating any form of MOR1 (25). These mice were insensitive to all μ agonists, including morphine and IBNtxA. IBNtxA analgesia could be rescued in these animals by expression of the 6TM OPRM1–exon 11 splice variant receptor (MOR1G), which was mediated by lentiviral transduction into spinal cord neurons after intrathecal injection. Moreover, MOR1G-transduced animals were insensitive to the analgesic actions of morphine. This essential result demonstrates that the MOR1G splice variant is both necessary and sufficient to confer IBNtxA analgesia. The approach employed by Lu et al. eliminates the potential for an ectopically expressed receptor to interact with the endogenously expressed isoform (25). It is also important to recognize that while IBNtxA analgesia was rescued, lentiviral injection does not necessarily recapitulate the pattern of expression of the endogenous Oprm1 gene (33, 34). Nonetheless, viral overexpression occurred in enough spinal neurons to confer analgesia at this level of the CNS.
Quantitation of splicing using RNA-sequencing (RNA-Seq)
Other studies have also indicated that IBNtxA can produce analgesia without constipation, respiratory depression, physical dependence, or reward behaviors (32). How does IBNtxA retain analgesia, yet does not produce these adverse effects? Furthermore, where exactly in the CNS does IBNtxA work? The answers to these questions have the potential to provide many mechanistic or functional neurobiological answers to long-standing questions. It may be that visceral tissues, as well as respiratory and forebrain centers, lack the splicing mechanisms that produce the 6TM isoform. These regions clearly have the capacity for generation and expression of typical OPRM1 7TM isoforms. The Pasternak group has previously addressed the regional expression of splice variants in the brains of several strains of mice using reverse transcription PCR (RT-PCR) (35, 36). However, splicing can now be examined in more detailed, quantitative, and comprehensive terms using deep RNA-Seq. This method sequences millions of short mRNA fragments and provides a fully quantitative count of the number of fragments associated with each exon of all the genes expressed in a particular tissue or brain region. In libraries prepared from polyA+ mRNA, most of the reads are contained within the borders of an exon (37) but some span the splice junctions and can be used to track splicing patterns for a transcript (Figure 1).
Figure 1. RNA-Seq analysis of mouse Oprm1 exons in Trpv1 lineage DRG.
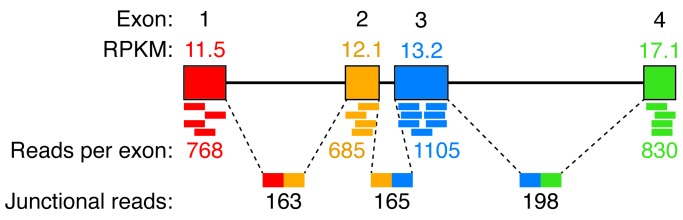
In the mouse DRG, cells expressing RFP under the Trpv1 promoter were separated by FACS and analyzed by RNA-Seq (37). Exons 1, 2, 3, and 4 were clearly identified (colored boxes) with approximately equal reads/kilobase of transcript/million bases sequenced (RPKM), although exon 4 shows a slightly increased RPKM, likely due to additional sequence present at the 3′ end of this exon. Measurements of junctional reads containing sequences from more than one exon provide quantitative assessments of mRNA splicing. Junctional reads were detected between each of the major exons, spanning the introns between them (dashed lines). Additional splicing of Oprm1 was not robustly detected. Notably, the 5′ exon 11 was detected with 5 reads (not shown); however, this is below what can be reliably measured by RNA-Seq.
A brief examination of mouse dorsal root ganglion (DRG) from our recent RNA-Seq provides an informative look at Oprm1 transcript splicing within the pain-sensing primary afferent neurons. In this tissue, using a dataset of approximately 150 million reads from TRPV1-positive nociceptors, a splicing pattern that is consistent with the canonical MOR1 can be detected. Additional upstream 5′ exons were not detected, and the ratio between exons 1 and 2 was approximately equal; an enrichment of exon 2 relative to exon 1 would be expected if there was extensive splicing of 5′ exons. The DRG considerations support a site of action of IBNtxA in the CNS, possibly on MOR1 receptors in the descending control circuits (38) where the splicing of Oprm1 transcripts is thought to be highly varied (39).
Examination of the sequence also raises the question of alternative translation, as the beginning of exon 2 contains a highly conserved Kozak consensus site in the same frame as an ATG in exon 1, indicating that transcriptionally truncated variants could be potentially translated from this start site. Additionally, several forms of MOR1 have been proposed to begin at the exon 2 translational start site. Notably, in the rat, exon 11–containing variants lead to an early stop codon, potentially requiring initiation at exon 2 to produce 6TM variants (39). Further analysis, either immunological or analysis by mass spectrometry, may be informative for N-terminal analyses of 6TM-MOR1 variants.
The future of opiate analgesia
The sustained efforts to understand the molecular biology, biochemistry, and conjoined pharmacology of opioid receptors are apparently still providing several new directions in the quest for a strong analgesic with a reduced side effect profile. The development of biased agonists for the δ- and κ-opioid receptors is in progress (40), and the exploration of splice-specific agonists, such as IBNtxA, is just beginning. The work by Lu et al. demonstrates that MOR1G acts as an inhibitory GPCR upon viral mediated expression in the spinal cord in a μ-opioid receptor–deficient background. Collectively, a series of observations from this same research group suggest a renewed life for the μ-opioid receptor and provide evidence that novel drugs may be designed that can make use of these processes to achieve safer and better analgesia.
Acknowledgments
This research was supported by the Intramural Research Program of the Clinical Center, NIH.
Footnotes
Conflict of interest: Michael J. Iadarola receives royalty payments from Sorrento Therapeutics on US Patent 8,338,457: Selective ablation of pain-sensing neurons by administration of a vanilloid agonist.
Reference information:J Clin Invest. 2015;125(7):2558–2561. doi:10.1172/JCI82060.
See the related article beginning on page 2626.
References
- 1.Pert CB, Snyder SH. Properties of opiate-receptor binding in rat brain. Proc Natl Acad Sci U S A. 1973;70(8):2243–2247. doi: 10.1073/pnas.70.8.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258(5536):577–580. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 3.Roberts JL, Seeburg PH, Shine J, Herbert E, Baxter JD, Goodman HM. Corticotropin and β-endorphin: construction and analysis of recombinant DNA complementary to mRNA for the common precursor. Proc Natl Acad Sci U S A. 1979;76(5):2153–2157. doi: 10.1073/pnas.76.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gubler U, Seeburg P, Hoffman BJ, Gage LP, Udenfriend S. Molecular cloning establishes proenkephalin as precursor of enkephalin-containing peptides. Nature. 1982;295(5846):206–208. doi: 10.1038/295206a0. [DOI] [PubMed] [Google Scholar]
- 5.Kakidani H, et al. Cloning and sequence analysis of cDNA for porcine β-neo-endorphin/dynorphin precursor. Nature. 1982;298(5871):245–249. doi: 10.1038/298245a0. [DOI] [PubMed] [Google Scholar]
- 6.Gubler U, Kilpatrick DL, Seeburg PH, Gage LP, Udenfriend S. Detection and partial characterization of proenkephalin mRNA. Proc Natl Acad Sci U S A. 1981;78(9):5484–5487. doi: 10.1073/pnas.78.9.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11(7):308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- 8.Bzdega T, Chin H, Kim H, Jung HH, Kozak CA, Klee WA. Regional expression and chromosomal localization of the Δ opiate receptor gene. Proc Natl Acad Sci U S A. 1993;90(20):9305–9309. doi: 10.1073/pnas.90.20.9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a Δ opioid receptor by functional expression. Science. 1992;258(5090):1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 10.Meng F, et al. Cloning and pharmacological characterization of a rat κ opioid receptor. Proc Natl Acad Sci U S A. 1993;90(21):9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol Pharmacol. 1993;44(1):8–12. [PubMed] [Google Scholar]
- 12.Zhu Y, et al. Retention of supraspinal Δ-like analgesia and loss of morphine tolerance in Δ opioid receptor knockout mice. Neuron. 1999;24(1):243–252. doi: 10.1016/S0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 13.Matthes HW, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature. 1996;383(6603):819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 14.Sora I, et al. Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A. 1997;94(4):1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485(7398):327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manglik A, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granier S, et al. Structure of the Δ-opioid receptor bound to naltrindole. Nature. 2012;485(7398):400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bardoni R, et al. Δ Opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron. 2014;81(6):1312–1327. doi: 10.1016/j.neuron.2014.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. National Institutes of Health Roadmap for Medical Research. NIH Common Fund Web site. http://commonfund.nih.gov/. Updated April 24, 2015. Accessed April 28, 2015.
- 20.Eddy NB, May EL. The search for a better analgesic. Science. 1973;181(4098):407–414. doi: 10.1126/science.181.4098.407. [DOI] [PubMed] [Google Scholar]
- 21.Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician. 2008;11(2 suppl):S133–S153. [PubMed] [Google Scholar]
- 22.Smith H. A comprehensive review of rapid-onset opioids for breakthrough pain. CNS Drugs. 2012;26(6):509–535. doi: 10.2165/11630580-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shackelford SA, et al. Prehospital pain medication use by U.S. forces in Afghanistan. Mil Med. 2015;180(3):304–309. doi: 10.7205/MILMED-D-14-00257. [DOI] [PubMed] [Google Scholar]
- 24.Wedmore IS, et al. Safety and efficacy of oral transmucosal fentanyl citrate for prehospital pain control on the battlefield. J Trauma Acute Care Surg. 2012;73(6 suppl 5):S490–S495. doi: 10.1097/TA.0b013e3182754674. [DOI] [PubMed] [Google Scholar]
- 25.Lu Z, Xu J, Rossi GC, Majumdar S, Pasternak GW, Pan Y-X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J Clin Invest. 2015;215(7):2626–2630. doi: 10.1172/JCI81070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408(6813):720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 27.Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314(3):1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 28.Rivero G, et al. Endomorphin-2: a biased agonist at the μ-opioid receptor. Mol Pharmacol. 2012;82(2):178–188. doi: 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35(7):308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Soergel DG, et al. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155(9):1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proc Natl Acad Sci U S A. 2009;106(12):4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majumdar S, et al. Truncated G protein-coupled μ opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Natl Acad Sci U S A. 2011;108(49):19778–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spike RC, Puskar Z, Sakamoto H, Stewart W, Watt C, Todd AJ. MOR-1-immunoreactive neurons in the dorsal horn of the rat spinal cord: evidence for nonsynaptic innervation by substance P-containing primary afferents and for selective activation by noxious thermal stimuli. Eur J Neurosci. 2002;15(8):1306–1316. doi: 10.1046/j.1460-9568.2002.01969.x. [DOI] [PubMed] [Google Scholar]
- 34.Iadarola MJ, Brady LS, Draisci G, Dubner R. Enhancement of dynorphin gene expression in spinal cord following experimental inflammation: stimulus specificity, behavioral parameters and opioid receptor binding. Pain. 1988;35(3):313–326. doi: 10.1016/0304-3959(88)90141-8. [DOI] [PubMed] [Google Scholar]
- 35.Xu J, et al. Stabilization of morphine tolerance with long-term dosing: association with selective upregulation of μ-opioid receptor splice variant mRNAs. Proc Natl Acad Sci U S A. 2015;112(1):279–284. doi: 10.1073/pnas.1419183112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, et al. Differential expressions of the alternatively spliced variant mRNAs of the micro opioid receptor gene, OPRM1, in brain regions of four inbred mouse strains. PLoS One. 2014;9(10):e111267. doi: 10.1371/journal.pone.0111267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goswami SC, et al. Molecular signatures of mouse TRPV1-lineage neurons revealed by RNA-Seq transcriptome analysis. J Pain. 2014;15(12):1338–1359. doi: 10.1016/j.jpain.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer DJ, Price DD. Central nervous system mechanisms of analgesia. Pain. 1976;2(4):379–404. doi: 10.1016/0304-3959(76)90080-4. [DOI] [PubMed] [Google Scholar]
- 39.Pasternak GW, Pan YX. μ Opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65(4):1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White KL, et al. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. 2015;352(1):98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]