

Abstract
Targeted mass spectrometry is an essential tool for detecting quantitative changes in low abundant proteins throughout the proteome. Although selected reaction monitoring (SRM) is the preferred method for quantifying peptides in complex samples, the process of designing SRM assays is laborious. Peptides have widely varying signal responses dictated by sequence-specific physiochemical properties; one major challenge is in selecting representative peptides to target as a proxy for protein abundance. Here we present PREGO, a software tool that predicts high-responding peptides for SRM experiments. PREGO predicts peptide responses with an artificial neural network trained using 11 minimally redundant, maximally relevant properties. Crucial to its success, PREGO is trained using fragment ion intensities of equimolar synthetic peptides extracted from data independent acquisition experiments. Because of similarities in instrumentation and the nature of data collection, relative peptide responses from data independent acquisition experiments are a suitable substitute for SRM experiments because they both make quantitative measurements from integrated fragment ion chromatograms. Using an SRM experiment containing 12,973 peptides from 724 synthetic proteins, PREGO exhibits a 40–85% improvement over previously published approaches at selecting high-responding peptides. These results also represent a dramatic improvement over the rules-based peptide selection approaches commonly used in the literature.
Targeted proteomics using selected reaction monitoring (SRM)1 and parallel reaction monitoring (PRM) is increasingly becoming the gold-standard method for peptide quantitation within complex biological matrices (1, 2). By focusing on monitoring only a handful of transitions (associated precursor and fragment ions) for targeted peptides, SRM experiments filter out background signals, which in turn increases the signal to noise ratio. SRM experiments are almost exclusively performed on triple-quadrupole instruments. These instruments can isolate single transitions as an ion beam and measure that beam with extremely sensitive ion-striking detectors. As a result, SRM experiments generally exhibit significantly more accurate quantitation when compared with similarly powered discovery based proteomics experiments, and frequently benefit from a much wider linear range of quantitation (3). SRM experiments often require less fractionation and can be run in shorter time on less expensive instrumentation. These factors allow researchers to greatly scale up the number of samples they can run, which in turn increases the power of their experiment.
However, the process of developing an effective SRM assay is often cumbersome, as subtle differences in peptide sequence can have a profound impact on the physiochemical properties and subsquent SRM responses of a peptide. To successfully develop an SRM assay for a protein of interest, unique peptide sequences must be chosen that also produce a high SRM signal (e.g. high-responding peptides). Once identified, these high-responding peptides are often synthesized or purchased, and independently analyzed to determine the most sensitive transition pairs. Finally, the selected peptide and transition pairs must be tested in complex mixtures to screen for transitions with chemical noise interference and to validate the sensitivity of the assay within a particular sample matrix. Peptides and transitions that survive this lengthy screening process can then undergo absolute quantitation by calibrating the signal intensity against standards of known quantity.
Although experimental methods have been developed to empirically determine a set of best responding peptides (4), these strategies can be time consuming and require analytical standards, which are currently unavailable for all proteins. More often than not, representative peptides are essentially chosen at random, using only a small number of criteria, such as having a reasonable length for detection in the mass spectrometer, a lack of methionine, and a preference for peptides containing proline (5). It is not uncommon for SRM assays to fail at the final validation steps simply because the peptides chosen in the first assay creation step happened to be unexpectedly poor responding peptides.
In an effort to speed up the process of generating robust assays, several groups (6–9) have designed approaches to predict sets of proteotypic peptides using machine-learning algorithms. Proteotypic peptides are peptides commonly identified in shotgun proteomics experiments for a variety of reasons including high signal, low interference, and search engine compatible fragmentation. Enhanced Signature Peptide (ESP) Predictor (7) was the first successful modification of this prediction approach to use proteotypic peptides as a proxy for high-responding peptides for SRM-based quantitation. In brief, Fusaro et al. built a training data set from data-dependent acquired (DDA) yeast peptides and a proxy for their response was quantitated using extracted precursor ion chromatograms (XICs). The authors calculated 550 physiochemical properties for each peptide based on sequence alone and built a random forest classifier to differentiate between the high and low response groups. Other peptide prediction tools follow the same general methodology for developing training data sets. CONSeQuence (8) applies several machine learning strategies and a pared down list of 50 distinct peptide properties. Alternately, Peptide Prediction with Abundance (9) (PPA) uses a back-propagation neural network (10) trained with 15 distinct peptide properties selected from ESP Predictor's 550. The authors of CONSeQuence and PPA found that their approaches outperformed the ESP Predictor on a variety of data sets.
As with most machine learning-based tools, the generality of the training set to real-world data is key to the effectiveness of the resulting prediction tool. Although MS1 intensities extracted from DDA data can be useful for predicting high-responding peptides (11, 12), several factors make them less than ideal for generalizing to SRM and PRM experiments. In particular, DDA peptides must be identified before being quantified and key biochemical features beneficial for targeted analysis of transitions can reduce overall identification rates by producing fragment spectra that are difficult to interpret with typical search engines. By building training data sets on precursor intensities alone these tools ignore the fact that targeted assays actually use fragment ions for quantification. We propose that constructing training sets from DIA fragment intensities (13) will produce machine-learning tools that are more effective at modeling peptides that produce detectible transitions, rather than just proteotypic peptides.
The use of digested proteins in training sets presents additional concerns. The observed variance in peptide intensities is confounded by variation in protein abundance. Converting peptide intensities to ranks can remove the dependence on varying protein levels at the cost of corrupting the training set with proteins that biochemically contain no high-responding peptides. PPA attempts to ease this concern by training with Intensity Based Absolute Quantitation values (14) for DDA peptides estimated from XICs. We hypothesize that constructing a training set from equimolar synthetic peptides removes most adverse effects of digestion from the training set, making it possible to construct a more generalizable tool.
EXPERIMENTAL PROCEDURES
Training Set Stable Isotope Peptides
A total of 1679 stable isotope labeled (SIL) peptides (C-terminal K* = Lys (U-13C6;U-15N2) or C-terminal R* = Arg (U-13C6;U-15N4)) were obtained as a crude (SpikeTide L) mixture from JPT Peptide Technologies GmbH (Berlin, Germany). All peptides are tryptic digestion products of human proteins that have been observed in previous shotgun DDA runs of human samples. This peptide selection may introduce a small bias toward peptides that can be interpreted with DDA, although significant fractionation was required to initially assign many of the peptides. Peptides were acquired with all cysteines alkylated to carbamidomethyl cysteine. In general, the training peptides are representative of normal peptides with one exception: the training data set does not contain peptides with a methionine. One aliquot of the peptide mixture (∼ 0.1 nmol of each peptide) was resuspended in 100 μl of 80% 0.1 m ammonium bicarbonate and 20% acetonitrile. The mixture was bath sonicated for 5 min, vortexed at 37 °C for 5 min. One microliter of the ∼1 picomole/μl solution was diluted in 99 μl of 0.1% formic acid for a 10 fmol/μl solution, which was spun down prior to transferring to a sample vial for liquid chromatography tandem MS (LC-MS/MS) analysis.
Training Set LC-MS/MS Analysis
A 1.5 μl (15 fmol runs) or 4.5 μl (45 fmol runs) aliquot of the SIL mixture was loaded onto a 2 cm × 150 μm Kasil-fritted trap packed with 4 μm Jupiter C12 90A material (Phenomenex, Torrance, CA). The sample was loaded and desalted using 5 μl of a 0.1% formic acid, 2% acetonitrile solution. The trap was brought on-line with the analytical column. The analytical column was a fused-silica capillary (75 μm inner diameter) with a tip pulled using a CO2 laser-based micropipette puller (P-2000; Sutter Instrument Company; Novato, CA). The analytical column was packed with 15 cm of 3 μm Reprosil-Pur C18-AQ beads (Dr. Maisch GmbH, Germany). The analytical column was coupled in-line to a Waters nanoAcquity UPLC pump and autosampler (Waters Corp, Milford, MA). Peptides were eluted off of the column at a flow rate of 300 nL/min using a 90 min gradient of 2–35% acetonitrile in 0.1% formic acid, followed by 35–60% acetonitrile in 0.1% formic acid over 5 min. Peptides were ionized by electrospray (2kV spray voltage) and emitted into a Q-Exactive HF mass spectrometer (Thermo Scientific; Bremen, Germany). Data were acquired using one of two acquisition methods: data-dependent acquisition (DDA) or data-independent acquisition (DIA).
Training Set DDA Acquisition
The DDA method acquires an MS scan analyzing 485–925 m/z with resolution 120,000 (at 200 m/z), automated gain control (AGC) target 3 × 106 charges, and maximum injection time 50 ms. Next, up to 20 MS/MS scans were triggered from the top 20 most intense precursors detected in the MS master scan. The MS/MS scans have resolution 15,000 (at 200 m/z), AGC target 1 × 105 charges, maximum injection time 25 ms, isolation width 1.5 m/z, normalized collision energy 27. Precursors with an intensity below 2 × 105, an unassigned charge state, charge state 1, or charge >5 were excluded. The dynamic exclusion time was 10 s, with isotope peaks of targeted precursors being excluded and the underfill ratio set to 5%.
Training Set DIA Acquisition
A full MS scan was acquired analyzing 495–905 m/z with resolution 60,000 (at 200 m/z), AGC target 3 × 106 charges, and maximum inject time 100 ms. After the MS scan, 20 MS/MS scans were acquired, each with a 20 m/z wide isolation window, resolution 30,000 (at 200 m/z), AGC target 1 × 106 charges, maximum injection time 55 ms, normalized collision energy 27, with the default charge state set to 2. The 20 MS/MS scans were contiguous and collectively cover the m/z range from 500–900 m/z. The cycle of 20 scans (center of isolation window) was as follows (m/z): 510.4819, 530.4910, 550.5001, 570.5092, 590.5183, 610.5274, 630.5365, 650.5456, 670.5547, 690.5638, 710.5729, 730.5820, 750.5911, 770.6002, 790.6093, 810.6183, 830.6274, 850.6365, 870.6456, and 890.6547. The entire cycle of MS and MS/MS scan acquisition takes roughly 2 s and was repeated throughout the LC-MS/MS analysis.
Training-Set Data Processing
The DDA data was searched using Comet 2014.02 rev. 2 against a database containing the heavy-labeled peptide sequences. Prior to searching with Comet, the MS/MS spectra had been processed using Hardklor (15) v. 2.16 and Bullseye (16) v. 1.30 to assign more accurate precursor matches based on analysis of MS spectra and remove MS/MS spectra without a matching MS1 precursor. The peptide-spectrum matches were processed with Percolator (17) v. 2.07 to assign q-values to peptide spectrum matches and peptide identifications. Bibliospec (18) v. 2.0 was used to combine the peptide-spectrum matches into a spectral library containing any spectra with q < 0.3. The score cutoff is extremely loose because the spectral library is simply used as an aide for manually choosing peaks during processing of the DIA data.
The DIA data were analyzed using the Skyline (19) software package. In Skyline, chromatograms were extracted for the +2 and/or +3 charged precursor of each peptide that fell within the analyzed 500–900 m/z range. For each peptide precursor, chromatograms were extracted for the M, M+1, and M+2 precursor ions from the MS data, and chromatograms for the y-ion series (ion 2 to last ion −1) were extracted from the MS/MS data. The chromatographic peaks for each peptide precursor were manually selected and integrated in each of the four DIA data sets acquired. The retention time of library matches from the DDA data were overlaid on the DIA data to aid in selecting the correct peak. Additionally, the mass measurement error (< 10 ppm), similarity in ratios of the area of the precursor peaks to the theoretical isotope distribution, and similarity in the ratios of the area of the extracted fragment ion chromatograms from the DIA data to matches in the spectral library were used to verify that the correct chromatographic peak was being integrated. In the vast majority of cases, there was a single, intense peak meeting all of these criteria. When this was not the case, the peptide precursor was discarded, resulting in a total of 1331 confidently detected peptides remaining. Fragment ions showing interference were also discarded.
SRM Testing Set and Training Cross Validation Set
The data presented in Stergachis et al. was used as a primary testing data set. A new SRM training cross validation data set was constructed using the protocols presented in Stergachis et al. Briefly, clones for GST fusion proteins from the pANT7_cGST clone collection (20) were synthesized in vitro using the Pierce 1-step Human Coupled in vitro protein synthesis kit (Thermo Scientific; Bremen, Germany). In instances where a cDNA clone was unavailable, recombinant proteins were purchased from a commercial source. GST tagged proteins were captured using glutathione Sepharose 4B beads (GE Healthcare Life Sciences; Pittsburgh, PA), and iteratively washed to remove nonspecific binders. Bead bound GST fusion proteins were individually denatured with 5 mm dithiothreitol (DTT) for 30 min at 60 °C and alkylated with 15 mm iodoacetamide for 30 min at room temperature. Proteins were then digested with 1 μg of sequencing grade modified porcine trypsin (Promega, Madison, WI) for 2 h at 37 °C.
Protein digests were resolved on a 12 cm × 150 μm analytical column packed with ReproSil-Pur 3 μm C18-AQ beads (Dr. Maisch GmbH, Germany). The analytical column was coupled in-line to a Waters nanoAcquity UPLC pump and autosampler (Waters Corp). Peptides were eluted off the column at a flow rate of 0.75 μl/min using 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) following this linear solvent schedule: 0–7 min, 95–60% A; 7.0–7.1 min, 60–32% A; 7.1–8.0 min, 32% A; 8.0–8.1 min, 32–5% A; 8.1–11.0 min, 5% A; 11.0–11.1 min, 5–95% A; 11.1–18.0 min, 95% A. Peptides are ionized by electrospray and emitted into a TSQ-Vantage triple quadrupole instrument (Thermo Scientific). Doubly charged, fully tryptic peptides of length 7 to 23 for each protein were analyzed using the Skyline software package. Peptide fragment chromatograms for the y-ion series (ion 3 to last ion −1) were extracted from the MS/MS data and quantified. Forty-four of the proteins were used for training cross validation to protect against over fitting. The 18 remaining proteins were reserved exclusively for a secondary testing data set and used only after training was complete.
Peptide Response Prediction
Peptide responses for peptides in the Stergachis et al. SRM testing data set were predicted using PPA, CONSeQuence, and ESP Predictor. PPA RC4 (available online at http://software.steenlab.org/rc4/PPA.php) was used using the default parameters (peptide mass from 600 to 6000 and minimum peptide length of 5). The artificial neural network and linear support vector machine components of CONSeQuence (available online at http://king.smith.man.ac.uk/CONSeQuence/) were run independent of the consensus binary score. The consensus binary score was not used because it produces only four discrete values, which made it impossible to compare against the other scoring systems. ESP Predictor version 3 (available online at http://www.broadinstitute.org/cancer/software/genepattern/esppredictor) is parameter-free.
RESULTS
Challenges in Predicting Peptide Responses
Peptide response factors within proteins vary widely: on average by over three orders of magnitude between the highest and lowest responding peptides. Stergachis et al. has presented previously an experimental method for determining the best responding peptides to monitor proteins in targeted experiments. This method was shown by synthesizing over 700 human transcription factors in vitro and generating SRM assays for all singly charged, monoisotopic y3 to yn−1 ions from virtually every tryptic peptide. Because of variations in translation, proteins in this experiment were not produced at the same level. However, all peptides within a given protein were guaranteed to be present at equimolar levels, and using this knowledge, the authors were able to determine which peptides produced the best SRM transitions for in vivo monitoring. In this work, we use the Stergachis et al. data set as an independent test set to validate our methods. Some potential limitations of this data set for benchmarking include that it was acquired only considering precursor charge state +2 peptides (that may bias against high basicity peptides and very long peptides), and that analyzed fragment ions were limited to only y-type ions. We feel that the benefits of the scale of this data set outweigh these limitations.
The Stergachis et al. data set provides an excellent testing ground for understanding the challenges in predicting peptide responses. Fig. 1 illustrates the range of peptide transition responses in the Stergachis et al. SRM data set. Although the median dynamic range of peptide responses within a protein was 3.4 orders of magnitude, some rare proteins shown response ranges of up to five or six orders of magnitude. An example distribution for CASZ1, a typical transcription factor with an apparent dynamic range of 4.1 orders of magnitude, is shown in supplemental Fig. S1. This wide diversity of responses underlines the need for a robust mechanism for choosing peptides to target. In this work, we leverage the Stergachis et al. data set containing 12,973 peptides from 724 proteins (with a median of 15 peptides per protein and a mode of 10) to test our approach for predicting peptide responses for SRMs and PRMs.
Fig. 1.
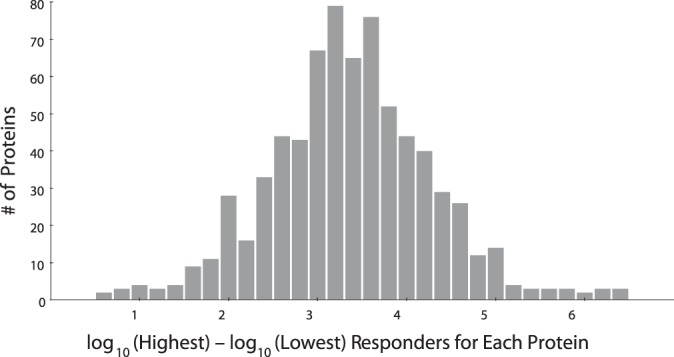
A histogram of the dynamic ranges calculated for 724 proteins. The dynamic range is estimated as the number of orders of magnitude separation for each protein. This value is calculated as the difference between the log10 intensities of the highest responding peptide and the lowest responding peptide. The median dynamic range is 3.4 orders of magnitude, with an interquartile range of 1.2 orders. All protein intensity data was drawn from the Stergachis et al. SRM testing data set.
Training Set Preparation
Training data sets that are generalizable to real world applications are critical for effective machine learning. However, creating an exhaustive targeted data set of equimolar peptides for training a peptide response prediction model is extremely time consuming as it would require very many SRM experiments to account for all potential transition ions for every peptide. We have developed a strategy for generating large-scale, realistic SRM and PRM-like training sets using DIA MS/MS experiments acquired on a QExactive-HF (Thermo Scientific) using HCD fragmentation. For the purposes of determining a training data set, DIA MS/MS has the advantage that all sequence specific fragments are measured, making it easy to identify the most promising transitions. Additionally, we used beam-type higher energy collisional dissociation (HCD) fragmentation to generate fragments, which is very similar to triple-quad fragmentation used in most SRM experiments (21). We derived the training set from the most intense singly charged y-type fragment intensity for each of 1679 stable isotope labeled peptide detections made by Skyline, given certain restrictions. Only singly charged y-type fragments were used because b-type fragments can lose carbon monoxide to form a-type fragments, resulting in both lowered response and increased variability. Also, typically the b-ion series undergoes multiple collisions in beam-type instruments and fragments to smaller product ions until it stops at the b2 ion. This fragment ion is frequently one of the most intense but least selective product ions in the spectrum. First, we filtered our list of potential signature y-type fragment ions to remove nonspecific y2 fragments. Then, for each acquisition, we removed the 2.5% worst fragment ions by mass accuracy in both directions (supplemental Fig. S2). At this point, we estimated the maximum y-type fragment for each peptide as a proxy for the maximum transition response.
Because peptide detections were made from two pairs of acquisitions at different amounts (∼45 fmol and 15 fmol on-column), we were able to use the distribution of parent-intensity quantitative ratios to indicate outlier peptides (supplemental Fig. S3). Based on this analysis, we removed 69 SIL peptides from the initial 1331 detected peptides that eluted earlier than 30 min or later than 85 min from further analysis. In our runs, early eluting peptides tended to saturate in ratio between 45 fmol and 15 fmol injections, suggesting that their intensities were unreliable. Peptides eluting after 85 min were excluded because our instrument tuning parameters made their intensities also unreliable. After removing these peptides, we recalculated the median ratio of the two pairs of acquisitions to be 2.45, slightly under the expected 45:15 fmol ratio. We estimated the overall intensity for each peptide as the average of the intensities from the 45 fmol acquisition and 2.45 times the 15 fmol intensities and removed the peptides with the 2.5% highest and 2.5% lowest ratios to compensate for peptides with unstable responses. This resulted in a final training data set of 1186 well-behaved peptides, which are presented in supplemental Table S1. Summary statistics about these peptides are presented in supplemental Fig. S4. Finally, we ranked the peptides in the training set based on these aggregate fragment ion intensities and linearly normalized the ranks to be between zero and one.
Physiochemical Property Selection and Artificial Neural Network Training
For each peptide sequence we calculated 550 physiochemical properties used by ESP Predictor, the large majority of which were derived from the Amino Acid Index Database (22). We point out that one potential source of variability is that cysteines used in this work (and in proteomics generally) are alkylated, whereas the majority of the Amino Acid Index Database properties assume cysteines are unmodified. We normalized the values for these properties to be between zero and one. We selected meaningful physiochemical properties using a minimum redundancy, maximum relevance (mRMR) algorithm (23, 24). For each property, we calculated the Pearson's correlation coefficient of ranked peptides with the property values derived from their respective peptide sequences. The property with the highest correlation was selected as a meaningful feature and all other properties that correlate with that feature at an absolute Pearson's correlation coefficient of >0.3 are removed. This process is iterated using the remaining properties until all properties that have any positive correlation to the intensity ranks are either selected or removed.
The mRMR algorithm produced 11 most relevant physiochemical properties. These properties and their correlation to the ranked training intensities are listed in Table I. As the mRMR algorithm chooses the most representative of several properties, the specific properties themselves are less important than their higher-level classification. Peptides with lower molecular weights correlated strongest with high transition intensities in our training set, followed by various structural and hydrophobicity properties.
Table I. Most relevant physiochemical peptide properties.
| Rank | Correlation Coefficienta | Peptide property | Property typeb |
|---|---|---|---|
| 1 | -0.53 | Peptide mass | Size |
| 2 | -0.36 | Average relative preference value at C1 (28) | Structural |
| 3 | -0.33 | Average activation Gibbs energy of unfolding, pH7.0 (29) | Hydrophobicity |
| 4 | -0.27 | Average hydrophobicity coefficient in RP-HPLC, C4 (30) | Hydrophobicity |
| 5 | -0.20 | Average normalized frequency of zeta R (31) | Structural |
| 6 | 0.20 | Average linker propensity from 1-linker data set (32) | Structural |
| 7 | 0.16 | Average hydrophobicity coefficient in RP-HPLC, C18 | Hydrophobicity |
| 8 | 0.15 | Average AA composition of EXT2 of single-spanning proteins (33) | Structural |
| 9 | -0.14 | Average normalized frequency of α-helix in all-α class (34) | Structural |
| 10 | 0.08 | Average relative population of conformational state A (35) | Structural |
| 11 | 0.07 | Average surface composition of AAs in intracellular proteins of thermophiles (36) | Structural |
a Peptide properties were iteratively selected from a pool of 550 total properties based on their Pearson's correlation with the intensity ranks in the training data set. Properties are sorted based on the absolute value of the correlation coefficient, which is an indication of their importance for classification. Negative correlations indicate inverse relationships. As each feature was selected, redundant features with interproperty correlation coefficients >0.3 were removed.
b Peptide properties were loosely categorized into three types, those corresponding with peptide size, secondary structure, and hydrophobicity.
The final training set consisted of the top 25% (high responders) and the bottom 25% (low responders) of peptides to promote differentiation between high and low responding peptides, where the expected output was the percentage intensity rank. We constructed a back-propagation neural network with 11 input neurons corresponding to the 11 mRMR-selected relevant physiochemical properties, eight hidden neurons in a single layer, and a single output neuron. We configured the neural network for a 10% learning rate and trained it to reach a minimum recall error level of 1%. Neural networks typically produce a score between zero and one, indicating the classification of the input feature set. Instead of using the neural network score directly, the PREGO score was assigned to:
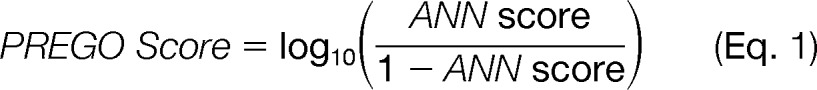 |
in an effort to stratify scores that clump around zero and one. This score is analogous to the log-likelihood ratio statistic for comparing two classification models. Pseudo code of the PREGO algorithm is presented in Fig. 2.
Fig. 2.

Algorithmic outline of the PREGO method. A, Algorithmic outline describing feature selection using an mRMR style algorithm to identify nonredundant features with maximum relevance. Feature sets with low redundancy often decrease the potential for over-training in machine learning algorithms. B, Algorithmic outline for neural network construction using the mRMR-selected feature set. C, Testing of the algorithm was performed using the Stergachis et al. SRM testing data set.
There are many decisions to make when picking a supervised machine learning architecture. As with PPA and CONSeQuence, we chose to implement an artificial neural network because “deep architectures” (like ANNs) tend to perform better than “shallow architectures” (e.g. support vector machines) on “deep learning” tasks (25). However, unlike the support vector machine approach to gradient descent, back-propagation gradient descent is random in nature, causing artificial neural networks to often converge on local minima, rather than global minima. Consequently, we trained 1000 different ANNs and cross validated them using 44 proteins selected from an exhaustive SRM data set modeled after the Stergachis et al. experiment. We selected the best model that maximized the area of the receiver operating characteristic (ROC) that compared the number of peptides picked per protein versus the number of proteins where at least one high-responding peptide was picked. For each protein, peptides were considered high responders if they produced a single most intense y-type fragment ion for each peptide in the top 20% of peptides from that protein. This approach also provides a buffer against over-fitting because we trained using DIA data and cross validated the training models with SRM data acquired in a completely different manor.
Evaluation of PREGO
We evaluated PREGO using the Stergachis et al. data set, which describes experimental SRM transition responses acquired for almost 13,000 peptides found in over 700 proteins. For consistency with our current practice we reprocessed this data set to quantitate using the only the single most intense fragment ion (y3 to yn−1), whereas the original publication used the sum of those ions. Fig. 3 shows PREGO scoring for CASZ1, a representative protein in this data set. CASZ1 has a Pearson's correlation coefficient of 0.65 when compared with the experimental intensity ranks, the mode of the correlation distribution across all proteins in the data set (supplemental Fig. S5). Although there is significant deviation in any individual measurement, PREGO scores are generally high in cases of high-responding peptides, and low with less responsive peptides. Supplemental Fig. S6 illustrates the range of PREGO scores for a variety of proteins that show similar trends with correlation coefficients ranging from 0.9 to 0.2.
Fig. 3.
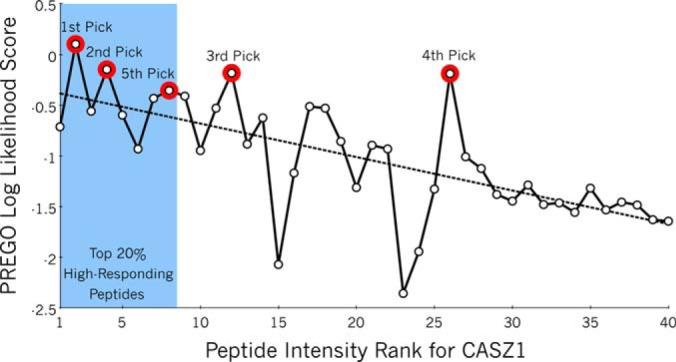
PREGO Scores for peptides in CASZ1. Peptides in CASZ1 (also known as cDNA FLJ20321) are ranked on their experimentally acquired transition fragment intensity from the Stergachis et al. SRM testing data set where the peptide with the strongest response is awarded a rank of one. The top 20% of peptides by intensity rank are considered “high-responding peptides” and are shaded in blue. The top five peptides chosen by PREGO are marked with red borders. Although there is large variation in predicting response intensities for any given peptide (solid line), there is a definite trend (dashed line) to score first ranked peptides somewhat higher than worse ranked peptides. Consequently, the highest scoring peptides picked by PREGO are often also high-responding peptides. CASZ1 represents a “typical” protein with a correlation score of 0.65.
We combined traces like those shown in Fig. 3 across all proteins in the Stergachis et al. data set. Fig. 4A depicts the distribution of PREGO scores for peptides at various ranks in all of the proteins, where the black line indicates the median score and the gray shaded area indicates the interquartile range. Following the trend shown in Fig. 3, there is wide scatter at each individual rank. However, the downward trend in scores as rank decreases suggests that PREGO is able to differentiate peptide responses in SRM experiments.
Fig. 4.

Score distributions for four scoring methods by peptide rank. A, The PREGO score distribution for peptides of descending rank across the entire Stergachis et al. SRM testing data set. The median ranks are annotated as dots, where the nearest-neighbor-smoothed trend is plotted as a black line. The interquartile range (Q1 to Q3) is shaded blue. In general, first ranked peptides with the highest responses tend to get higher scores than those of lower ranks, as indicated by the downward trend from left to right. The B, PPA score distribution as well as the CONSeQuence; C, artificial neural network (ANN); and D, support vector machine (SVM) score distributions all show weaker downward trends.
Fig. 4B shows a similarly generated scoring profile for PPA on the same set of proteins. Although there is a slight downward trend in the median, PPA assigns high scores to peptides at all ranks. The spreading shape of the distribution suggests that PPA is more likely to assign low scores to low responding peptides. For any given protein, PPA eliminates some of these low responding peptides from the pool of options and thus increases the odds for choosing a high-responding peptide. CONSeQuence score distributions using both the artificial neural network option and the SVM option are depicted in Fig. 4C and 4D, respectively. In this data set, CONSeQuence produces a slight downward trend in scores with poorer responding ranks, although the scatter in the distributions overwhelms any major trends.
Although it is important that response prediction scoring schemes correlate with experimental peptide intensities, these algorithms will mainly be used to select multiple peptides to quantitate a protein in the hopes that at least one produces a strong response. The approaches need not identify the highest responding peptide every time; to be effective they must be able to select at least one relatively strong responding peptide in a handful of guesses. Fig. 5A asks the question: “If we selected N peptides for any given protein, would at least one of those peptides show high response?” We defined high response as being in the top 20% of peptides for each protein by rank-response. Given these criteria, on average PREGO correctly selects a high-responding peptide 57% of the time on the first selection. Similarly, if two peptides per protein are selected, then at least one is a high responder 80% of the time, and on average selecting three peptides produces a high responder 90% of the time. At each of these three stages, PREGO selects high responders ∼40% to 85% more often than the best competing methods.
Fig. 5.
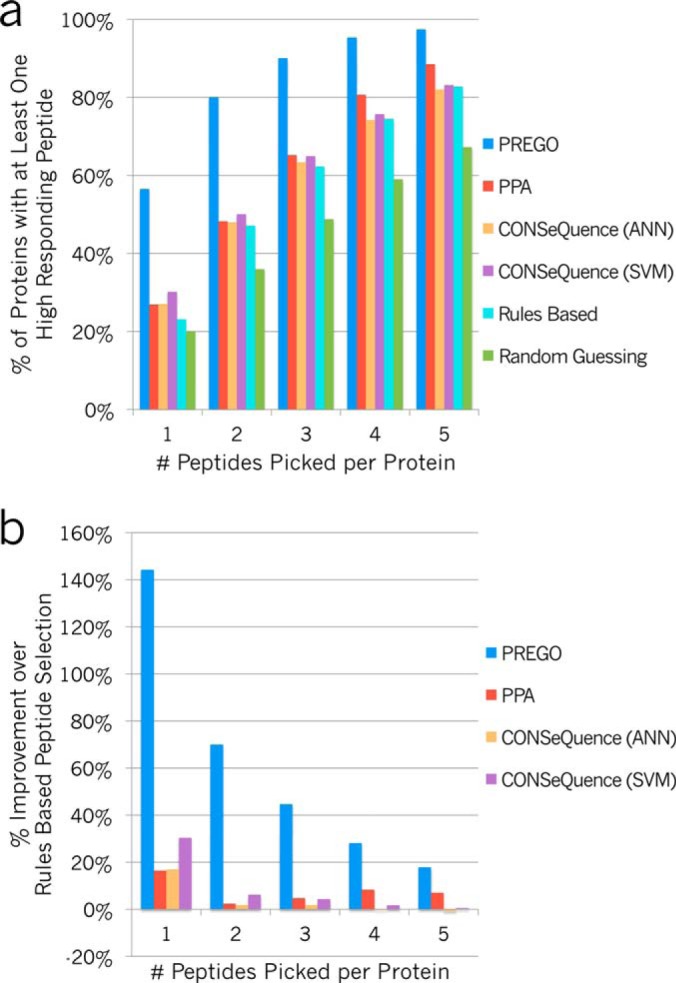
Percentage of proteins with at least one high-responding peptide, given N peptides picked. A, PREGO (blue), PPA (red), CONSeQuence artificial neural network (ANN, orange), and support vector machine (SVM, purple) machine learning-based scorers are compared with randomly guessing to select peptides (green) and the simple scoring function described in Equation 2 (cyan) based on common rules in the literature. Scorers are graded based on the likelihood that for any given protein, they could predict at least one high-responding peptide given N guesses. This is analogous to the strategy of picking N peptides to produce at least one useful peptide for each protein. For example, in Fig. 3 the top 1–5 peptides picked in CASZ1 have red borders and the high-responding peptides are shaded in blue. B, The same four learning-based scorers as a percentage improvement over rules based peptide selection. PREGO is dramatically better than the other approaches tested here at predicting high-responding peptides given five or fewer chances. All scoring data is based on the Stergachis et al. SRM testing data set.
As a baseline, Fig. 5A includes statistical calculations for selecting peptides entirely at random. However, typically scientists select peptides to build SRM and PRM assays by employing several simple selection rules and choosing randomly among the peptides that pass those rules. We built a simple scoring scheme to capture the Bereman et al. rules strategy that has bonuses for prolines (that produce strong fragmentation signatures) and penalties for methionine (that can be oxidized), asparagine/glutamine (that can be deamidated), glutamine/glutamic acid in the N-terminal position (that can cyclize to form pyroglutamic acid), and carbamidomethyl-cysteine in the N-terminal position (that can also cyclize). The rules-based “score” is a summation of values across all of the n amino acids in a peptide:
 |
Not surprisingly this strategy performs somewhat better than the baseline of randomly guessing. Fig. 5B illustrates the relative improvement of PREGO and the other various trained approaches over the rules based approach. All of the trained approaches improve over the rules based approach when only considering the top peptide. However, it is rare that scientists choose only a single peptide per protein for targeted assays. As one chooses more peptides at random, there is an increasing chance that at least one is a high-responding peptide and that correspondingly makes it increasingly harder to do a better job. An unexpected result is that when choosing two or more peptides from the Stergachis et al. data set, simply using the Bereman et al. rules performs essentially equivalently to the PPA and CONSeQuence methods. PREGO, on the other hand, continues to show increased performance over the rules based approach when choosing a typical number of peptides for targeted assays. supplemental Fig. S7 shows similar results using the reserved 18 proteins from the secondary testing SRM experiment collected separately.
DISCUSSION
It is important to note that in the situation of predicting peptides for building SRM and PRM assays any level of success is still success. The factors that determine peptide response are largely unknown and are likely staggering in number and complexity. Consequently, the vast majority of labs generating targeted assays do so by selecting peptides virtually at random using some variation of the rules described in Bereman et al. Improvement over these rules is the main measuring stick that peptide response prediction algorithms should be compared with.
Despite dramatically different training sets and machine learning architectures, PPA and both CONSeQuence scoring systems produce essentially identical success rates. We show that these software tools perform somewhat better than randomly selecting tryptic peptides for SRM assays, but not significantly better than using a rules-based random guessing approach for estimating peptide response characteristics. This suggests that there may be a glass ceiling for predicting SRM response behavior based on peptide responses in large-scale DDA data sets. Our results indicate that the PREGO algorithm produces a dramatic improvement over these other methods for building SRM assays.
Although the algorithmic improvements we propose likely provide some incremental improvement, we suspect that the large majority of PREGO's success stems from our training data set selection. In particular, we believe that training from DIA data sets using the QExactiveHF allows us to more closely represent data acquisition strategies employed by traditional SRM triple-quad instruments. In addition, DIA allows us to more accurately predict transition response directly from peptide fragmentation, instead of assuming that precursor intensities equate with fragment intensities. We find that there is an order of magnitude variation between product and precursor intensities (supplemental Fig. S8), which suggests that training using transition responses ought to be more accurate than training from precursors alone. Another key improvement is that PREGO ensures robust generalization by cross validating the DIA trained artificial neural network with SRM data. As different mass spectrometers and LC conditions can have a profound effect on peptide ionization, training using multiple diverse types of data from different sources is essential.
We also note that the underperformance of PPA and CONSeQuence may be partially driven by two aspects of our evaluation approach. First, data acquisition in the testing data set was restricted to only doubly charged precursor ions, and second, peptide response was evaluated using only the single most intense y-type fragment ion from each peptide. These aspects represent important practical considerations commonly employed in SRM assays and were incorporated into the training of PREGO but not in PPA or CONSeQuence.
Peptide response prediction can also be used to improve peptide-centric DIA search engines. Search engines that take this approach to querying DIA data sets can benefit from increased sensitivity using an SRM-like data analysis workflow. However, by individually considering every peptide for all proteins in a database, the peptide-centric approach suffers from a significantly increased false discovery rate that must be accounted for using multiple hypothesis testing corrections, which consequently decrease any sensitivity gains. Instead of looking for every possible peptide, PREGO can drastically help narrow down the search space by first considering only a handful of high-responding peptides per protein. A peptide-centric DIA search engine then only needs to look for low-responding peptides if high-responders are seen.
Critical Evaluation
We make one major assumption in the construction of our DIA training data: we assume that crude peptides in our mixture are essentially at equimolar concentrations. We make this assumption because developing a training set from purified peptides would be prohibitively expensive. JPT estimates that these peptides are between 20 and 90% pure, suggesting that there is somewhat less than fivefold variation in their original concentrations. We believe that, although this variation is significant, the unknown level of variation in proteoforms present for each gene product would overwhelm it if we were to use biological samples, such as with the PPA or CONSeQuence methods. We also believe that the benefits of removing the assumption that high ranked peptides in each protein produce equivalently high fragment ion intensities outweighs any detriments in using crude peptides. On the other hand, training using the single most intense y-type fragment ion for each peptide might bias PREGO toward preferring peptides with dominant fragmentation pathways. Also, the most intense fragment ion by DIA might differ from the most intense fragment ion by SRM where collision energies can be tuned to produce the most reliable and easy to detect fragmentation on a peptide-by-peptide basis.
Similarly, varying efficiencies in tryptic digestion are also not accounted for with synthetic peptides. This may be an advantage from the standpoint of machine learning in that training goals are focused solely on identifying peptide sequences that produce strong signals rather than being complicated by trying to interpret multiple layered sources of variation at the same time. The effects of incomplete digestion are difficult to ascertain in this experiment because the Stergachis et al. SRM data set only assayed 1445 peptides with missed cleavages (1.2%). However, incomplete digestion can be a significant concern when interpreting particular classes of peptides, for example phosphopeptides. In the future additional layers of focused training or filtering may help account for digestion efficiency.
It is important to note that although PREGO performs better than alternative methods, there is still considerable variability in the scores produced for each peptide. This is primarily because peptide transition response is the product of many complex factors, only some of which can be captured using amino acid frequency-based physiochemical properties. The gold standard for predicted peptide response remains as experimental evidence derived from synthetic proteins. The utility of PREGO is primarily in situations where experimental data from controlled systems is expensive, time-consuming, or even impossible to generate. Considerable room for improvement still remains with future prediction methods to use more diverse training data sets and more complex properties crafted for modern proteomics methods that consider secondary and tertiary gas-phase structure and interactions.
CONCLUSIONS
We present a new method, PREGO, to predicting high-responding peptides to aid in generating SRM and PRM assays. Our approach uses DIA experimental data of equimolar synthetic peptides to train an artificial neural network using 11 features selected with a Pearson correlation-based minimum redundancy, maximum relevance algorithm. We have validated our software using a massive SRM data set measuring virtually every possible tryptic peptide from over 700 proteins.
We designed PREGO to make it easy to train new neural network models based on future data sets. We expect that as comprehensive DIA or PRM experiments of synthetic peptides are performed, the resulting data sets could be used to improve the accuracy of the approach. New models can be constructed based on specific experimental conditions; in particular we imagine designing models to predict PTM modified peptide responses, such as those of captured phosphopeptides using immobilized metal affinity chromatography or titanium dioxide enrichment. All that is required to retrain PREGO is a tab-delimited text file containing two columns: peptide sequences and experimental intensities. PREGO can score peptides for predicted response levels using a text file containing a single column of sequences.
Although PREGO can be used for predicting the best responding SRM peptide; it makes no attempt to predict the best responding transition. Other modeling software, such as the thermodynamic peptide fragmentation model presented by Zhang (26, 27) will be required to make those predictions. Here we see inexpensive synthetic crude peptides as another answer. Because of the variability in actual abundance, it is hard to estimate specific best responding SRM peptides from a massively parallel crude mixture. However, we intend to use PREGO to predict generally which peptides will be worth targeting and using inexpensively purchased synthetic crude peptides to identify preferred y-type ion transitions from MS/MS experiments. These issues are rendered moot with regards to PRM experiments because in that methodology all fragment ions are measured.
PREGO is written in Java and is available as an external tool for Skyline. We have also released source code and cross platform binaries for PREGO on GitHub at https://github.com/briansearle/intensity_predictor under the Apache 2 license. The MS/MS data files used to train PREGO are available in mzML standard format at http://proteome.gs.washington.edu/SearleMCP and in RAW format at https://chorusproject.org/anonymous/download/experiment/-8935943952383739133. The exhaustive SRM training cross validation data is available on PanoramaWeb at https://panoramaweb.org/labkey/PREGO_manuscript.url.
Supplementary Material
Acknowledgments
We thank D.R. Mani (The Broad Institute of MIT and Harvard) and Vincent Fusaro (Harvard Medical School) for helpful discussions about ESP Predictor. We also thank Richard S. Johnson for help with the mass spectrometry experiments and Vagisha Sharma for help with PanoramaWeb.
Footnotes
Author contributions: B.C.S. and M.J.M. designed research; B.C.S., J.D.E., J.G.B., and A.B.S. performed research; B.C.S. analyzed data; B.C.S., J.D.E., J.G.B., A.B.S., and M.J.M. wrote the paper.
* This work is supported in part by National Institutes of Health grants P41 GM103533 and R01 GM107142.
 This article contains supplemental Figs. S1 to S8 and Table S1.
This article contains supplemental Figs. S1 to S8 and Table S1.
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- DDA
- data-dependent acquisition
- DIA
- data-independent acquisition
- ESP
- enhanced signature peptide
- mRMR
- minimum redundancy, maximum relevance
- PPA
- peptide prediction with abundance
- PRM
- parallel reaction monitoring
- SIL
- stable isotope labeled
- XIC
- extracted ion chromatograms.
REFERENCES
- 1. Marx V. (2013) Targeted proteomics. Nat. Methods 10, 19–22 [DOI] [PubMed] [Google Scholar]
- 2. Liebler D. C., Zimmerman L. J. (2013) Targeted quantitation of proteins by mass spectrometry. Biochemistry 52, 3797–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Picotti P., Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls, and future directions. Nat. Methods 9, 555–566 [DOI] [PubMed] [Google Scholar]
- 4. Stergachis A. B., MacLean B., Lee K., Stamatoyannopoulos J. A., MacCoss M. J. (2011) Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods 8, 1041–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bereman M. S., MacLean B., Tomazela D. M., Liebler D. C., MacCoss M.J. (2012) The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 12, 1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mallick P., Schirle M., Chen S. S., Flory M. R., Lee H., Martin D., Ranish J., Raught B., Schmitt R., Werner T., Kuster B., Aebersold R. (2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25, 125–131 [DOI] [PubMed] [Google Scholar]
- 7. Fusaro V. A., Mani D. R., Mesirov J. P., Carr S. A. (2009) Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 27, 190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eyers C. E., Lawless C., Wedge D. C., Lau K. W., Gaskell S. J., Hubbard S. J. (2011) CONSeQuence: Prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol. Cell. Proteomics 10, M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muntel J., Boswell S. A., Tang S., Ahmed S., Wapinski I., Foley G., Steen H., Springer M. (2015) Abundance-based classifier for the prediction of mass spectrometric peptide detectability upon enrichment. Mol. Cell. Proteomics 14, 430–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rumelhart D. E., Hinton G. E., Williams R. J. (1986) Learning representations by back-propagating errors. Nature 323, 533–536 [Google Scholar]
- 11. Prakash A., Tomazela D. M., Frewen B., Maclean B., Merrihew G., Peterman S., Maccoss M. J. (2009) Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 8, 2733–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mead J. A., Bianco L., Ottone V., Barton C., Kay R. G., Lilley K. S., Bond N. J., Bessant C. (2009) MRMaid, the web-based tool for designing multiple reaction monitoring (MRM) transitions. Mol. Cell. Proteomics 8, 696–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Egertson J. D., MacLean B., Johnson R., Xuan Y., MacCoss M. J. (2015) Multiplexed peptide analysis using data independent acquisition and skyline. Nat. Protoc. 10, 887–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 15. Hoopmann M. R., Finney G. L., MacCoss M. J. (2007) High speed data reduction, feature selection, and MS/MS spectrum quality assessment of shotgun proteomics datasets using high-resolution mass spectrometry. Anal. Chem. 79, 5630–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsieh E. J., Hoopmann M.R., MacLean B., MacCoss M. J. (2010) Comparison of database search strategies for high precursor mass accuracy MS/MS data. Proteome Res. 9, 1138–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 18. Frewen B. E., Merrihew G. E., Wu C. C., Noble W. S., MacCoss M. J. (2006) Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal Chem. 78, 5678–5684 [DOI] [PubMed] [Google Scholar]
- 19. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. The pANT7_cGST Clone Collection, https://dnasu.org/DNASU/Home.do.
- 21. de Graaf E. L., Altelaar A. F., van Breukelen B., Mohammed S., Heck AJ. (2011) Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 [DOI] [PubMed] [Google Scholar]
- 22. Kawashima S., Kanehisa M. (2000) AAindex: amino acid index database. Nucleic Acids Res. 28, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding C., Peng H. J. (2005) Minimum redundancy feature selection from microarray gene expression data. Bioinform. Comput. Biol. 3, 185–205 [DOI] [PubMed] [Google Scholar]
- 24. Peng H., Long F., Ding C. (2005) Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 27, 1226–1238 [DOI] [PubMed] [Google Scholar]
- 25. Bengio Y., LeCun L. (2007) Scaling Learning Algorithms towards AI in Bottou L, Chapelle O, DeCoste D, Weston J. (Eds.) Large-scale kernel machines (pp 321–360) Cambridge, MA, MIT Press [Google Scholar]
- 26. Zhang Z. (2004) Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 3908–3922 [DOI] [PubMed] [Google Scholar]
- 27. Zhang Z. (2005) Prediction of low-energy collision-induced dissociation spectra of peptides with three or more charges. Anal. Chem. 77, 6364–6373 [DOI] [PubMed] [Google Scholar]
- 28. Richardson J. S., Richardson D. C. (1988) Amino acid preferences for specific locations at the ends of alpha helices. Science 240, 1648–1652 [DOI] [PubMed] [Google Scholar]
- 29. Yutani K., Ogasahara K., Tsujita T., Sugino Y. (1987) Dependence of conformational stability on hydrophobicity of the amino acid residue in a series of variant proteins substituted at a unique position of tryptophan synthase alpha subunit. Proc. Natl. Acad. Sci. U.S.A. 84, 4441–4444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilce M. C., Aguilar M. I., Hearn M. T. (1995) Physicochemical basis of amino acid hydrophobicity scales: evaluation of four new scales of amino acid hydrophobicity coefficients derived from RP-HPLC of peptides. Anal. Chem. 67, 1210–1219 [Google Scholar]
- 31. Maxfield F. R., Scheraga H. A. (1976) Status of empirical methods for the prediction of protein backbone topography. Biochemistry 15, 5138–5153 [DOI] [PubMed] [Google Scholar]
- 32. George R. A., Heringa J. (2002) An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 15, 871–879 [DOI] [PubMed] [Google Scholar]
- 33. Nakashima H., Nishikawa K. (1992) The amino acid composition is different between the cytoplasmic and extracellular sides in membrane proteins. FEBS Lett. 303, 141–146 [DOI] [PubMed] [Google Scholar]
- 34. Palau J., Argos P., Puigdomenech P. (1981) Protein secondary structure: Studies on the limits of prediction accuracy. Int. J. Peptide Protein Res. 19, 394–401 [PubMed] [Google Scholar]
- 35. Vasquez M., Nemethy G., Scheraga H. A. (1983) Computed conformational states of the 20 naturally occurring amino acid residues and of the prototype residue α-aminobutyric acid. Macromolecules 16, 1043–1049 [Google Scholar]
- 36. Fukuchi S., Nishikawa K. (2001) Protein surface amino acid compositions distinctively differ between thermophilic and mesophilic bacteria. J. Mol. Biol. 309, 835–843 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.