

Abstract
The interferon-inducible protein IFI16 has emerged as a critical antiviral factor and sensor of viral DNA. IFI16 binds nuclear viral DNA, triggering expression of antiviral cytokines during infection with herpesviruses. The knowledge of the mechanisms and protein interactions through which IFI16 exerts its antiviral functions remains limited. Here, we provide the first characterization of endogenous IFI16 interactions following infection with the prominent human pathogen herpes simplex virus 1 (HSV-1). By integrating proteomics and virology approaches, we identified and validated IFI16 interactions with both viral and host proteins that are involved in HSV-1 immunosuppressive mechanisms and host antiviral responses. We discover that during early HSV-1 infection, IFI16 is recruited to sub-nuclear puncta and subsequently targeted for degradation. We observed that the HSV-1 E3 ubiquitin ligase ICP0 is necessary, but not sufficient, for the proteasom e-mediated degradation of IFI16 following infection. We substantiate that this ICP0-mediated mechanism suppresses IFI16-dependent immune responses. Utilizing an HSV-1 strain that lacks ICP0 ubiquitin ligase activity provided a system for studying IFI16-dependent cytokine responses to HSV-1, as IFI16 levels were maintained throughout infection. We next defined temporal IFI16 interactions during this immune signaling response. We discovered and validated interactions with the viral protein ICP8 and cellular ND10 nuclear body components, sites at which HSV-1 DNA is present during infection. These interactions may be critical for IFI16 to bind to nuclear viral DNA. Altogether, our results provide critical insights into both viral inhibition of IFI16 and interactions that can contribute to IFI16 antiviral functions.
The ability of mammalian cells to distinguish self from non-self is paramount for triggering host immune defenses in response to viral infection. To this end, cells intrinsically express highly specialized receptors purposed with “sensing” viral nucleic acids. Upon binding to their viral cognate ligand, these cellular receptors initiate intracellular immune signaling cascades, culminating in the production and secretion of cytokines, such as type I interferons (IFNs)1 (1). Subsequently, these cytokines stimulate antiviral gene programs in neighboring cells and mobilize effectors of the innate and adaptive arms of the host immune system. These cytokine functions are critical to abate viral replication and spread at the site of infection.
The mechanisms underlying the initial detection of and signaling to viral nucleic acids during infection remain to be fully defined. As viruses are intracellular obligate host parasites, their RNA or DNA genomes are derived directly from cellular nucleotide pools. Thus, there are limited features that distinguish viral and cellular nucleic acids. However, because RNA viruses lack certain characteristic eukaryotic RNA moieties, such as 5′-monophosphates, 5′-linked N-7-methylated guanosine caps, or 2′-O-methylations, some viral RNA sensing mechanisms have been readily characterized (2–4). In contrast, chemical or structural moieties that differentiate viral and cellular DNA have not been universally established. Until recently, viral DNA sensing mechanisms were thought to rely on compartmentalization, functioning exclusively within cellular organelles typically devoid of host DNA. In agreement, to date, nearly all identified DNA sensors have been shown to function within the cytosol or endosomes (5–11). However, these cytoplasmic sensing events do not fully explain the recognition of viruses that deposit their DNA and replicate in the nucleus. The genomes of these DNA viruses are protected within a proteinaceous capsid while traversing the cytosolic or endosomal compartments and are almost ubiquitously targeted to the nucleus for replication (12). As many important human pathogens, such as herpesviruses and adenoviruses, are nuclear replicating DNA viruses, defining mechanisms of viral DNA sensing is critically important to understanding host immunity and the development of novel antiviral therapeutics.
We and others have recently characterized the interferon-inducible protein IFI16 as the first known sensor of nuclear viral DNA (8, 9, 11, 13–17). IFI16 is able to bind to the genomic DNA of several different nuclear replicating herpesviruses, including herpes simplex virus 1 (HSV-1) (8, 9, 11, 14, 18, 19) and human cytomegalovirus (HCMV) (13, 17), during infection and to coordinate the production of antiviral cytokines. Thus, these recent findings challenge the paradigm that DNA sensing is excluded from the nucleus to prevent superfluous immune activation by the host cell genome. Interestingly, despite its nuclear localization, IFI16 requires the central components of a cytosolic immune signaling axis composed of stimulator of interferon genes (STING) (20), TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK-1) (21), and IFN response factor 3 (IRF3) (22) to elicit double-stranded DNA-dependent immune signaling (8, 14, 15, 17, 23). However, the mechanisms driving nuclear-cytoplasmic communication between IFI16 and these integral pathway components remain undefined. Several groups have observed that IFI16 retains its nuclear localization, remaining physically separated from the aforementioned pathway components early in infection (14, 17, 18). However, others have observed the active export of a subset of IFI16 from the nucleus upon binding herpesvirus double-stranded DNA, concomitantly assembling a multiprotein “inflammasome” complex that stimulates the secretion of proinflammatory cytokines IL-1β and IL-18 (11, 16, 24, 25). These discrepancies highlight the need for further studies regarding the functions and behaviors of IFI16 in the context of infection. In addition to its roles in immune and inflammatory signaling, IFI16 has also been implicated in other antiviral functions. In line with its original demonstrated function as a regulator of transcription (26), IFI16 can suppress HSV-1 and HCMV transcriptional activity and hence their replicative capacity (19, 27, 28). Furthermore, IFI16 levels correlate with the nucleosomal load and abundance of repressive epigenetic markers found on chromatinized HSV-1 genomes during infection (27). Altogether, these studies establish IFI16 as a critical antiviral factor against human herpesviruses. However, the molecular details of its multiple purported antiviral functions are widely debated and overall poorly understood.
Thus far, studying essential immune signaling pathways has been challenging as herpesviruses encode viral gene products that function to counteract cellular antiviral factors and suppress immune responses. Recent studies have delineated some of the viral strategies targeting IFI16 and IFI16-dependent pathways (14, 16–18). For instance, we recently reported that the HCMV protein pUL83 physically associates with IFI16 to block immune signaling (17). Furthermore, others have shown that IFI16 is degraded during HSV-1 infection (14, 16, 18), corresponding with an attenuation of antiviral cytokine expression. The ubiquitin ligase activity of the HSV-1 protein ICP0 was initially implicated in promoting IFI16 destabilization (14). However, a subsequent study demonstrated that, under certain conditions, an ICP0-deficient virus could still induce the degradation of IFI16, asserting that ICP0 was not required for this process (18). Given these contradictory reports, the modulation of IFI16 functions during HSV-1 infection remains to be further investigated. We have recently characterized the functional protein interaction networks of IFI16 and the other members of the pyrin domain (PY)- and hematopoietic IFN-inducible nuclear antigen with 200 amino acid repeats domain (HIN)-containing protein family (Absent in melanoma 2 (AIM2), IFN-inducible protein X (IFIX), and Myeloid cell nuclear differentiation antigen (MNDA)) in uninfected cells. This provided insights into the housekeeping functions of this important family of proteins and its regulation (29). However, the IFI16 protein interactions in the context of infection and therefore the means through which it exerts its antiviral functions have not yet been characterized.
Here, we used a hybrid proteomics and virology approach to perform the first characterization of the protein interaction network of endogenous IFI16 following HSV-1 infection in primary human fibroblasts. Given the contradictory reports on ICP0 functions, we characterized the differential cellular responses to HSV-1 viruses that diverge in ICP0 functionalities. Our results suggest that HSV-1 rapidly blocks IFI16-mediated immune responses during infection by catalyzing its degradation in part via the contribution of ICP0. Furthermore, we substantiate that the ubiquitin ligase activity of ICP0 is critical for suppressing host immune responses. To unbiasedly probe the molecular details underpinning this important cell-virus interplay, we used immunoaffinity purification and mass spectrometry to define endogenous IFI16 protein interactions in response to wild-type (WT) HSV-1 infection as well as following infection with a virus that lacks the ability to inhibit IFI16 (ICP0-mutant HSV-1). Interactions identified in uninfected and WT HSV-1-infected cells point to IFI16 housekeeping functions as well as associations used by the virus for immunoevasion, such as with ICP0. Indeed, we observed active recruitment of IFI16 to nuclear ICP0-containing puncta preceding its rapid proteasome- and ICP0-modulated degradation at early stages of infection. Furthermore, our studies following infection with the ICP0 mutant HSV-1 determined that active IFI16 associates with ND10 nuclear bodies where it can recognize and bind to viral DNA. Our findings significantly expand the current knowledge regarding IFI16 functions during HSV-1 infection, providing mechanistic insights into its regulation by HSV-1 as well as a resource of protein interactions that is expected to aid future functional studies on this important antiviral factor.
EXPERIMENTAL PROCEDURES
Cell Culture and Viruses
Primary human foreskin fibroblast (HFF) cells, inducible HEK293 cells (a gift from Dr. Loren W. Runnels, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School), and Vero cells were cultured in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. The complementing cell line E11 (ICP4+ ICP27+) was additionally cultured with appropriate supplements as described (30). Wild-type HSV-1 (strain 17+) and ICP0-RF (strain 17+; a gift from Dr. Saul Silverstein, Columbia University, and Dr. Bernard Roizman, University of Chicago) viruses were grown and titered on U2OS cells, whereas d106 (strain KOS; a gift from Dr. Neal DeLuca, University of Pittsburgh) virus was propagated and titered on Vero-derived E11 cells. To propagate virus, culture supernatant and cell-associated virus were collected from infected cells exhibiting a nearly 100% cytopathic effect, buffered with MNT (200 mm MES, 30 mm Tris-HCl, 100 mm NaCl, pH 7.4), sonicated, centrifuged, and titered by plaque assay. To infect, virus was diluted in DMEM containing 2% (v/v) FBS and incubated on cells at the indicated m.o.i. for 1 h at 37 °C to allow for virus adsorption. Cell monolayers were washed once with phosphate-buffered saline (PBS), overlaid with DMEM containing 2% (v/v) FBS, and incubated at 37 °C for the indicated lengths of time postinfection.
Antibodies and Reagents
For immunoaffinity purification and Western blotting of endogenous IFI16, a 1:1 (w/w) mixture of two monoclonal α-IFI16 antibodies was used (ab50004 and ab55328, Abcam). The antibodies additionally used for immunoaffinity purifications, reciprocal co-immunoprecipitations, Western blotting, and immunofluorescence were α-IgG (Sigma), α-ICP0 (H1A027-100, Virusys Corp.), α-ICP8 (sc-53329, Santa Cruz Biotechnology), α-catalytic subunit of DNA protein kinase complex (DNA-PKcs) (sc-5282, Santa Cruz Biotechnology), α-tubulin (T6199, Sigma-Aldrich), α-PML (sc-9862, Santa Cruz Biotechnology), α-IFI16 (sc-6050, Santa Cruz Biotechnology; for immunofluorescence only), and α-small ubiquitin-like modifier 2/3 (ab3742, Abcam). The proteasome inhibitor (S)-MG132 was obtained from Cayman Chemical (10012628), and the Protein A/G PLUS-agarose beads used for the reciprocal co-immunoprecipitations were obtained from Santa Cruz Biotechnology (sc-2003). Lipofectamine 2000 (Life Technologies) was used to transfect the indicated DNA constructs into HFF cells or HEK293T cells.
Cryogenic Cell Lysis and Isolation of IFI16 Protein Complexes by Immunoaffinity Purification
Assessment of IFI16 protein interactions was performed by immunoaffinity purification in conjunction with nano-LC-MS/MS analysis as described previously (31). All IFI16 isolations were performed in biological triplicates. Cells were infected with the indicated virus strain for the indicated length of time, washed and scraped in PBS, resuspended in freezing buffer (20 mm Na-HEPES, 1.2% polyvinylpyrrolidone (w/v), pH 7.4), and flash frozen in liquid nitrogen. Frozen cell material was ground with a Retch MM301 Mixer Mill (Retch, Newtown, PA) for 1.5 min at 30.0 Hz for 10 rounds and cooled in liquid nitrogen in between rounds as described (32). The ground cell powder was resuspended in 14 ml of lysis buffer (20 mm K-HEPES, pH 7.4, 0.11 m KOAc, 2 mm MgCl2, 0.1% Tween 20 (v/v), 1 μm ZnCl2, 1 μm CaCl2, 0.6% Triton X-100, 200 mm NaCl, 100 units/ml Benzonase for cell lysis (Pierce), protease inhibitor mixture (Sigma), phosphate inhibitor mixtures 2 and 3 (Sigma)) and incubated at room temperature for 10 min for effective endonuclease digestion. We selected Benzonase to use in our lysis buffer as it digests both DNA and RNA and is more effective in a wider range of buffer conditions than DNase. A PT 10–35 GT Polytron (Kinematica, Bohemia, NY) was used to homogenize the samples for 30 s at 20,000 rpm, and the suspensions were centrifuged at 7,000 × g for 10 min at 4 °C. For each indicated infection and time period, the clarified lysate was equally divided into immunoaffinity isolations for IFI16 and IgG. The pellet fraction was solubilized in SDS sample buffer for analysis of protein solubilization efficiency by Western blotting. Per isolation, 7 mg of M-270 epoxy magnetic beads (Life Technologies) were conjugated as described (33) with either a 1:1 mixture (w/w) of α-IFI16 monoclonal antibodies or α-IgG antibody, added to lysates, and incubated for 1 h at 4 °C. Subsequently, beads were washed six times with lysis buffer, and protein complexes were eluted in lithium dodecyl sulfate sample buffer (Life Technologies) by incubating the beads at 70 °C for 10 min. To reduce the protein complexes, 100 mm dithiothreitol was added to each eluate, and samples were heated at 70 °C for 10 min. SDS sample buffer was added to the postisolation suspensions for analysis of unbound flow-through protein and immunoisolation efficiency.
Sample Preparation and Mass Spectrometry Analysis
Eluted immunoisolates were alkylated with 50 mm iodoacetamide for 30 min at room temperature. Proteins were partially resolved by SDS-PAGE on a 4–12% Bis-Tris NuPAGE gel, stained with SimplyBlue Coomassie Safe Stain (Life Technologies), and processed through in-gel protein digestion as described (34). Specifically, gel lanes were cut into slices 1 mm thick; pooled into six total fractions; subjected to a series of destaining, washing, dehydration, and hydration steps; and subsequently digested with 12.5 ng/μl trypsin (Promega, Madison, WI) overnight at 37 °C as detailed previously (35). Peptides were then extracted by incubating the gel pieces in 0.5% formic acid for 4 h at room temperature followed by incubation in 0.5% formic acid and 50% acetonitrile for 2 h. The extracted peptides were concentrated by vacuum centrifugation until each of the six pooled fractions per immunoaffinity purification amounted to 10 μl. Peptides were desalted using a StageTip protocol modified from descriptions elsewhere (36, 37) by implementing a single plug poly(styrene-divinylbenzene) reverse phase sulfonate approach and vacuum-centrifuged to 8 μl. Subsequently, 4 μl of each peptide fraction was analyzed by mass spectrometry (nano-LC-MS/MS) using the Dionex Ultimate 3000 nanoRSLC system (Dionex Corp., Sunnyvale, CA) coupled on line to an ESI-LTQ-Orbitrap Velos (Thermo Fisher Scientific, San Jose, CA) as described (31). Peptides were separated by reverse phase chromatography (Acclaim PepMap RSLC, 1.8 μm × 75 μm × 25 cm) at a flow rate of 250 nl/min under a 90-min discontinuous gradient of acetonitrile consisting of 4–40% B over 90 min (mobile phase A, 0.1% formic acid in water; mobile phase B, 0.1% formic acid in 97% acetonitrile). The mass spectrometer was set to data-dependent acquisition mode, and peptide precursors were subjected to collision-induced dissociation (CID) MS/MS fragmentation in the linear ion trap of the 15 most abundant precursor ions. The following parameters were set: enabled Fourier transform MS predictive automatic gain control target value of 1E6 (500-ms maximum ion injection time), ion trap MS/MS target value of 1E4 (100-ms maximum ion injection time), enabled dynamic exclusion (repeat count of 1 and exclusion duration of 70 s), and enabled lock mass (mass list, 371.101233). A single MS scan consisted of an m/z range of 350–1700 and resolution of 60,000; CID fragmentation was set to an isolation width of 2.0 thomsons, normalized collision energy of 30%, and activation time of 10 ms.
Database Searching and Protein Assignment Analysis
MS/MS spectra obtained from raw files of each immunoaffinity purification were searched and extracted using Proteome Discoverer (v.1.4) and then submitted to SEQUEST HT (v.1.3) for searching against forward and reverse peptide entries in the UniProt FASTA database (22,630 entries consisting of human, herpesvirus, and common contaminant sequences; downloaded in August of 2013). Search parameters consisted of full trypsin specificity, maximum of two missed cleavages, ion precursor mass tolerance of 10 ppm, fragment ion mass tolerance of 0.5 Da, fixed modification including cysteine carbamidomethylation, and variable modifications including methionine oxidation and serine, threonine, and tyrosine phosphorylation. Phosphopeptides were included in search parameters as we have previously demonstrated the phosphorylation of IFI16 in other cell types (epithelial cells and T lymphocytes) (9, 29). Peptide spectrum matches from SEQUEST were imported into Scaffold (v.4.4.0, Proteome Software, Inc., Portland, OR) for validation with the Bayesian model local false discovery rate algorithm and researched with X!Tandem (GPM 2010.12.1.1) with the following additional variable modifications: oxidation of tryptophan (+16 Da), pyro-Glu formation of N-terminal glutamine and glutamate (+17.0265 and +18.0106 Da, respectively), deamidation of asparagine and glutamine (+0.9840 Da), and acetylation of lysine (+42.0106 Da). Following parsimony rules, peptide spectrum matches were assembled into protein categories. Peptide and protein confidences were set to 95 and 99%, respectively, to achieve a protein global false discovery rate of <1%. Protein groups were reported with a minimum of two unique peptides per protein in at least one biological replicate. Protein isoforms were characterized by Scaffold with unique peptides. Resulting proteins were exported as label-free “unweighted spectrum counts” for subsequent analysis using the Significance Analysis of INTeractions (SAINT) algorithm (38).
Spectral count matrices were assembled for endogenous IFI16 immunoaffinity purifications (IPs) relative to each respective IgG control IP. For the biological triplicates from each biological condition, the probability scores assigned by SAINT were averaged. All bait-prey interactions with an average probability cutoff score of 0.85 and gene ontology annotations with predicted nuclear localization were accepted for further analysis as putative specific interactions (supplemental Tables S1 and S2). Given the limited knowledge regarding IFI16 interactions, this score threshold was selected based on our previous studies (29, 39) and the distribution of average SAINT probability scores across all experimental conditions (supplemental Fig. S3, top) as a balance between stringency and maintenance of interactions. To further filter out false-positive interactions with IFI16, the data set was compared with negative control isolations available in the web-based CRAPome repository (40). Based on the distribution of CRAPome appearance frequencies for all identified IFI16 interactions (supplemental Fig. S3, bottom), proteins with greater than 20% appearance within the 411 deposited experiments were filtered out prior to final analysis. The proteins filtered as likely nonspecific associations are listed in supplemental Table S3.
Network Assembly of Functional IFI16 Protein Interactions
The filtered, enriched co-isolated proteins were submitted to the web-based STRING database (41) to generate functional interaction networks. Default parameters were enabled with the exception of text mining. Putative interactions not identified within the STRING database were manually included and are available in supplemental Tables S1 and S2. STRING networks were imported into Cytoscape (v.2.8.3) (42, 43). Node shapes were used to indicate the presence or absence of a protein in distinct IP conditions. Node colors were used to indicate log2-transformed normalized spectral count -fold changes between uninfected and WT HSV-1-infected cells or between 3 and 8 h postinfection with ICP0-RF HSV-1. The co-isolated proteins were further clustered according to their known biological functions and gene ontology terms (e.g. transcriptional regulation and immune signaling).
Validation of IFI16 Interactions via Reciprocal Co-immunopurifications
One 15-cm dish of confluent HFF cells was harvested after infection with the indicated virus strain at the indicated length of time postinfection and lysed in 1.5 ml of lysis buffer. The lysate was divided into two separate isolations (IgG and either ICP0 or ICP8), and 5 μl of Protein A/G-agarose beads were incubated per IP for 1 h at 4 °C for preclearing. Subsequently, cell lysates were incubated with either α-IgG antibody or α-ICP0/ICP8 antibody for 1 h at 4 °C followed by the addition of 20 μl of Protein A/G beads per isolation for 1 h at 4 °C. The beads were washed once with lysis buffer and three times with PBS, resuspended in 40 μl of lithium dodecyl sulfate sample buffer, and eluted by heating at 70 °C for 10 min. Co-isolated proteins were analyzed by Western blotting.
Immunocytochemistry and Fluorescence Microscopy
HFF cells were seeded on 13-mm Number 1.5 glass coverslips and infected with the indicated HSV-1 virus strains for the indicated lengths of time. All the following steps were performed at room temperature. Cells were fixed in 2% (v/v) paraformaldehyde in PBS for 15 min, permeabilized with 0.1% (v/v) Triton-X in PBS for 15 min, and blocked with 2% (w/v) BSA and 2.5% human serum in PBS-T (0.2% (v/v) Tween 20 in PBS). The fixed cells were sequentially incubated with primary antibody for 1 h in PBS-T, washed three times with PBS-T for 5 min each, and incubated with secondary antibody conjugated to either Alexa Fluor 488, 568, or 633 (Life Technologies) in PBS-T. Cell nuclei were stained using 1 μg/ml 4′,6-diamidino-2-phenylindole for 10 min. The coverslips were mounted with Aqua-PolyMount (Polysciences, Inc.), and images were acquired under a 63× immersion oil objecting using a Leica SP5 confocal microscope (Leica Microsystems).
RNA Isolation and Quantitative RT-PCR
Total cellular RNA was isolated from cell cultures using the RNeasy kit (Qiagen) according to the manufacturer's instructions. One microgram of RNA was reverse transcribed to cDNA with the RETROscript kit (Life Technologies). Quantitative PCR was conducted on the cDNA with gene-specific primer sets (supplemental Table S4) and SYBR Green PCR Master Mix (Life Technologies) using an ABI 7900HT. Relative quantification of PCR products was performed by normalizing to β-actin with the ΔΔCT method.
Lentivirus-mediated Short Hairpin RNA (shRNA) Knockdown of IFI16 in HFFs
To produce lentivirus in the HEK293T packaging cell line (ATCC CRL-11268), the IFI16 target shRNA sequence (supplemental Table S4) was cloned into pLKO.1 and co-transfected with the lentiviral packaging vectors psPAX2 and pMD2.G using Lipofectamine 2000. Cell supernatant was harvested at 40, 56, and 72 h post-transfection; filtered through a 0.45-μm membrane; and used to transduce HFF cells for 3 days. Target HFF cells were subjected to selection in 2 μg/ml puromycin (Invitrogen) for at least 1 week and assayed for knockdown efficiency by Western blotting.
RESULTS AND DISCUSSION
HSV-1 Infection Triggers Proteasome-dependent Degradation of IFI16
The ultimate goal of our study is to provide insights into not only the mechanisms by which IFI16 abates nuclear replicating herpesviruses, but also the strategies herpesviruses use to impede IFI16-mediated functions. We and others have reported that during HSV-1 infection IFI16 binds viral genomic double-stranded DNA in the nucleus and subsequently mediates cellular immune signaling (9, 14). Furthermore, viruses can trigger substantial alterations in cellular proteomes to preclude host antiviral functions, including the immune response. Given the critical antiviral activity of IFI16 and the standing questions regarding its regulation, we first probed IFI16 expression over the course of HSV-1 infection. In support of recent reports (14, 16, 18), we observed a rapid decrease in IFI16 protein levels beginning at early stages of HSV-1 infection in primary human fibroblasts (Fig. 1A). By 6 h postinfection (hpi), IFI16 was scarcely detected and remained at these levels through late infection time points. Additionally, this loss of IFI16 correlated with the expression of the HSV-1 protein ICP0.
Fig. 1.

IFI16 is degraded in a proteasome-dependent manner during HSV-1 infection. A, protein levels of IFI16 and ICP0 at different time points of infection with WT HSV-1 (m.o.i. = 10) by Western blotting. B, protein levels of IFI16 and ICP8 during early periods of infection (m.o.i. = 10) with WT HSV-1, UV-irradiated HSV-1, or no infection in HFFs. Uninf., uninfected. C, mRNA expression levels of ifi16 (circles) and icp0 (triangles) in HFF cells infected with WT HSV-1 (m.o.i. = 10) up to 8 hpi as determined by quantitative PCR analysis in HFF cells. Error bars represent S.E. D, Western blot analysis of protein levels of IFI16, DNA-PKcs, ICP0, and ICP8 in uninfected or WT HSV-1-infected fibroblasts up to 8 hpi (m.o.i. = 10) in the presence or absence of the proteasome inhibitor MG132. U, uninfected.
To determine whether the observed IFI16 degradation is due to a viral protein synthesized de novo upon infection, we exposed fibroblasts to UV-irradiated virions, which are no longer competent for viral gene expression. Similar to uninfected fibroblasts, exposure to UV-irradiated virions did not result in an observable loss of IFI16 (Fig. 1B). The absence of the HSV-1 early gene ICP8 confirmed the inability of UV-irradiated virions to express viral gene products. IFI16 destabilization occurred exclusively following infection with replication-competent HSV-1, indicating that viral gene expression is required to trigger IFI16 degradation.
As HSV-1 is known to co-opt the host transcriptional machinery and manipulate the expression of cellular genes during infection, we next assessed the stability of IFI16 at the mRNA transcript level throughout infection. Interestingly, we observed relatively steady IFI16 transcript levels within the first 8 h of infection (Fig. 1C), suggesting that IFI16 levels are not regulated at the transcript level upon infection. Considering that HSV-1 can promote the ubiquitylation and proteasome-dependent degradation of cellular proteins upon infection, we tested whether IFI16 depletion requires proteasome activity. To accomplish this, infected fibroblasts were treated with the proteasome inhibitor MG132. Importantly, to avoid the reported inhibitory MG132-dependent effects on the progression of viral infection (44), medium was supplemented with MG132 at 1 h after viral adsorption. Inhibition of proteasomal activity rescued IFI16 in HSV-1-infected cells, mimicking its protein levels observed in uninfected cells (Fig. 1D). As a control, we also observed the rescue of DNA-PKcs, a protein known to be proteasomally degraded during HSV-1 infection (45). Furthermore, equivalent ICP0 and ICP8 staining between DMSO- and MG132-treated cells indicated that drug treatment did not affect viral gene expression kinetics. Altogether, our results suggest that productive HSV-1 infection triggers a targeted and proteasome-dependent degradation of IFI16.
Endogenous IFI16 Protein Interactions during HSV-1 Infection of Primary Human Fibroblasts
The proteasome dependence and rapidity of the observed IFI16 degradation implicate an early acting mechanism during infection. To gain insights into the means through which HSV-1 targets IFI16, we next sought to characterize its protein interactions during early infection. Endogenous IFI16 was immunoaffinity-purified from primary human fibroblasts that were either uninfected or infected with WT HSV-1 (Fig. 2A) in biological triplicates. As IFI16 is known to bind DNA, to limit the identification of indirect interactions via DNA, all IPs were performed in the presence of Benzonase, which more efficiently digests both RNA and DNA in a wider range of lysis buffer conditions than conventional DNase I. The isolation of each of the three known IFI16 isoforms was evident by SDS-PAGE separation (Fig. 2B). Co-isolated proteins were identified by dissecting and subjecting the entire gel lanes to digestion with trypsin followed by nano-LC-MS/MS analyses on an LTQ-Orbitrap Velos (supplemental Tables S1 and S2). The SAINT algorithm (38) was used to assess interaction specificity through comparison of IFI16 and parallel IgG control isolations from either uninfected or infected cells. The distributions of average assigned SAINT probability scores (supplemental Fig. S3, top) revealed that a scoring threshold of 0.85 provided a relatively stable list of prominent IFI16 interactions at high stringency. This is critical as nonspecific associations to magnetic beads, IgG molecules, and the target protein complexes of interest are all known to contribute to background within immunoaffinity isolation experiments (46). To further increase the stringency of the specificity filtering, we also compared the observed IFI16 interactions with the CRAPome database, a repository of negative control isolations collected across a wide variety of experimental conditions (40). A vast majority of the identified IF116 interactions appeared in fewer than 20% of the 411 total CRAPome-deposited control experiments (supplemental Fig. S3, bottom), validating the stringency of our isolation conditions and providing an additional scoring threshold for eliminating likely nonspecific, sticky, or overly abundant interactions. The proteins with greater than 20% appearance in the CRAPome database were eliminated (supplemental Table S3) from our final list of IFI16 interactions. Our analysis led to the identification of numerous previously unreported associations of IFI16 with both viral and cellular proteins during HSV-1 infection (Fig. 2C and supplemental Fig. S1). Among the cellular associations were proteins involved in transcriptional and chromatin regulation, in agreement with previously observed housekeeping functions of IFI16. Furthermore, we detected IFI16 interactions with immune signaling factors, including the known DNA sensor LRRFIP1 (47), suggesting the possible coordinated function of these critical antiviral proteins.
Fig. 2.

Endogenous IFI16 has dynamic interactions with viral and host proteins following HSV-1 infection. A, proteomic workflow to map protein interactions with endogenous IFI16 in uninfected and WT HSV-1-infected HFFs. The specificity of interaction was assessed using SAINT and the CRAPome database. B, immunoisolates from IFI16 and control IgG IPs were resolved by SDS-PAGE and stained with Coomassie Blue. IFI16 isoforms are indicated by a bracket. C, filtered interactions with IFI16 were assembled using STRING and visualized in Cytoscape. Gene shapes indicate the detection of an interaction in uninfected (Uninfected; diamond), WT HSV-1 infection (square), or both (circle). Node colors indicate spectral enrichment of an interaction in one of the conditions. D, validation of the IFI16-ICP0 interaction by reciprocal co-immunopurification using α-ICP0 antibody and visualized by Western blotting. HFF cells were infected with WT HSV-1 (m.o.i. = 10), harvested at 3 hpi, and incubated with Protein A/G-agarose beads conjugated to either control α-IgG or α-ICP0 antibodies. Uninf., uninfected.
Importantly, our results indicate that IFI16 rapidly associates with viral proteins during infection as 19 viral proteins passed our stringent specificity filtering criteria. Of these IFI16 interactions, several are constituents of the HSV-1 DNA replication complex. The viral replication fork helicase (UL5) along with viral DNA-dependent DNA polymerase catalytic subunit (UL30) and its processivity factor (UL42) were all enriched relative to IgG control spectral counts (∼7-, 7-, and 3.5-fold enriched, respectively). In addition to viral genome replication factors, we observed interactions between IFI16 and several critical regulators of viral gene expression, including ICP22, UL13, and ICP0. ICP22 (∼6-fold enriched) is a viral immediate-early protein involved in modulation of cellular RNA polymerase II activities for both promoting viral gene expression and antagonizing host gene expression. Some of these ICP22 functions are dependent on phosphorylation via the viral serine/threonine kinase UL13 (∼10-fold enriched). Finally, the immediate-early transactivator ICP0 was the most spectrally abundant SAINT-filtered viral protein interacting with IFI16 (∼10-fold enriched). ICP0 has critical roles in regulating viral and host gene expression as well as subverting host immune and apoptotic responses. These ICP0 functions are accomplished in part through the ubiquitin ligase activity of its N-terminal ring finger domain (RF) that mediates the proteasome-dependent degradation of target cellular proteins in the nucleus. Thus far, identified ICP0 substrates include DNA-PKcs (45); ubiquitin ligases RNF8 and RNF168 (48); centromere components CENP-A, -B, and -C (49–51); and major constituents of subnuclear ND10 bodies PML and SP100 (52, 53). The proteasome dependence and kinetics of our observed IFI16 degradation (Fig. 1) are similar to that of the above targeted cellular factors, implicating a role of ICP0. We therefore performed a reciprocal immunoaffinity isolation of ICP0 during HSV-1 infection in fibroblasts. Our results confirmed this interaction as we specifically observed endogenous IFI16 in ICP0 but not in IgG immunoisolates (Fig. 2D).
The HSV-1 ICP0 Ubiquitin Ligase Contributes to but Is Not Sufficient for IFI16 Degradation
Our identification of a physical interaction between IFI16 and the HSV-1 protein ICP0 during early stages of infection suggests one potential mechanism for IFI16 degradation. Indeed, during the course of performing our studies, two reports implicated ICP0 in promoting IFI16 destabilization upon infection (14, 16), although these studies did not assess the physical association of these proteins. However, another study asserted that IFI16 degradation was independent of ICP0 function (18). Therefore, the impact of ICP0 on IFI16 remains to be fully understood. Given our identification of their association, we proceeded to investigate their roles during infection using two mutant viruses, ICP0-RF and d106 (Fig. 3A). The ICP0-RF possesses two point mutations (C116A and C156G) within the RF domain of ICP0, abrogating its E3 ubiquitin ligase activity (54). In contrast, the d106 mutant expresses a fully functional ICP0 but no other HSV-1 immediate-early transactivators, rendering it replication-incompetent (30). As these viruses differ with respect to ICP0, we compared their impact on IFI16 levels. We infected primary human fibroblasts with either WT, ICP0-RF, or d106 viruses at equal multiplicities of infection and monitored IFI16 levels throughout 20 h of infection by Western blotting (Fig. 3B). In agreement with previously characterized ICP0 functions (52, 53), we observed a rapid loss of PML in both the WT and d106 infections, confirming the sufficiency of ICP0 for the degradation of its target host proteins. Interestingly, however, the d106 virus triggered a partial and kinetically slower loss of IFI16 relative to the WT virus. Furthermore, although IFI16 levels were decreased during the early stages of infection with d106 (3 and 6 hpi), a rescue was observed at a late time point (20 hpi), suggesting a possible inhibition or loss of the ICP0-IFI16 interaction late in infection or an IFN-mediated effect on IFI16 levels. In stark contrast, the ICP0-RF infection did not result in IFI16 degradation. In fact, IFI16 levels appeared to be slightly induced at 3 and 6 h postinfection. Likewise, PML and its isoforms were resistant to degradation over the course of ICP0-RF infection. All three viruses expressed comparable amounts of ICP0 (active or inactive) at each of the selected time points. These data suggest that the E3 ubiquitin ligase activity of ICP0 may be required for the destabilization of IFI16.
Fig. 3.

ICP0 contributes to, but is not sufficient for, the degradation of IFI16 during HSV-1 infection. A, schematic of WT HSV-1 genome and the immediate-early gene mutations present in the ICP0-RF and d106 strains. The genome encodes unique long (UL) and unique short (US) sequences (black line) and inverted repeat sequences (IRL and IRS) flanked by long (TRL) and short (TRL) terminal repeat sequences (white boxes). The immediate-early genes are depicted (black arrows showing direction of transcription). The ICP0-RF strain is deficient in functional ICP0 protein and contains C116G and C156A substitutions in each copy of the ICP0 coding sequence. The d106 strain is deficient in functional ICP4, -22, -27, and -47 proteins and contains a transgene cassette with a GFP reporter gene under the control of the HCMV immediate-early promoter (HCMVIEp) substituted into a partial ICP27 deletion (smaller solid gray box). Deletions (larger solid gray boxes) in ICP4 and the promoter element region of ICP22 and -47, TAATGARAT, result in the loss of these functional immediate-early proteins. B, protein levels of IFI16, ICP0, and PML in HFF cells after infection with WT HSV-1, ICP0-RF, or d106 at various hpi (m.o.i. = 10) were measured by Western blot. PML isoforms are highlighted with a bracket. U, uninfected. C and D, protein levels of IFI16-eGFP, ICP0, and PML in IFI16-eGFP inducible HEK293 cells (IFI16−/+) (C) with and without the transient expression of ICP0. Non-expressing ICP0 cells (ICP0−) were transiently transfected with mCherry (D) and uninfected or infected with WT or ICP0-RF HSV-1 at 4 hpi (m.o.i. = 10). E, protein levels of endogenous IFI16, ICP0, and DNA-PKcs in HFF cells uninfected or infected with WT or ICP0-RF HSV-1 at 4 hpi and at the indicated m.o.i. Uninf., uninfected.
Thus, we next assessed the sufficiency of ICP0 in promoting IFI16 proteasomal degradation. To do so, we constructed a HEK293 cell-based system allowing for the tetracycline-dependent induction of an integrated, full-length IFI16 transgene (herein HEK293-IFI16-GFP) (9, 29). As these cells lack endogenous IFI16, they can function as a minimalistic system for reconstituting and studying IFI16-dependent pathways and functions. First, we stimulated HEK293-IFI16-GFP cells to express IFI16, and then transiently transfected them with either ICP0- or control mCherry-expressing plasmid. Although the expression of ICP0 alone was sufficient to trigger the degradation of endogenous PML, it had a minimal effect on the protein levels of the IFI16 construct (Fig. 3C). To ensure that these results were not an artifact of our IFI16-inducible cell system, we next infected IFI16-expressing cells with the WT or ICP0-RF viruses. In agreement with our observations in primary fibroblasts, the IFI16 construct was substantially degraded by 4 hpi in WT, but not ICP0-RF, infection (Fig. 3D). Endogenous PML displayed a trend similar to that of IFI16. Thus, our results suggest that ICP0 is necessary, but not sufficient, for IFI16 degradation. This implicates the contribution of an additional mechanism of degradation distinct from other ICP0 targets, such as PML.
Previous studies have established that HSV-1 mutants lacking ICP0 ubiquitin ligase activity are kinetically delayed in viral gene expression due to their failure to degrade repressive cellular factors (54). Thus, it is possible that subsequent viral gene products not expressed in ICP0-RF infection may also contribute to IFI16 degradation. Experimentally, these effects can be overcome by using a greater virus-to-cell ratio. Indeed, when we infected primary fibroblasts at a greater ICP0-RF multiplicity of infection, we observed greater degradation of IFI16 relative to lower multiplicities at 6 hpi (Fig. 3E). However, even at high multiplicity, the extent of ICP0-RF-triggered IFI16 degradation was less than that of a lower multiplicity WT infection. Importantly, degradation of DNA-PKcs is not influenced by the amount of ICP0-RF virus used. This further substantiates that although the mechanisms mediating DNA-PKcs or PML stability are entirely dependent upon ICP0 E3 ubiquitin ligase, those regulating IFI16 stability may be dependent on both ICP0 ubiquitin ligase activity and other viral or cellular factors.
IFI16 Is Recruited to ICP0-containing Puncta during Early HSV-1 Infection
To gain further insights into the interplay between IFI16 and ICP0, we next examined their spatial organization during early HSV-1 infection. By immunofluorescence microscopy, IFI16 displays diffuse nuclear localization with nucleolar enrichment in uninfected fibroblasts (Fig. 4A). Interestingly, viral infection led to significant changes in IFI16 subnuclear localization. WT HSV-1 infection triggered co-localization of IFI16 and ICP0 in discrete nuclear punctate structures at 3 hpi. In agreement with its proteasome-dependent degradation (Figs. 1 and 3), IFI16 signals were no longer detected by 6 hpi. Concurrently with this loss of IFI16, we observed dispersion of ICP0 nuclear puncta and its subsequent translocation into the cytoplasm. Similar to WT HSV-1, ICP0-RF and d106 infections both induced the reorganization of IFI16 into ICP0-containing puncta. These findings suggest that ICP0 ubiquitin ligase activity is not required for recruitment of IFI16 to these foci. In contrast to WT infection, IFI16 remained localized within subnuclear ICP0 foci through 6 hpi. This is consistent with the inability of the ICP0-RF and d106 viruses to entirely degrade IFI16 as observed by Western blotting (Fig. 3B). Unexpectedly, fibroblasts treated with MG132 did not display a redistribution of IFI16 during WT HSV-1 infection (Fig. 4B). In these cells, IFI16 maintained its nucleoplasmic and nucleolar staining through 6 hpi despite the normal expression and punctate organization of ICP0 (Fig. 4B). Thus, MG132 may block a yet undetermined viral or cellular process required to stimulate IFI16 relocalization upon HSV-1 infection. Nevertheless, our results demonstrate that IFI16 is reorganized into subnuclear puncta that contain ICP0 and that in the presence of ICP0 ubiquitin ligase and proteasome activities, IFI16 is consequently degraded.
Fig. 4.
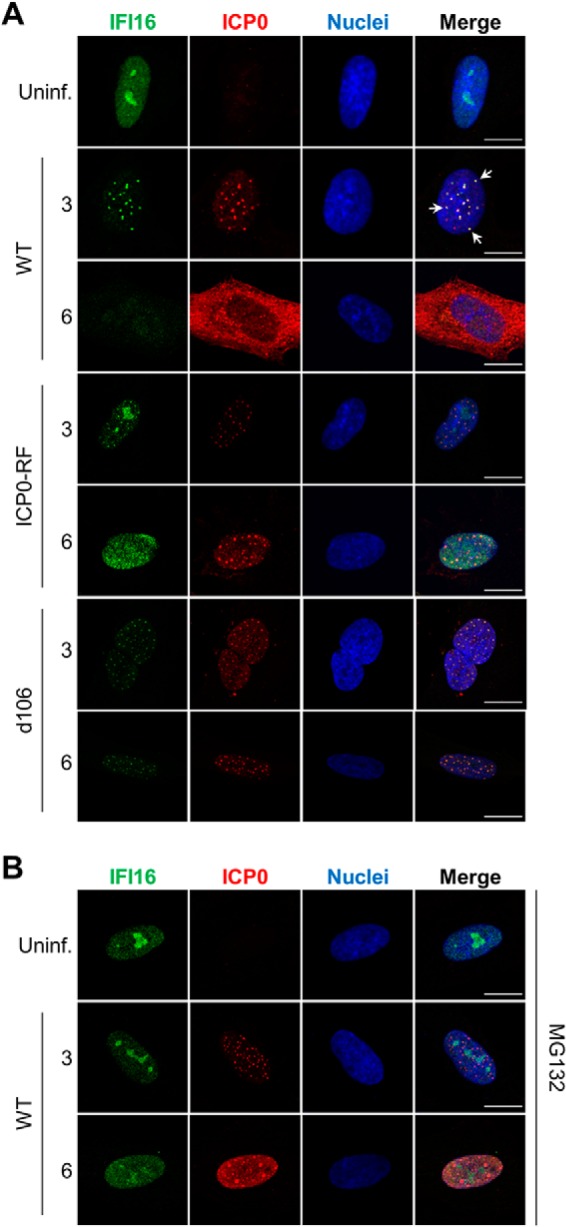
IFI16 co-localizes with ICP0 in discrete nuclear puncta during early stages of HSV-1 infection. A, representative immunofluorescence images of fixed HFF cells stained for IFI16 and ICP0 after infection with WT HSV-1, ICP0-RF, or d106 (m.o.i. = 1). B, cells were pretreated with the proteasome inhibitor MG132 and then analyzed as in A. Uninf., uninfected. Co-localization is indicated with white arrows. Scale bars, 5 μm.
HSV-1 ICP0 Activity Suppresses Antiviral Cytokine Expression during Infection
As our data suggest that ICP0 may contribute to the suppression of IFI16-dependent functions during HSV-1 infection, we next sought to establish a cell-virus system in which IFI16 remains active throughout infection, allowing it to trigger the expression of antiviral cytokines in response to HSV-1 infection. This system could be exploited to study the means through which IFI16 mediates its antiviral functions. Therefore, primary HFFs were infected with WT HSV-1 and assayed for their ability to produce antiviral cytokines in response to infection. In parallel, we assessed infections with the ICP0-RF and d106 mutants. For WT HSV-1 infection, we observed only moderate induction of IRF3- and NF-κB-dependent cytokines, including IFN-β, CXCL10, and CCL5/RANTES, and relatively no discernible induction of interferon-stimulated genes (ISGs) ISG54/IFIT2 and ISG56/IFIT1 (Fig. 5A) when compared with uninfected HFFs. In contrast, ICP0-RF infection induced a cytokine and ISG response far greater than either WT or d106 infections. This is in agreement with E3 ubiquitin ligase activity of the ICP0 ring finger domain functioning to repress the host immune response. Interestingly, however, the d106 infection induced moderate but significant ISG56 and ISG54 expression but not IFN-β, CXCL10, or CCL5 expression. These results imply that although ICP0 plays a substantial role in disarming host cell immune activities, other viral gene products additionally contribute to the subversion of such cellular functions.
Fig. 5.
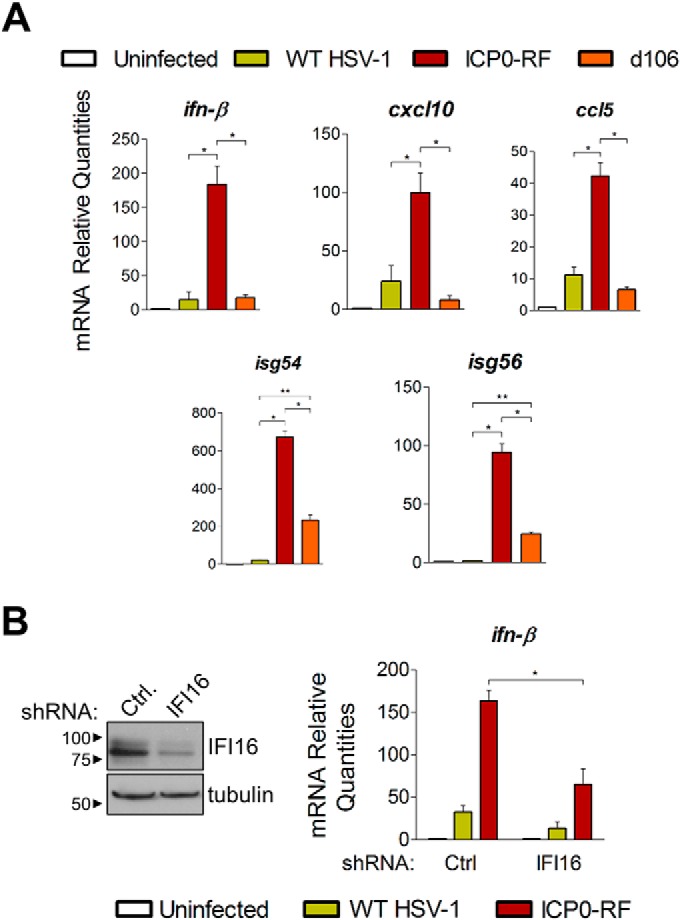
IFI16 is required for host immune response after HSV-1 infection and is attenuated by ICP0. A, mRNA levels of cytokines in HFF cells uninfected or infected with WT HSV-1, ICP0-RF, or d106 at 6 hpi (m.o.i. = 10). Results were measured by quantitative PCR and are shown as averages (n = 2), and error bars represent S.E. (p ≤ 0.05 (*) and p < 0.01 (**) by Student's t test). B, mRNA levels measured as in A of ifn-β in HFFs stably knocked down for IFI16 by lentivirus-mediated shRNA transduction (right). IFI16 knockdown was confirmed by Western blotting (left). Ctrl, control.
IFI16 Is Required for the Induction of Cellular Immune Responses during HSV-1 Infection
Having determined that infected cells effectively detect and trigger immune responses to the ICP0-RF virus, we next assessed the requirement of IFI16. We utilized lentivirus-mediated RNA interference to generate primary human fibroblasts stably expressing an shRNA specifically targeting IFI16. We confirmed by Western blotting that IFI16 protein levels are strongly reduced in shIFI16-expressing cells relative to a control shRNA cell line (Fig. 5B, left panel). Strikingly, when infected with the ICP0-RF virus, IFI16-depleted fibroblasts were markedly attenuated in their ability to induce IFN-β expression relative to control shRNA-expressing cells (Fig. 5B, right panel). These results further substantiate that IFI16 is a sensor of viral DNA within the nucleus and is integral in orchestrating early immune responses to a variety of human pathogens.
Defining Virus and Host Protein Interactions Mediating Antiviral IFI16 Functions
Our results indicate that the infection with the ICP0-RF virus provides an environment in which IFI16 is not inhibited by ICP0 and is necessary for the ability of infected cells to elicit antiviral cytokine expression. Therefore, although the IFI16 interactions in cells infected with WT HSV-1 helped to reveal viral immunosuppressive mechanisms, we predicted that studying it following infection with ICP0-RF virus would help to identify interactions specifically enriched while IFI16 exerts its antiviral functions. Given the slightly delayed replication kinetics of ICP0-RF virus when compared with WT HSV-1, we monitored endogenous IFI16 interactions at two early time points of infection (3 and 8 hpi) (Fig. 6A). The lysis buffer conditions, IgG control experiments, and assessment of specificity were performed as in our studies in WT HSV-1-infected fibroblasts. Interestingly, numerous interactions were observed to be enriched when compared with those identified in uninfected or WT HSV-1-infected cells (Fig. 6B and supplemental Fig. S2, node colors). Among these were interactions with viral proteins, including proteins involved in regulation of viral gene expression (UL46, UL48, ICP4, US1/ICP22, UL13, and ICP0) and proteins modulating viral DNA replication and nucleotide metabolism (ICP8, UL39, UL40, and UL50). Associations with cellular proteins were also temporally regulated with distinct changes in interactions with chromatin and transcription regulatory factors. An increase in the IFI16 association with NF-κB repressing factor, a negative regulator of NFκB-mediated transcription (55), is observed later in infection. This association may reflect a role for IFI16 in transcriptionally derepressing NFκB target genes, as we observed an IFI16-dependent modulation of their expression (Fig. 5). Importantly, a subset of IFI16 interactions was observed as uniquely specific following ICP0-RF virus infection (Fig. 6B, diamond shapes). Of particular interest was the viral protein ICP8, also known as DNA-binding protein. ICP8 is well established as an essential regulator of viral gene expression during the early stages of the HSV-1 life cycle as it is involved in the formation of nuclear prereplicative sites and viral replication compartments (for a review, see Ref. 56). These prereplicative sites have a nuclear punctate appearance similar to that which we observed for IFI16 following infection (Fig. 4). Therefore, we sought to confirm the IFI16-ICP8 interaction using reciprocal isolation (Fig. 6C). Indeed, IFI16 was specifically isolated with ICP8 but was not observed in the IgG control isolation following infection with ICP0-RF HSV-1. These results suggest that IFI16 may be targeted to sites of viral replication following HSV-1 infection, which would provide opportunity both for binding to viral DNA and, in view of IFI16 transcription regulatory function, for modulation of viral gene expression. In support of this model, our interactome also revealed the association of IFI16 with PML and SP100, known components of ND10 nuclear bodies (Fig. 6B). These interactions were enriched as the ICP0-RF infection progressed when compared with uninfected and WT HSV-1-infected cells.
Fig. 6.

Immunoaffinity purifications of IFI16 during ICP0-RF infection reveal novel interactions, including with PML-containing ND10 bodies. A, immunoisolates of IFI16 and control IgG IPs from HFF cells infected with ICP0-RF at 3 and 8 hpi were resolved by SDS-PAGE and stained with Coomassie Blue. B, SAINT-filtered interactions with IFI16 were assembled using STRING and visualized as a network in Cytoscape. Gene shapes indicate the detection of an interaction uniquely in ICP0-RF HSV-1 infection (diamond) or in common with at least one other condition (uninfected = mock or WT HSV-1 infection; circle). Node colors indicate spectral enrichment of an interaction at 3 or 8 h post ICP0-RF HSV-1 infection. C, validation of the IFI16-ICP8 interaction by reciprocal co-immunopurification using α-ICP8 antibody and visualization by Western blotting. HFF cells were infected with WT HSV-1 (m.o.i. = 10), harvested at 8 hpi, and incubated with Protein A/G-agarose beads conjugated to either control α-IgG or α-ICP8 antibodies. Uninf., uninfected.
IFI16 Is Targeted to PML Nuclear Bodies following Infection
As ND10 nuclear bodies are known as important cellular restriction factors during early stages of HSV-1 infection, we were particularly intrigued by the interactions of IFI16 with the integral ND10 body components PML and SP100 in ICP0-RF-infected fibroblasts. Previous studies have established that ND10 bodies associate with incoming genomes of nuclear replicating DNA viruses, such as HSV-1, to epigenetically suppress viral gene expression (57–64). As a countermeasure, HSV-1 ICP0 localizes to ND10 bodies immediately upon infection, targeting PML and SP100 for proteasomal degradation to disperse ND10 constituents and other restrictive factors (52, 53, 65–68). These virus-host interactions are critical determinants of viral replication proficiency. As we observed both a physical interaction and strong co-localization between ICP0 and IFI16, we next examined the relative spatial organization of IFI16 and PML (representative of ND10 bodies) during early HSV-1 infection. By immunofluorescence microscopy, we observed minimal co-localization between IFI16 and PML in uninfected primary fibroblasts (Fig. 7, row 1). Upon WT HSV-1 infection, we observed a dramatic dispersion of ND10 puncta and nearly complete loss of PML signal by 3 hpi. Interestingly, these events occur prior to the recruitment of IFI16 from the nucleoplasm and nucleoli to the observed subnuclear puncta (Fig. 7, row 2). By 6 hpi, both IFI16 and PML signals were scarcely detected (Fig. 7, row 3). These results are consistent with our prior observation that PML degradation kinetics are more rapid than those of IFI16 (Fig. 3B). In contrast to WT HSV-1, the ICP0-RF virus could neither degrade PML and IFI16, nor disperse ND10 bodies (Fig. 7, rows 4–6). Furthermore, we observed a concomitant decrease in IFI16 nucleolar localization with an increase in PML co-localization within ND10 structures between 3 and 6 hpi. Interestingly, at 6 hpi, a significant subset of ICP0-RF-infected cells displayed substantial co-localization between elongated, filamentous IFI16 structures and coalesced, misshapen PML/ND10 structures. Altogether, these immunofluorescence data suggest that in the absence of ICP0 ubiquitin ligase activity, HSV-1 triggers the active recruitment of IFI16 to ND10 bodies, leading to the formation of large IFI16- and PML-containing nuclear aggregates. Aggregate formation may represent the initiation of IFI16 and PML antiviral functions. Although previous studies have implicated activation of cytoplasmic IFI16 inflammasomes in response to herpesvirus infections (11, 16, 24, 25), we did not identify any IFI16-associated inflammasome components by mass spectrometry. Thus, aggregation may be mediating IFI16- and PML-dependent apoptosis, transcriptional repression, or interferon signaling. Further studies are necessary.
Fig. 7.
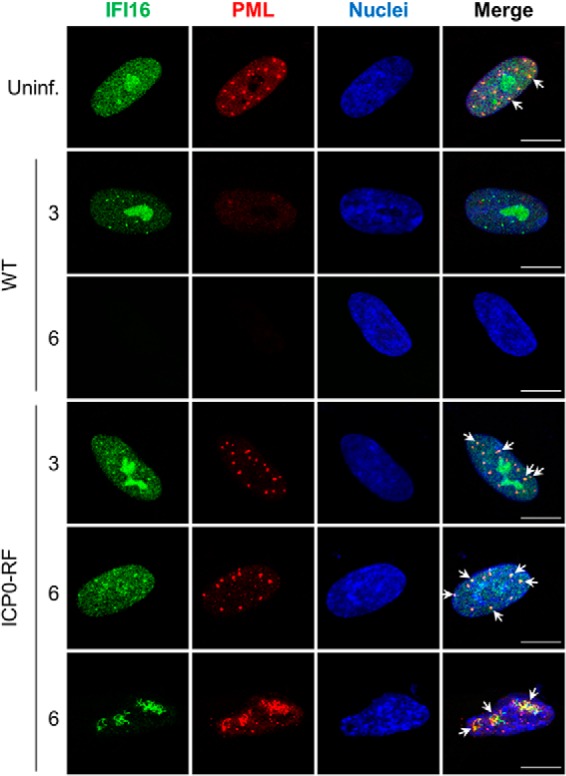
IFI16 is targeted to PML nuclear bodies following HSV-1 infection. Representative immunofluorescence images of HFF cells stained for IFI16 and PML in uninfected cells (Uninf.) or after infection with either WT HSV-1 or ICP0-RF at 3 and 6 hpi (m.o.i. = 1) are shown. Co-localization is indicated with white arrows. Scale bars, 5 μm.
Conclusions
The ability of mammalian cells to recognize DNA viruses, and subsequently trigger immune signaling, relies on the functions of viral DNA-“sensing” proteins, such as IFI16. Although several DNA sensors have been recently characterized, IFI16 is the first shown to function within the nucleus, the site of replication for the majority of DNA viruses, including those of the prevalent Herpesviridae family. This discovery has generated a significant level of interest as it has broad implications for understanding how the host immune system recognizes pathogens. Specifically, it challenges the long-standing dogma that recognition of viral DNA relies primarily on its spatial, subcellular separation from cellular DNA. Nevertheless, this concept of nuclear sensing raises numerous still unanswered questions. Are the viral and cellular DNA still separated within distinct subnuclear sites? What molecular and structural properties confer the specific recognition of viral DNA? And, importantly, how are the immune signals propagated from the nucleus? Interestingly, several previous studies including ours (14, 17) have shown that immune signaling following sensing of nuclear replicating herpesviruses still requires the endoplasmic reticulum protein STING, a known hub for DNA sensing (20). Therefore, it remains to be determined how nuclear IFI16 communicates with STING within the endoplasmic reticulum. An important requirement for starting to elucidate these mechanisms of immune signaling is defining IFI16 protein interactions in the context of viral infection.
Here, we show that IFI16 is rapidly degraded in a proteasome-dependent manner in HSV-1-infected primary human fibroblasts. Furthermore, we demonstrate that this degradation is dependent on viral gene products synthesized de novo upon infection. Although other studies have observed this HSV-1-dependent IFI16 degradation, they did not define the involved processes. To gain insight into these mechanisms, we then assessed endogenous IFI16 interactions during early HSV-1 infection using an affinity purification-MS/MS approach. From the resulting high confidence interaction network, we identified a physical association between IFI16 and the viral E3 ubiquitin ligase ICP0, a viral protein critical for dismantling restrictive cellular processes through the degradation of target cellular proteins. ICP0 appears to be insufficient to degrade IFI16, as demonstrated in our IFI16-inducible HEK293 system. Furthermore, the loss of ICP0 E3 ubiquitin ligase activity in the ICP0-RF mutant rescues the majority, but not the entirety, of IFI16 expression. In fact, a slight IFI16 degradation was observed when using a greater amount (i.e. higher m.o.i.) of ICP0-RF virus to infect fibroblasts. Thus, although ICP0 activity does substantially influence IFI16 stability during infection, there are likely other viral or cellular mechanisms that also contribute. Interestingly, we observed recruitment of IFI16 to subnuclear ICP0-containing punctate structures at early stages of infection prior to its degradation. Previous reports have demonstrated that the cellular immune signaling factor IRF3 which is required for antiviral gene induction in response to viral DNA, is similarly sequestered in nuclear structures during early HSV-1 infection (69). It may be that an initial host response involves the recruitment of cellular antiviral factors to these nuclear puncta but is counteracted by HSV-1 via the expression of ICP0, which can target these cellular factors for degradation. This model is substantiated by our findings regarding IFI16 interactions following infection with ICP0-RF virus. In this infection environment in which ICP0 is not active and IFI16 is mediating the antiviral cytokine response, IFI16 interacts with the core components of subnuclear ND10 bodies, PML and SP100. PML is a well known antiviral factor, accumulating on deposited nuclear viral genomes and transcriptionally repressing viral gene expression (59, 66). Its defense functions are normally inhibited by ICP0, which triggers the degradation of PML and SP100, and the dispersion of ND10 bodies (52). In the absence of ICP0, the members of ND10 bodies become components of viral replication centers. Therefore, our studies following ICP0-RF HSV-1 infection provided the opportunity to identify this interaction of IFI16 that likely represents an important mechanism for promoting antiviral defense responses. In agreement with this, our interactome study also identified a previously unrecognized interaction between IFI16 and the essential viral protein ICP8, which localizes to subnuclear viral replication centers for genome replication (56). Our finding of the localization of IFI16 to these subnuclear bodies may highlight a critical aspect in its dual antiviral functions: sensing of viral DNA and transcriptional regulation of viral gene expression. Indeed, at the time point of infection with ICP0-RF HSV-1 that corresponds to the association of IFI16 with PML and its localization to these nuclear puncta, we observe an IFI16-dependent induction in antiviral cytokines.
Altogether, our results provide mechanistic insights into the regulation of IFI16 during HSV-1 infection with a focus on our discovered interactions with ICP0 and PML-containing ND10 bodies. These findings have broad implications for understanding how the mammalian immune system expands its range of surveillance to include the recognition of nuclear-replicating viruses. Similar to recent drug design efforts for RNA virus inhibition, our results could help identify targets for novel therapeutic strategies against DNA viruses. Additionally, our interactome studies provide a powerful resource for future studies of the multiple functions of IFI16 in the context of viral infection and host immune response.
Supplementary Material
Acknowledgments
We graciously thank Dr. Saul Silverstein and Dr. Bernard Roizman for providing us with the ICP0-RF HSV-1 virus, Dr. Neal DeLuca for the d106 HSV-1 virus and Vero-derived E11 cells, and Dr. Loren W. Runnels for the inducible HEK293 cells. We are grateful to Todd M. Greco for assistance with mass spectrometry analyses and to Gary Laevsky for technical support (Microscopy Facility, Princeton University).
Footnotes
Author contributions: I.M.C. designed research; B.A.D., K.K.L., and A.J. performed research; B.A.D., K.K.L., A.J., and I.M.C. analyzed data; B.A.D., K.K.L., and I.M.C. wrote the paper.
* This work was supported, in whole or in part, by National Institutes of Health Grants DP1DA026192, R21AI102187, and R21HD073044 (to I. M. C.). This work was also supported by Human Frontier Science Program Organization Award RGY0079/2009-C (to I. M. C.) and by American Heart Association Predoctoral Fellowship 14PRE18890044 (to B. A. D.).
 This article contains supplemental Tables S1–S4 and Figs. S1–S3.
This article contains supplemental Tables S1–S4 and Figs. S1–S3.
1 The abbreviations used are:
- IFN
- interferon
- CRAPome
- contaminant repository for affinity purification
- d106
- replication-incompetent HSV-1 strain expressing a functional ICP0 but no other HSV-1 immediate-early transactivators
- HEK
- human embryonic kidney
- HFF
- primary human foreskin fibroblast
- HSV-1
- herpes simplex virus 1
- ICP0-RF
- HSV-1 strain lacking ICP0 E3 ubiquitin ligase activity
- IFI16
- interferon-inducible protein
- ND10
- nuclear domain 10
- PML
- promyelocytic leukemia
- SAINT
- Significance Analysis of INTeractions
- WT
- wild type
- HCMV
- human cytomegalovirus
- STING
- stimulator of interferon genes
- m.o.i.
- multiplicity of infection
- DNA-PKcs
- catalytic subunit of DNA protein kinase complex
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- IP
- immunoaffinity purification
- shRNA
- short hairpin RNA
- hpi
- h postinfection
- RF
- ring finger
- RANTES
- regulated on activation normal T cell expressed and secreted
- ISG
- interferon-stimulated gene.
REFERENCES
- 1. Ishikawa H., Ma Z., Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pichlmair A., Lassnig C., Eberle C. A., Górna M. W., Baumann C. L., Burkard T. R., Bürckstümmer T., Stefanovic A., Krieger S., Bennett K. L., Rülicke T., Weber F., Colinge J., Müller M., Superti-Furga G. (2011) IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 12, 624–630 [DOI] [PubMed] [Google Scholar]
- 3. Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B. W., Ziebuhr J., Szretter K. J., Baker S. C., Barchet W., Diamond M. S., Siddell S. G., Ludewig B., Thiel V. (2011) Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abbas Y. M., Pichlmair A., Górna M. W., Superti-Furga G., Nagar B. (2013) Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494, 60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., Fitzgerald K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun L., Wu J., Du F., Chen X., Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., Bowie A. G. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li T., Diner B. A., Chen J., Cristea I. M. (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. U.S.A. 109, 10558–10563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 [DOI] [PubMed] [Google Scholar]
- 11. Kerur N., Veettil M. V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., Chandran B. (2011) IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rathinam V. A., Fitzgerald K. A. (2011) Innate immune sensing of DNA viruses. Virology 411, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cristea I. M., Moorman N. J., Terhune S. S., Cuevas C. D., O'Keefe E. S., Rout M. P., Chait B. T., Shenk T. (2010) Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84, 7803–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orzalli M. H., DeLuca N. A., Knipe D. M. (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A. 109, E3008–E3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jakobsen M. R., Bak R. O., Andersen A., Berg R. K., Jensen S. B., Tengchuan J., Jin T., Laustsen A., Hansen K., Ostergaard L., Fitzgerald K. A., Xiao T. S., Mikkelsen J. G., Mogensen T. H., Paludan S. R. (2013) IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U.S.A. 110, E4571–E4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson K. E., Chikoti L., Chandran B. (2013) Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 87, 5005–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li T., Chen J., Cristea I. M. (2013) Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuchet-Lourenço D., Anderson G., Sloan E., Orr A., Everett R. D. (2013) The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 87, 13422–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson K. E., Bottero V., Flaherty S., Dutta S., Singh V. V., Chandran B. (2014) IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 10, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishikawa H., Barber G. N. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka Y., Chen Z. J. (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stetson D. B., Medzhitov R. (2006) Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 [DOI] [PubMed] [Google Scholar]
- 23. Paludan S. R., Bowie A. G. (2013) Immune sensing of DNA. Immunity 38, 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ansari M. A., Singh V. V., Dutta S., Veettil M. V., Dutta D., Chikoti L., Lu J., Everly D., Chandran B. (2013) Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 87, 8606–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singh V. V., Kerur N., Bottero V., Dutta S., Chakraborty S., Ansari M. A., Paudel N., Chikoti L., Chandran B. (2013) Kaposi's sarcoma-associated herpesvirus latency in endothelial and B cells activates γ interferon-inducible protein 16-mediated inflammasomes. J. Virol. 87, 4417–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnstone R. W., Kerry J. A., Trapani J. A. (1998) The human interferon-inducible protein, IFI 16, is a repressor of transcription. J. Biol. Chem. 273, 17172–17177 [DOI] [PubMed] [Google Scholar]
- 27. Orzalli M. H., Conwell S. E., Berrios C., DeCaprio J. A., Knipe D. M. (2013) Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. U.S.A. 110, E4492–E4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gariano G. R., Dell'Oste V., Bronzini M., Gatti D., Luganini A., De Andrea M., Gribaudo G., Gariglio M., Landolfo S. (2012) The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 8, e1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diner B. A., Li T., Greco T. M., Crow M. S., Fuesler J. A., Wang J., Cristea I. M. (2015) The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol. Syst. Biol. 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Samaniego L. A., Neiderhiser L., DeLuca N. A. (1998) Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72, 3307–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsai Y. C., Greco T. M., Boonmee A., Miteva Y., Cristea I. M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteomics 11, 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cristea I. M., Chait B. T. (2011) Affinity purification of protein complexes. Cold Spring Harb. Protoc. 2011, pdb.prot5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cristea I. M., Chait B. T. (2011) Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harb. Protoc. 2011, pdb.prot5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Greco T. M., Yu F., Guise A. J., Cristea I. M. (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 10, M110.004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greco T. M., Miteva Y., Conlon F. L., Cristea I. M. (2012) Complementary proteomic analysis of protein complexes. Methods Mol. Biol. 917, 391–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 37. Kulak N. A., Pichler G., Paron I., Nagaraj N., Mann M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 [DOI] [PubMed] [Google Scholar]
- 38. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. La Frazia S., Amici C., Santoro M. G. (2006) Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: role of nuclear factor-κB. Antivir. Ther. 11, 995–1004 [PubMed] [Google Scholar]
- 45. Parkinson J., Lees-Miller S. P., Everett R. D. (1999) Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73, 650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miteva Y. V., Budayeva H. G., Cristea I. M. (2013) Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 85, 749–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang P., An H., Liu X., Wen M., Zheng Y., Rui Y., Cao X. (2010) The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol. 11, 487–494 [DOI] [PubMed] [Google Scholar]
- 48. Lilley C. E., Chaurushiya M. S., Boutell C., Landry S., Suh J., Panier S., Everett R. D., Stewart G. S., Durocher D., Weitzman M. D. (2010) A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29, 943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Everett R. D., Earnshaw W. C., Findlay J., Lomonte P. (1999) Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18, 1526–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lomonte P., Morency E. (2007) Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581, 658–662 [DOI] [PubMed] [Google Scholar]
- 51. Lomonte P., Sullivan K. F., Everett R. D. (2001) Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276, 5829–5835 [DOI] [PubMed] [Google Scholar]
- 52. Chelbi-Alix M. K., de Thé H. (1999) Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18, 935–941 [DOI] [PubMed] [Google Scholar]
- 53. Everett R. D., Freemont P., Saitoh H., Dasso M., Orr A., Kathoria M., Parkinson J. (1998) The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72, 6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lium E. K., Silverstein S. (1997) Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential α27 gene. J. Virol. 71, 8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang K. H., Wang C. H., Lin C. H., Kuo H. P. (2014) NF-κB repressing factor downregulates basal expression and mycobacterium tuberculosis induced IP-10 and IL-8 synthesis via interference with NF-κB in monocytes. J. Biomed. Sci. 21, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weller S. K., Coen D. M. (2012) Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 4, a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ishov A. M., Maul G. G. (1996) The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maul G. G., Ishov A. M., Everett R. D. (1996) Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217, 67–75 [DOI] [PubMed] [Google Scholar]
- 59. Everett R. D., Murray J. (2005) ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79, 5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Everett R. D., Murray J., Orr A., Preston C. M. (2007) Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81, 10991–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liang Y., Vogel J. L., Narayanan A., Peng H., Kristie T. M. (2009) Inhibition of the histone demethylase LSD1 blocks α-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15, 1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lukashchuk V., Everett R. D. (2010) Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84, 4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Glass M., Everett R. D. (2013) Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 87, 2174–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gu H., Liang Y., Mandel G., Roizman B. (2005) Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U.S.A. 102, 7571–7576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Everett R. D., Parada C., Gripon P., Sirma H., Orr A. (2008) Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Everett R. D., Rechter S., Papior P., Tavalai N., Stamminger T., Orr A. (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80, 7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lomonte P., Thomas J., Texier P., Caron C., Khochbin S., Epstein A. L. (2004) Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 78, 6744–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gu H., Roizman B. (2007) Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U.S.A. 104, 17134–17139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Melroe G. T., Silva L., Schaffer P. A., Knipe D. M. (2007) Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-β induction. Virology 360, 305–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.