

Abstract
The goal of next-level bottom-up membrane proteomics is protein function investigation, via high-coverage high-throughput peptide-centric quantitation of expression, modifications and dynamic structures at systems scale. Yet efficient digestion of mammalian membrane proteins presents a daunting barrier, and prevalent day-long urea–trypsin in-solution digestion proved insufficient to reach this goal. Many efforts contributed incremental advances over past years, but involved protein denaturation that disconnected measurement from functional states. Beyond denaturation, the recent discovery of structure/proteomics omni-compatible detergent n-dodecyl-β-d-maltopyranoside, combined with pepsin and PNGase F columns, enabled breakthroughs in membrane protein digestion: a 2010 DDM-low-TCEP (DLT) method for H/D-exchange (HDX) using human G protein-coupled receptor, and a 2015 flow/detergent-facilitated protease and de-PTM digestions (FDD) for integrative deep sequencing and quantitation using full-length human ion channel complex. Distinguishing protein solubilization from denaturation, protease digestion reliability from theoretical specificity, and reduction from alkylation, these methods shifted day(s)-long paradigms into minutes, and afforded fully automatable (HDX)-protein-peptide-(tandem mass tag)-HPLC pipelines to instantly measure functional proteins at deep coverage, high peptide reproducibility, low artifacts and minimal leakage. Promoting—not destroying—structures and activities harnessed membrane proteins for the next-level streamlined functional proteomics. This review analyzes recent advances in membrane protein digestion methods and highlights critical discoveries for future proteomics.
The goal of proteomics has grown out of matured protein identification toward the next level of function discovery, particularly including membrane proteins, through quantitative and structural proteomics at high coverage, high throughput and systems scale. Bottom-up membrane proteomics provides two paths to spearhead toward this goal. Path 1, to identify and quantify, at higher accuracy, proteins present in a mixture based on sampling fragments of each (modification-bearing or not), often aiming to find protein candidates for new biomarkers or drug targets. Path 1 represents the predominant pursuit in current membrane and global proteomics research and reviews. Path 2, to define, at atomic precision, the complete molecular identities of signaling-pivot membrane proteins—that are established high-priority therapeutic targets—under functional states and to comprehensively quantify their dynamic changes upon stimuli (Fig. 1). Path 2 includes concrete quantitation of pan-post-translational modification (PTM)1 percent site occupancy and structural labels such as H/D-exchange (HDX), both requiring reproducible peptides that cover near-full protein sequence. This approach complements crystallography, electron microscopy (EM), NMR, and top-down MS, can discover critical details and landscapes beyond current reach, yet is largely untapped—for technical hurdles (1, 2), thus this review focuses on Path 2. As downstream state-of-the-art HPLC and mass spectrometry technologies grow mature, generic, and widely accessible, digestion sample preparation methods for membrane proteins increasingly delimit the capacity of bottom-up proteomics.
Fig. 1.

Bottom-up quantitative membrane proteomics may serve as a hub that connects various structure and function technologies, to accelerate discovery of the structure–function mechanisms of signaling TM proteins for better therapeutics (Path 2). Breaking the barriers against direct proteomic analysis of functional-state membrane proteins, at high peptide reproducibility and coverage, is key to reaching this goal. Brackets indicate optional but often preferred steps.
Abundant in human genome and pivotal in cell signaling, transmembrane (TM) proteins are coveted therapeutic targets (3), yet tremendously difficult to study at all levels (1)—including proteomics (2, 4, 5). Nearly 25% of 29,375 unique protein sequences in human proteome contain one or more TM helices (3). Prominently, TM neurotransmitter receptors such as G protein-coupled receptors (GPCR) and ligand-gated ion channels (LGIC)—with most members yet to tap—exceed 50% of current therapeutic targets (3). However, hydrophobic and prone to aggregation, membrane proteins have long vexed bottom-up proteomics with under-representation in every metric, including: sequence coverage, peptide spectrum matches (PSMs), unique peptides, and peptide reproducibility (2, 4, 5)—regardless of global lysates or highly purified samples—contrasting facile soluble proteins.
Aggregation, though originated from hydrophobic TM domains, engages entire proteins and diminishes overall protease access, thus the chances to capture non-TM peptides at authentic quantity from aggregated samples are proven slim and unpredictable, and this fact cannot be reversed by simply changing database size. Global analysis without the fair share of membrane proteins is hardly global, most readily detectable soluble proteins do not make useful new drug targets, and sporadic sketchy touches on TM receptors, known high-priority targets, generate no concrete structure–function roadmap to guide therapeutic discovery. Therefore, for the next-level functional proteomics aiming to improve health, the technical barriers of membrane proteomics must be solved.
Most digestion methods emphasized finding more proteins from mixtures, yet few addressed comprehensive peptide reproducibility, let alone both. Although identifying the same membrane proteins—represented by two or more unique peptides—between independent digestions and sample states was routinely achieved in shotgun proteomics, until recently (6, 7), comprehensively reproducing peptide forms and abundances, which matters for peptide-centric peak-area or tandem mass tag (TMT)-based quantitation of expression, modification, and labels, remained out of reach (2, 4, 5).
The challenges in detecting peptides for membrane proteins are twofold: (1) forming peptides, and (2) detecting formed peptides. Using prevalent overnight urea–trypsin in-solution digestion, extreme three-dimensional 8-day HPLC separations benchmarked sequence coverage at 20–30% for membrane proteins (8), demonstrating deficient peptide formation pre-HPLC is a major unmet need, although many recent efforts to delve deeper focused on HPLC, mass spectrometer, and tandem MS strategies downstream to digestion (8, 9). Further, unaddressed PTMs of mammalian membrane proteins (such as extensive glycosylation at high site occupancy (10)), may prevent sequence identification, even if peptides are formed. Both hydrophobic TM domains and intricate PTMs, hallmarks of human signaling TM proteins, are important for functions. Detergents, inherited from protein extraction (typically SDS and Triton X-100), severely challenged downstream digestion and/or HPLC steps in global proteomics.
The “urea–trypsin solution” tradition contains three elements—urea denaturation, trypsin, and in-solution format—that each and all, deserve critical thinking for membrane proteins. Myriad resorts have been proposed to address membrane protein gel-free digestions for various needs over past decades (11, 12), yet attempts to denature and subdue proved ineffective. This review will focus on advances since 2009 for prospects to support the next-level streamlined high-coverage, high-throughput quantitative membrane proteomics (Table I).
Table I. Advances of membrane protein gel-free digestion methods. MSP, membrane scaffold protein; FDD, flow/detergent-facilitated protease and de-PTM digestions for deep sequencing. st, StageTip; iodoaa, iodoacetamide; ovnt, overnight (14 h); N-glyco, N-glycosylation; med, medium; id, identify; qt, quantify; hp, human proteome search.
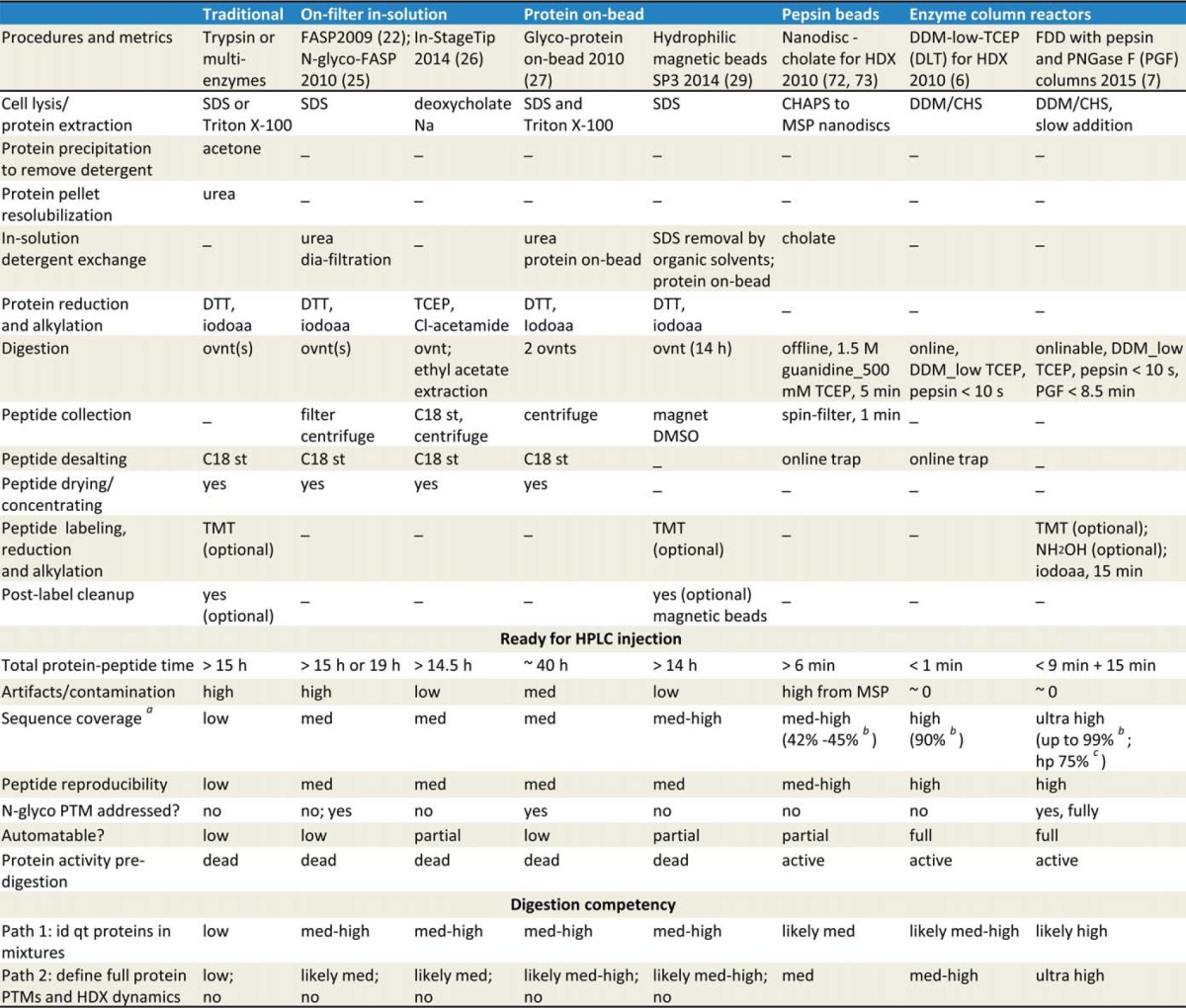
a Sequence coverage also depends on HPLC method and sequence database used for search.
b Purified protein searched against specified sequence.
c Identified by searching against human proteome (original, downloaded May, 23, 2013 from Ensembl) at peptide FDR <1%, using SEQUEST-HT and Percolator in PD 1.4. Signaling peptide sequence was not removed (a “/95%” correction, assuming a 20-residue signaling peptide for a total 500-residue protein). MSP, membrane scaffold protein; FDD, flow/detergent-facilitated protease and de-PTM digestions for deep sequencing; st, StageTip; iodoaa, iodoacetamide; ovnt, overnight (14 h); N-glyco, N-glycosylation; med, medium; id, identify; qt, quantify; hp, human proteome search.
In-solution Digestion and its Limits
Although prevalent, urea–trypsin in-solution digestion is neither complete nor predictable (13) for membrane proteins, and poses a weak link to future proteomics. Typical workflow includes: protein extraction by boiling 4–7% SDS, acetone or acid precipitation to remove SDS, resolubilization in 8 m urea, DTT reduction, iodoacetamide alkylation, in-solution digestion with trypsin in 2 m urea overnight, peptide desalting with C18 to remove the high urea, peptide drying to remove C18 elution solvent and concentrate (often causing oxidation), and redissolving in HPLC sample buffer. Pronounced problems are: serious unpredictable protein loss to aggregation (14), high oxidation artifacts and contaminations introduced by laborious processing (15), biases over protease specificity and abundance (13, 16), low digestion efficiency, coverage, and peptide reproducibility (2, 4).
Previous efforts to improve membrane protein digestion have focused on proteases with strict specificity, such as trypsin, Lys-C, Glu-C, Glu-N, and Asp-N, aiming to concentrate the quantity of peptides in defined theoretical forms. However, applied with denaturation and/or protease-deficient in-solution conditions, the advantage of this strategy was masked by large loss of accessible membrane proteins to aggregation (including re-aggregation), as evidenced by previous sub-25% coverage of purified human GPCR β2-adrenergic receptor (β2AR) using specific or nonspecific proteases in urea (6), and by common low numbers of peptides identified for Cys-loop LGICs from multiple rat or mouse brains using high-urea Lys-C and/or trypsin in-solution digestions. Further, in-solution digestion requires low protease-to-substrate ratio (1:50 or lower, m/m) to reduce contamination, thereby operates near-entirely in the enzyme-deficient, diffusion-limited, low-efficiency region (7); slow reaction rates of compromised trypsin in urea amplify such deviation from ideal. In-solution trypsin (2 m urea) digestion has also been extended by combining with other enzymes such as Lys-C (8 m urea) (17), pepsin (18), Asp-N (19), and PNGase F (pre-digestion (19)). Increasing evidence suggests multispecificity, often via using multiple proteases, is necessary for reliable PSM quantitation of lysates (13, 20). Pressure cycling technology speeded tandem urea-solution Lys-C/trypsin digestion of tissue lysate into 6 h, and strict cycle control enhanced peptide reproducibility (21).
Alternatives to Traditional In-solution Digestion
On-filter In-solution Digestions (FASP and in-StageTip)
Avoiding complete precipitation/resolubilization, the filter-based in-solution digestion method FASP (22, 23) achieved landmark improvement in digestion yield (up to 50%) by keeping membrane proteins in solutions during SDS removal. Following SDS protein solubilization, FASP applied dialysis (dial)-filtration with small molecular-weight-cut-off filters to gradually replace SDS in solution with high urea, performed in-solution digestion(s) overnight on filter, then separated peptides from enzyme and undigested proteins via centrifugation. FASP and its derivatives that added deoxycholic acid (2014 eFASP (24)) or further extended the on-filter reactor for reactions such as deglycosylation (2010 N-glyco-FASP (25)), have been widely applied since 2009. However, ∼50% protein sample resists digestion likely because of inevitable re-aggregation upon detergent depletion, and although overall protein identification is improved, peptide reproducibility remains low (23). The laborious manual operation is prone to contamination and sample loss. Abandoning SDS, the latest version of FASP, 2014 in-StageTip (26), used smaller tips sealed with C18 disk that doubles as micro-filter besides desalting, and combined protein deoxycholate solubilization, reduction, alkylation, and overnight in-solution digestion in these tips in 96-well format. However, this method still mandates manual processing, hours of digestion, and centrifugation (which interrupt automation streamline), and inherits all problems intrinsic to the denaturation-trypsin in-solution paradigm; ionic and high in cmc (critical micelle concentration; 1% m/v, 4.2× cmc was 25 mm), sodium deoxycholate incurred an extra step of post-digestion removal with ethyl acetate extraction and centrifugation (26), further obstructing automation.
Protein On-bead Digestions
Fixing membrane proteins on beads following specific affinity-enrichment (27, 28) effectively alleviated interprotein aggregation, such as during detergent removal (27), and greatly improved digestion and identification of membrane proteins using Lys-C and trypsin urea solutions (27). However, information of unbound proteins is lost. In the 2014 Single-Pot Solid-Phase-Enhanced Sample Preparation (SP3) method, hydrophilic magnetic beads adsorbed proteins and peptides less discriminatingly, and simplified solution separation for pre-digestion SDS removal and post-digestion peptide cleanup (29). However, common to both protein on-bead strategies, inevitable intraprotein aggregation may limit digestion access, and peptides remain mixed with protease solution.
Immobilized Enzyme Reactors for Fast Reproducible Digestion
Numerous micro-reactors of enzymes, including trypsin (30–32), trypsin/Lys-C (33), pepsin (34, 35), and PNGase F (36–38), adsorbed or covalent-bonded on various solid supports, have been described for peptides and/or glycans studies, mainly for purposes of faster reaction and easy product separation. They demonstrated advantages over in-solution methods, but mostly using simple soluble proteins in high denaturants such as urea, guanidine, or organic solvent.
Covalently immobilized pepsin columns emerged in 2002 for HDX to rapidly digest proteins at low pH low temperature to minimize D-label back-exchange, without complicating spectra with protease peaks (34). With high urea or guanidine denaturant (1.5–3 M, some studies also added 500 mm TCEP) at pH 2.5, pepsin column successfully digested most soluble proteins tested at high (over 90%) coverage and high peptide reproducibility within seconds-minutes, and is widely adopted in HDX (34, 35, 39, 40). HPLC-grade pepsin column's advantages are multifold and attributable to its operation as a high-surface-concentration plug-flow reactor, in retrospect (7). First, immobilizing pepsin on POROS beads can increase effective surface concentration to over-1-mm scale (34), near-thousand-fold higher than the 2 μm (or 0.1 mg/ml, 50 kDa) protein typically applied, thereby allowing pepsin column to operate well within the abundance-unbiased high-efficiency region, unrestricted by total sample size (7). Second, flow drive overcomes the problems of product inhibition caused by slow diffusion intrinsic to in-solution incubation, and affords a precise control of the fast product formation from nonspecific protease, which is unconceivable to in-solution and beads-based methods. Third, typical column size, flow rate (1–2 mm i.d. × 20 mm, 25–200 μl/min (6)) and pressure fit with HPLC system for full automation. Trypsin POROS column also showed efficient real-time digestion of soluble proteins online in organic solvent (30–32). However, until 2010 (6), protease columns met no success with membrane proteins, and for years GPCR β2AR coverage remained below 25% (6, 41), preventing useful HDX mapping.
Alternative Detergents for Protein Extraction and Digestion
Other advances were achieved by changing detergents. Traditional detergents for protein extraction in proteomics, such as ionic SDS and poly-disperse Triton X-100, inhibit protease digestion and/or peptide HPLC MS, but are difficult to remove. Alternatively, novel protease-compatible use-and-shred detergents such as RapiGest (42), could increase membrane protein sequence coverage from 10–20% to about 30% in global proteomics (42), and are increasingly applied with success in place of, or after (hours of dial-filtration), SDS or Triton extraction since 2007. However, these detergents vary solubilization power with proteins (some instantly precipitate proteins such as cytochrome c oxidase, CcO), require chemical cleavage post-digestion—difficult for fast full automation, and some cleaved products contain amines that interfere with TMT. A latest acid-cleavable analog of SDS, anionic MaSDeS, showed SDS-like strong solubilization power on tissues, though it is severely denaturing and forms amine products (43, 44). Amphi-polymers (amphipols) can support protein extraction and digestion, but most aggregate below pH 3–4 and require removal pre-HPLC by precipitation and centrifugation (45), interrupting automatable workflow. Several recent studies compared numerous detergents for digestion, but most remained confined to industrial poly-disperse and ionic detergents that deactivated proteins and required removal; other attempts combined harsh detergents, organic solvents, high urea, high pressure, and high temperature (46).
Despite these incremental advances in membrane protein coverage, obvious obstacles endured. First, hydrophobicity and aggregation persisted. Second, PTMs remained mostly unaddressed. Third, all these methods incur laborious manual operation, centrifugation (except for magnetic beads) and hours of processing before HPLC injection, interrupt automation, and remain unable to meet the minutes' time window allowed by HDX pipeline. Central to these method designs are the common observation and belief that: Detergents are incompatible with the protein-peptide-RP HPLC-ESI MS/MS pipeline, and must be avoided or removed at some point pre-HPLC. Further, the concepts of protein solubilization versus denaturation, and protease digestion reliability versus theoretical specificity, were frankly undistinguished. Consequently, Path 2 (Fig. 1), direct pan-PTM and HDX structural mapping of native functional human membrane proteins—the established high-priority therapeutic targets urgently awaiting precise structure-function roadmaps—remained a formidable field to bottom-up proteomics.
Detergent Selection for Functional Proteomics: The Schrödinger's Cat Scare
For protein states, we see what the method presents. As a primary tool to retrieve TM proteins from lipid bilayers for solution-based analysis, detergents present proteins in detergent-protein micelles, and inevitably—more or less—affect their function and structure integrity. Rather than presolubilization cross-linking that may disturb the system and complicate analysis, functional proteomics predominantly counts on detergents to preserve proteins' authentic conformation and non-covalent interacting network, during solubilization and enrichment. Thus ideal detergents ought to: (1) solubilize membrane proteins, (2) maintain proteins' native structure and interactome, and (3) reduce downstream cleanup that loses samples to re-aggregation and biases against hydrophobic components.
Although current proteomics literature frequently mixed detergents' solubilization strength with denaturation severity (47), they are in fact two distinct concepts—a premise for functional proteomics. Detergents vary vastly in their effects on protein activity despite solubilization (Table II), ranging from severe (SDS), partial (CHAPS), mild (Triton X-100), to minimal denaturation. Typically 2–5× cmc is required to keep proteins soluble, and more in extraction because of more proteins and lipids in membranes. At such high amounts, ionic (including zwitterionic) and poly-disperse detergents overwhelm peptide reversed-phase (RP) HPLC and ESI, via forming positive fixed charges or strong adducts at acidic pH. Current norm believes detergents must be removed before digestion or HPLC (4, 5, 22), at unavoidable sample loss.
Table II. Detergent selection for functional membrane proteomics. cmc, critical micelle concentration; AN, aggregation number; dtg, detergent; n/a, not available. DM, n-decyl-β-d-maltopyranoside; OG, n-octyl-β-d-glucoside. FA, steroid-based facial amphiphiles; FA-3, type 3 (2 maltosides); FA-4, type 4 (3 maltosides); FA-5, type 5 (3 glucosides). BP-1, beta-strand peptide type 1, (N) acetyl-(octyl)G-S-l-S-(N-methyl)l-d-(octyl)G-d-NH2.
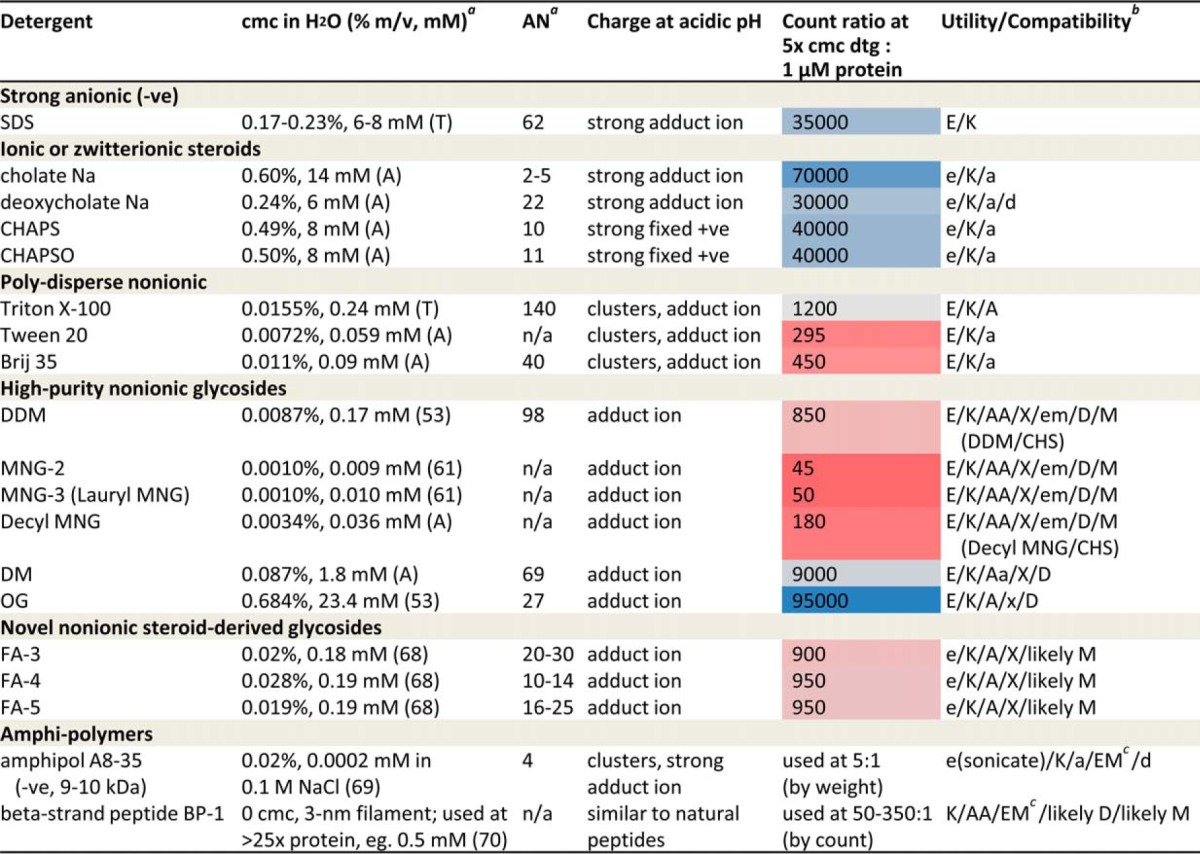
a Source for cmc and aggregation number is Anatrace (A), Thermo Pierce (T), or specified reference.
b Tissue-, cell- and protein-dependent, shown are general trends, based on Refs (6, 7, 24, 45, 49, 51–53, 61, 66–70), detergent charge state, count ratio over protein, PDB database, and experiences with CcO, hGPCR β2AR and hLGIC GABAAR. Utility/Compatibility: Extraction from membrane (E; success rate E > e; e: requiring sonication, or effective for mammalian and insect cells but less effective for bacterial cells), Keeping protein soluble (K), Activity maintenance (A; AA > Aa > A > a). Crystallography (X; X > x), EM (EM > em; em: often difficult to tell protein TM domains from detergent micelles), Digestion (D; D > d), and peptide RP HPLC MS (M; M > m). Unmarked means no or usually unconsidered.
c After exhaustively replacing other detergents.
cmc, critical micelle concentration; AN, aggregation number; dtg, detergent; n/a, not available; DM, n-decyl-β-D-maltopyranoside; OG, n-octyl-β-D-glucoside; FA, steroid-based facial amphiphiles; FA-3, type 3 (2 maltosides); FA-4, type 4 (3 maltosides); FA-5, type 5 (3 glucosides); BP-1, beta-strand peptide type 1, (N) acetyl-(octyl)G-S-L-S-(N-methyl)L-D-(octyl)G-D-NH2.
Rigorous membrane protein functional interactome studies typically compared multiple detergents—as they produced vastly variable, partially overlapping protein populations—and emphasized proteins that overlapped or were unique (48, 49). Subsequent analyses resorted to detergent removal by overnight RapiGest exchange-cleavage (48) or by protein precipitation-resolubilization (49), and digestion with trypsin solution (48) or beads (49).
Broadly defined as “a reagent to wipe away” and chemically also termed surface-active agent (surfactant), detergent is characterized with being amphiphilic, and the hydrophilic domain can assume various shapes: one pole of a rod (SDS, Triton X-100), one face of a bulky molecule (CHAPS, deoxycholate), or one side of a linear molecule (amphipols) (Table II).
High-purity n-dodecyl-β-d-maltopyranoside (DDM) was synthesized and first shown to be unusually effective for both solubilizing and stabilizing active membrane proteins—attributable to its micelle size and rigid tight-packing nonionic head group—by the Ferguson–Miller group in the 1980s (50–53), using animal tissues then cell cultures. Subsequently, DDM-based extraction and/or purification have critically enabled high-resolution crystallization of most of the over-500 diverse membrane proteins in the Protein Data Bank, including GPCRs, multisubunit ion channels, neurotransmitter transporters, and respiratory complexes (54–60); DDM's recent “X”-shaped dimeric derivative, maltose-neopentyl glycols (MNG) type 3 (MNG-3, lauryl MNG), stabilized hGPCR-Gs complex for crystallization (55, 61). For mammalian signaling TM proteins native to ∼20–25%-cholesterol lipid bilayer habitats (by weight and by count) (62–64), the Stevens group found adding cholesteryl hemisuccinate (CHS) to DDM forms wider bicelle-like DDM/CHS micelles that crucially stabilize human GPCRs for activity and crystallization (54, 65). DDM/CHS also proved effective to extract and purify active synaptic hetero-pentamer hGABAAR from HEK293 (66); seconds of DDM/CHS sonication afforded over 90% solubilization of HEK293 membranes (66) and no visible pellet post-centrifuge for rat brains (personal communication, Feb 2015). Decyl MNG/CHS stabilized hLGIC (β3)5 GABAAR for crystallization (67). By contrast, traditional ionic or poly-disperse detergents, such as SDS, CHAPS, cholate, deoxycholate, C12E9, Triton X-100, Tween 20, and Brij 35, as well as nonionic DM, OG, and digitonin, failed to emulate despite decades of pursuit (51–53, 68). Ionic CHAPS and cholate consistently deactivated hGABAAR (66) and mammalian CcO (52) by several-fold compared with DDM and DDM/CHS. Latest nonionic detergents that combined two to three DDM's head groups or three glucosides with a steroid body markedly stabilized proteins for crystallization (68). By replacing other detergents, various amphipols and beta-strand peptides also emerged effective to chaperone proteins for high-resolution EM (69, 70).
However, these detergent-driven advances in membrane protein technology since 1980 circulated largely among the crystallography community, not proteomics. Inheriting industrial-grade detergents such as SDS and Triton X-100, historically applied for cheap price, proteomics has suffered huge penalties in quality and cost. Using ESI MS experiments of intact model protein solutions (infusion and flow injection), a pioneering 1994 study reported several nonionic, zwitterionic, and anionic detergents tolerable by ESI MS, in acetic acid/acetonitrile/H2O spray (top 7: n-dodecyl glucoside, n-hexyl glucoside, CHAPS, cholate, and three equally scored DDM, OG, and octyl thioglucoside), and in H2O spray (top 5: n-dodecyl glucoside, n-hexyl glucoside, OG, n-octyl sucrose, and n-dodecyl sucrose) (71).
We further considered detergent compatibility with protein extraction, protein activity, protease activity, RP-HPLC, data-dependent MS/MS competition and peptide labeling. We propose that beyond tolerance by intact protein MS, detergents may be cultivated as a tool for both membrane protein extraction/purification and digestion modules in proteomic pipeline. Detergents as a tool for membrane protein extraction and purification (discussed above) emphasize maintaining protein native conformations and activity at high yield (often with additives), and providing near-equal grounds of protein states to correlate with other technologies such as crystallography, EM, and function assays (Fig. 1). Detergents as a tool for digestion put emphasis on maintaining protease activity, solubilizing and stabilizing substrates against inter- and intraprotein aggregation over a wide pH range, and minimizing interference with labels (such as HDX, oxidation and TMT), peptide RP HPLC, or ESI MS/MS, but less on native conformations, as digestion often desires acidic pH and proteins are dissected rapidly. Detergents as a tool for united proteomic pipelines emphasize optimal compatibility with all modules.
Based on detergent properties—including charge state at acidic pH, mono- or poly-dispersion, and cmc (count ratio over protein at 5× cmc:1 μm protein)—and their documented performances in membrane protein extraction, activity maintenance, and high-resolution structures, particularly experiences gained from CcO, hGPCR, and hLGIC complexes, Table II analyzed both common and recent detergents for their utility/compatibility potentials to various steps in integrated functional membrane proteomic pipelines.
Direct Flow/DDM-facilitated Digestions for HDX, Deep Sequencing, and Quantitation
Discovery of Omnicompatible Detergents as a Pivotal Tool to Fully Automate Deep and Direct Membrane Proteomics
Breakthroughs in membrane protein digestion started in 2010 for HDX dynamic structure studies, arguably the ultimate digestion challenge, by including detergents that complied with structure/protease/HPLC MS (6), or by applying membrane scaffold proteins (MSP) that kept proteins in nanodiscs followed by cholate (which entailed high-pressure ultra-performance liquid chromatography, UPLC) (72, 73), to solve aggregation. Thinking outside the box of denaturation anddetergent removal, we discovered that nonionic low-cmc amine-free DDM—the most protein nature-promoting and crystallization-successful detergent to date—was completely compatible with structure/protease/TMT/HPLC MS/MS, and devised DDM-based protein extraction, purification and digestion methods that cleared all these challenges of membrane proteins (6, 7, 66). DDM-low-TCEP (DLT, for HDX) and flow/detergent-facilitated protease and de-PTM digestions (FDD, for deep sequencing and quantitation) both completed peptide preparations within seconds-minutes, at robust coverage and peptide reproducibility supporting full automation, demonstrated by using hGPCR (6) and hLGIC complex (7), respectively.
Advantages of DDM as a tool for membrane proteomics are multifold and pivotal to achieving fully automated deep and direct proteomic analyses of membrane proteins in physiological states. First, for protein extraction/purification, DDM combined with CHS to mimic cholesterol in human cells, can extract membrane proteins at high activity, and high efficiency when applied with co-micelle-promoting slow addition or sonication (66). DDM and its derivatives that critically enabled the purification and crystallization of myriad active membrane proteins (50, 51, 54–57, 61, 74, 75), present proteins in functional states in solution, thus providing a common ground for proteomics to bridge functions directly with protein PTMs, dynamic conformations, and static structures resolved by crystallography (Fig. 1). Second, as a tool for digestion, DDM preserves—not destroys—the activity and solubility of both enzymes and protein substrates, thus facilitates effective catalytic contact and unbiased flow of hydrophobic species during digestion, and obviates problematic re-aggregation, high urea, subsequent desalting, and reconcentrating. DDM is effective over a wide pH range, thus can broadly simplify diverse digestion formats such as: enzyme column, protein on-bead, in-solution, and in-nanodisc, by replacing conventional detergents, urea or guanidine. Third, for overall pipelines, amine-free nonionic DDM supports downstream HPLC, MS/MS competition and TMT reactions, can be included throughout the cell lysis-protein extraction-(HDX)-digestion-(TMT)-HPLC-MS/MS process without removal, thus confers the workflows with no interruption, no sample leakage and high sensitivity. DDM at 0.4–1 mm (0.02–0.05%, 2.3–5.7× cmc) over 1 μm proteins (0.04–0.28 mg/ml) was well tolerated (6, 7).
At even lower cmc (∼9, ∼10, and 36 μm) (61), nonionic MNG-2, MNG-3, and decyl MNG are expected compatible with RP HPLC MS as well; likewise are the low-cmc nonionic facial amphiphiles (Fig. 2B and Table II). MNG-3 greatly stabilized protein complex in solution for crystallization (61), we found its membrane solubilization power for HEK293 was comparable to DDM basal (66), thus MNG can serve as a valuable control to cross-check DDM extraction results and as another tool for digestion.
Fig. 2.

General approaches of fully automatable flow-and-detergent facilitated membrane proteomics. A, Workflows of flow/detergent-facilitated protease and de-PTM digestions (FDD) and DDM-low-TCEP (DLT) for membrane protein samples, B, structures of protein activity/HDX/TMT/RP-HPLC/MS omni-compatible detergents and additives, and C, alternative modes of FDD. Instrument images were from www.thermoscientific.com.
DDM-low-TCEP Digestion for HDX
Instead of conventional 3 m urea, 1.5 m guanidine, or 500 mm TCEP, the 2010 DLT method developed a digestion solution of 0.02% (m/v) DDM, low 15 mm TCEP and 100 mm NaH2PO4-HCl pH 2.4 (final 7.5 mm TCEP), to quench membrane protein HDX (DDM/CHS/glycerol protein buffer in H2O or D2O) for direct digestion, online in a fully automated workflow (6). DLT with pepsin column achieved the first comprehensive HDX profile of 7TM hGPCR at near 90% coverage, using a PTM-deprived minimalistic β2AR developed for crystallization (6). These peptides were each reproduced by several hundred independent digestions of apo and various ligand-bound β2AR from multiple batches of purifications (Ref (6) and unpublished data). The DDM solution also successfully reproduced high coverage for other GPCRs in other laboratories (76).
However, devised for direct HDX, DLT did not address extensive PTMs and Cys, as they did not pose a problem in the β2AR crystallizable construct: All PTM-bearing domains were either genetically truncated (intracellular C-terminal tail and loop 3) or deglycosylated (two adjacent extracellular-domain N-glycosylations) in purification (6). Although 7.5 mm TCEP allowed detecting peptides bearing free Cys, without alkylation, they were the weakest, and the C–C linked versions were detected at higher ion counts (6). Also designed with a desalting trap and high-flow-rate HPLC and ESI (50 μl/min), DLT-HDX's peptide signals were close to baseline in HPLC-ion count chromatograms, and did not appeal to broader applications (Fig. 3A).
Fig. 3.
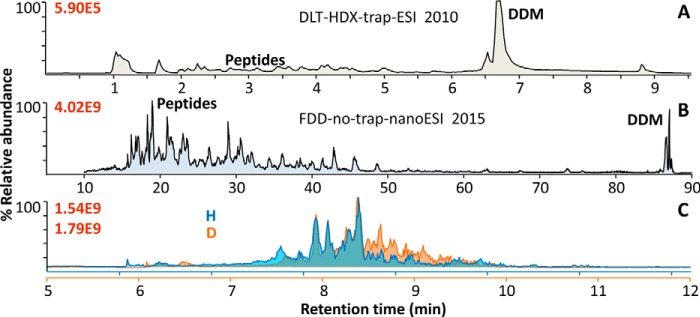
At much higher peptide ion sensitivity, the 2015 FDD-no-trap-nESI method proved DDM can be a superior tool for broad nESI-based membrane proteomics. Representative HPLC MS traces of (A) 2010 DLT-HDX-trap-ESI of hGPCR apo β2AR, typical to the data for (6), B, FDD-no-trap-nESI of hGABAAR, and (C) independent digests of H/D versions of hGABAAR using FDD-no-trap-nESI's HDX module showing good reproducibility. All spectra were acquired with orbitrap analyzers in Thermo Exactive (A), Q-Exactive (B), or LTQ-orbitrap XL (C) mass spectrometer. Experiments used similar concentrations of protein and detergent, but A loaded several-fold more total sample to HPLC than B and C. Figure was adapted from (7).
Flow/DDM-facilitated Protease and De-PTM Digestions for Deep Sequencing and Quantitation
Because DDM obviated high salt, by removing the desalting trap and adopting capillary HPLC column and nanoESI, a no-trap-nanoESI upgrade of DLT remarkably increased peptide ion sensitivity (66). When applied to full-length hGABAAR hallmarked with Cys-loops, it produced up to 80–84% coverage with a 75-min HPLC gradient (66), but gaps of over 15% persisted at Cys and potential PTM sites.
Addressing PTMs and Cys, the 2015 FDD method further combined immobilized pepsin column, PNGase F column, Cys-selective reduction and alkylation, and greatly improved peptide metrics. A novel integrated digestion paradigm, FDD's components each targeted the key challenges for human signaling TM proteins—hydrophobicity and intricate PTMs—inaccessible for decades by conventional methods. (1) FDD's tandem flow reactor format achieved fast, effective, reproducible, and controllable reactions for both proteolysis and de-PTM; (2) DDM resolved aggregation caused by TM hydrophobicity and promoted unbiased flow through all reactors; (3) de-PTM PNGase F column solved thorough mapping of N-glycosylation sites that turned out to crosstalk with other PTMs and critical for functions. At minutes of total preparation time, FDD achieved up to 99% coverage in one run, high peptide reproducibility and low artifacts for all subunits of hGABAAR/HEK293 (7) (Fig. 4A and Fig. 3C). Importantly, the FDD-no-trap-nESI platform increased peptide ion sensitivity by four orders of magnitude from 2010 DLT-trap-ESI (Fig. 3), and discovered DDM is no longer a compromise, but a superior tool broadly applicable to nESI-based proteomics. FDD further demonstrated direct TMT labeling and analysis (7) can be integrated in a fully automatable membrane proteomic workflow (Table I and Fig. 2).
Fig. 4.

Protein sequence coverage identified from direct FDD-pepsin digestion and analysis of DDM/CHS-enriched hGABAAR solution, by searching against. A, the target sequences of (α1)2(β3)2(γ2L)1 GABAAR (SEQUEST in Proteome Discoverer 1. 3), or (B) the original human proteome (May 23, 2013 Ensembl, SEQUEST-HT in Proteome Discoverer 1.4), both at no protease specificity and filtered to peptide FDR <1% by Percolator. Each thin line represents one PSM (apple green, α1 subunit; blue, β3; orange, γ2L); bold lines indicate domains (dark blue, TM; red, intracellular loop; black, extracellular Cys-loop). B, shows the % sequence coverage (blue bar) for the top 34 proteins (ranked by PSMs and unique peptides), and their percent PSM (red bar) of the total PSMs of all proteins identified by three or more valid unique peptides; (m) designates membrane residence, as annotated in human proteome database. N-terminal signaling peptide sequences were not removed. A was adapted from (7). These proteins were mainly cell-originated survivors of two affinity screenings by membrane isolation and by buffer washing during anti-FLAG enrichment, not contaminant from processing.
FDD presents multifold advantages for bottom-up membrane proteomics, and critically supports the next-level quantitative-structural integration. First, FDD transformed the days-long aggregation-plagued paradigm into immediate digestion and measurement of active protein solutions—in seconds–minutes total peptide preparation time—at unprecedented coverage, reproducibility, and authenticity. This allowed both the minutes-windowed HDX and the deep PTM/interactome mapping to look at the near-identical states of proteins and peptides, thus broke barriers against integrating quantitative-structural membrane proteomics. Second, FDD eliminated precipitation, filtration and centrifugation central to the denaturation-, tip- or filter-based in-solution traditions. This enabled a robust, instant, versatile protein-(HDX)-peptide-(TMT)-HPLC MS streamline for uninterrupted automation. Together, FDD made Path 2 immediately achievable (Fig. 1) and may improve Path 1 in speed, depth, accuracy, sensitivity, and throughput (Table I).
Striking peptide metric increases in DLT and FDD are not artifacts of database search, but reflect real changes in peptide production. First, digestion methods—urea-based trypsin or pepsin versus DLT-pepsin digestion of GPCR (6), and DDM-facilitated pepsin solution, beads versus flow reactor format (66)—were compared on the equal grounds of target sequence search and parallel experiments. Second, spectra search against human proteome maintained the high peptide number and coverage of target proteins (7) (Fig. 4B). Third, identified proteins showed average sequence coverage higher than current ∼25% benchmark (8, 42) (Fig. 4B). Further, compared one-to-one with 7TM hGPCR and hGABAAR, most proteins in cell lysates are technically easier, thus unlikely to pose further obstacles.
Concrete reach to PTMs—including N-glycosylation on Cys-loop and endogenous M-oxidation—proved crucial for discovering function clues inaccessible to methods with sporadic coverage and severe artifacts (7). Extensive PTMs in human signaling TM proteins, such as high-occupancy N-glycosylation, had remained largely a target of observation, but rarely exploited as a means to aid overall peptide formation and detection, partly for fear that PTM enzymes overwhelm real samples (though PNGase F was used to release enriched glycopeptides (77)). Predigestion PNGase F solution incubation indeed identified more proteins in yeast cell wall (19). But emerging complicated N-glycosylation scenarios in mammalian TM proteins suggest, treating peptides post-digestion is more likely efficient and complete than treating proteins (7, 66).
Extended Applications of FDD Format
As a general platform to prepare membrane protein peptides, FDD's elements—enzyme flow reactors, omnicompatible detergents, and de-PTM—are flexible to adapt to nearly all levels of proteomics and support myriad enzymes and new detergents (Fig. 2C). Both the DDM/CHS-extraction and FDD digestion strategies are compatible with upstream enrichment of cell fractions, proteins, protein nanodiscs, downstream peptide enrichment (DDM supports IMAC (6, 54, 57, 74, 75)), fractionation, TMT, and HPLC. Excitingly, besides DDM/CHS, MNGs and several recent detergents superb for crystallography and EM likely fit FDD as well (Table II), suggesting the proposed proteomics-EM-crystallography-function integration is not an exception, but an increasingly achievable new paradigm for membrane proteomics (Fig. 1). This flexibility also allows matching digestion detergent with the one used in protein preparation to avoid multiple HPLC peaks.
Membrane Protein Digestion: Protease Reliability beyond Theoretical Specificity
Confounded by membrane proteins' unique hydrophobicity and tendency to aggregate, protease's empirical reliability has proved distinct from its theoretical specificity over primary sequence. How to apply the protease appears to matter more than theoretical specificity for achieving reliable digestion. Several recent digestion advances can be interpreted via improving substrate–protease contacts, such as the protease-mild substrate-solubilizing RapiGest (42), and the pressure cycling technology to improve reaction mixing in urea (21), though all under the detergent-removal/avoiding paradigm. For this purpose, an HPLC-grade flow-propelled high-concentration immobilized enzyme micro-column reactor is arguably the most effective. Omnicompatible detergents provided the bridge for membrane proteins to take advantage of this format, to reach complete reproducible digestion and seconds–minutes scale under ambient physiological solutions, temperature, and pressure, obviating any brutal force.
For specific proteases, effective substrate-protease contacts shall help overcome the deviation from theoretical specificity, and finally achieve the “peptide concentrating” strategy described above. DDM-facilitated trypsin beads already showed complete digestion of a multihelix protein within minutes (supplemental Fig. S1A) contrasting overnight in urea solution, thus DDM-facilitated trypsin column is expected effective with membrane proteins too (Fig. 2C). Applying multiple specific proteases in the format of HPLC-grade tandem columns or mixed-beads column (minutes) shall be more efficient than combining days of individual urea-solution digestions (Fig. 2C).
Challenging conventional belief, fast multi-specific protease proved able to be highly reproducible and reliable—when applied in the DDM-facilitated HPLC-grade enzyme column format (6, 7). Reproducible peptide ladders provide desired redundancy and bypass specific proteases' cleavage gaps. Pepsin column rapidly generated comprehensive coverage including TM domains, with redundant overlapping peptides four to 20 residues long, useful for HDX and PTM mapping (6, 7). Nepenthesin-1, a recent pepsin alternative, has different multispecificities, max activity at pH 2.5, low autolysis, low tolerance to denaturants, yet stability in basic pH (78, 79)—making it suitable for high-yield immobilization and nondenaturant digestion.
Fast and preferring hydrophobic sites, HPLC-grade columns of acidic multispecific proteases such as pepsin and nepenthesin-1 (78–80) offered a robust, efficient (seconds) alternative to combining multiple solutions of specific proteases (days), and can be favored over trypsin particularly: (1) when TM receptors such as hGABAAR frequently contain K/R over 50 residues apart in extracellular domain, but also dense K/R (1–2 residues apart) in intracellular domain: Seeing these K/R-dense peptides relies on trypsin's unreliability—missing K/R cleavage, a ∼15% chance in urea solution (81), (2) when K/R-PTMs interfere with trypsin cleavage impeding peptide-centric quantitation, (3) when proteins such as myoglobin resist trypsin digestion (supplemental Fig. S1B Lane 3), and (4) when acidic digestion is desired to minimize nonenzymatic deamidation (82) and scrambling of native PTMs such as C-C bonds (83). The large number of peptides is within capacity of matured HPLC separation.
CONCLUSIONS
Shedding the decade-long cliché of denaturation, detergent removal/avoidance and laborious processing, the detergent- and reactor-based digestion strategies demystified the technical challenges of membrane proteins, shifted the day(s)-long paradigm into seconds–minutes scale, and cleared barriers against real-time integration in proteomic pipelines. These digestion methods empower membrane proteomics to grow beyond protein identification to the next level: integrative structure-function mechanisms of membrane proteins, via direct quantitative PTM and structural mapping in their physiological states, at unprecedented coverage, throughput, sensitivity, and accuracy (Fig. 1). At the surge of ambition and efforts to tackle membrane protein-central human proteome, signaling interactome, and dynamic structural mechanisms of TM receptors for better therapeutics, they afford a timely tool box of solutions to accelerate bottom-up proteomics to achieve these goals.
Supplementary Material
Acknowledgments
I thank Dr. Shelagh M. Ferguson-Miller for discussions on detergents and this manuscript. I thank Dr. Keith W. Miller for providing a CYP11B1 sample used in supplemental figure.
Footnotes
Author Contributions: X.Z. conceived and wrote paper.
* This work was supported in part by the National Institute of General Medical Sciences (GM 58448, K.W.M.).
 This article contains supplemental Fig. S1.
This article contains supplemental Fig. S1.
1 The abbreviations used are:
- PTM
- post-translational modification
- HDX
- H/D exchange
- TM
- transmembrane
- DDM
- n-dodecyl-β-D-maltopyranoside
- TCEP
- Tris-2-carboxyethylphosphine
- TMT
- tandem mass tag
- DLT
- DDM-low-TCEP digestion for HDX
- FDD
- flow/detergent-facilitated protease and de-PTM digestions for deep sequencing
- h
- human
- LGIC
- ligand-gated ion channel
- GPCR
- G protein-coupled receptor
- GABAAR
- gamma-aminobutyric acid type A receptor
- β2AR
- beta-2 adrenergic receptor
- HEK293
- human embryonic kidney cell line 293
- PSM
- peptide spectrum match
- FDR
- false discovery rate
- C-C
- disulfide-bonded Cys-loop
- Mox
- Methionine oxidation
- RP
- reversed-phase
- UPLC
- ultra-performance liquid chromatography
- FASP
- filter-aided sample preparation
- dial-filtration
- dialysis-filtration
- SP3
- single-pot solid-phase-enhanced sample preparation
- EM
- electron microscopy
- amphipols
- amphi-polymers
- MSP
- membrane scaffold proteins
- PGF
- PNGase F
- cmc
- critical micelle concentration
- AN
- aggregation number
- CHS
- cholesteryl hemisuccinate
- MNG
- maltose-neopentyl glycol
- FA
- steroid-based facial amphiphiles
- BP
- beta-strand peptide
- DM
- n-decyl-β-D-maltopyranoside
- OG
- n-octyl-β-D-glucoside.
REFERENCES
- 1. Baker M. (2010) Making membrane proteins for structures: A trillion tiny tweaks. Nat. Methods 7, 429–434 [DOI] [PubMed] [Google Scholar]
- 2. Yates J. R., 3rd (2013) The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J. Am. Chem. Soc. 135, 1629–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pieper U., Schlessinger A., Kloppmann E., Chang G. A., Chou J. J., Dumont M. E., Fox B. G., Fromme P., Hendrickson W. A., Malkowski M. G., Rees D. C., Stokes D. L., Stowell M. H., Wiener M. C., Rost B., Stroud R. M., Stevens R. C., Sali A. (2013) Coordinating the impact of structural genomics on the human alpha-helical transmembrane proteome. Nat. Struct. Mol. Biol. 20, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whitelegge J. P. (2013) Integral membrane proteins and bilayer proteomics. Anal. Chem. 85, 2558–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meissner F., Mann M. (2014) Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat. Immunol. 15, 112–117 [DOI] [PubMed] [Google Scholar]
- 6. Zhang X., Chien E. Y., Chalmers M. J., Pascal B. D., Gatchalian J., Stevens R. C., Griffin P. R. (2010) Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Anal. Chem. 82, 1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang X. (2015) Instant integrated ultradeep quantitative-structural membrane proteomics discovered PTM signatures for human Cys-loop receptor subunit bias. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou F., Lu Y., Ficarro S. B., Adelmant G., Jiang W., Luckey C. J., Marto J. A. (2013) Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molinari M. (2007) N-glycan structure dictates extension of protein folding or onset of disposal. Nat. Chem. Biol. 3, 313–320 [DOI] [PubMed] [Google Scholar]
- 11. Vowinckel J., Capuano F., Campbell K., Deery M. J., Lilley K. S., Ralser M. (2013) The beauty of being (label)-free: sample preparation methods for SWATH-MS and next-generation targeted proteomics. F1000Res 2, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Switzar L., Giera M., Niessen W. M. (2013) Protein digestion: an overview of the available techniques and recent developments. J. Proteome Res. 12, 1067–1077 [DOI] [PubMed] [Google Scholar]
- 13. Peng M., Taouatas N., Cappadona S., van Breukelen B., Mohammed S., Scholten A., Heck A. J. (2012) Protease bias in absolute protein quantitation. Nat. Methods 9, 524–525 [DOI] [PubMed] [Google Scholar]
- 14. Griffin N. M., Schnitzer J. E. (2011) Overcoming key technological challenges in using mass spectrometry for mapping cell surfaces in tissues. Mol. Cell. Proteomics 10, R110 000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., Nesvizhskii A. I. (2013) The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fonslow B. R., Stein B. D., Webb K. J., Xu T., Choi J., Park S. K., Yates J. R., 3rd (2013) Digestion and depletion of abundant proteins improves proteomic coverage. Nat. Methods 10, 54–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell. Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Golizeh M., Sleno L. (2013) Optimized proteomic analysis of rat liver microsomes using dual enzyme digestion with 2D-LC-MS/MS. J. Proteomics 82, 166–178 [DOI] [PubMed] [Google Scholar]
- 19. Bailey U. M., Schulz B. L. (2013) Deglycosylation systematically improves N-glycoprotein identification in liquid chromatography-tandem mass spectrometry proteomics for analysis of cell wall stress responses in Saccharomyces cerevisiae lacking Alg3p. J. Chromatogr. 923–924, 16–21 [DOI] [PubMed] [Google Scholar]
- 20. Glatter T., Ludwig C., Ahrne E., Aebersold R., Heck A. J., Schmidt A. (2012) Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 11, 5145–5156 [DOI] [PubMed] [Google Scholar]
- 21. Guo T., Kouvonen P., Koh C. C., Gillet L. C., Wolski W. E., Rost H. L., Rosenberger G., Collins B. C., Blum L. C., Gillessen S., Joerger M., Jochum W., Aebersold R. (2015) Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med., Epub Mar 02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 23. Wisniewski J. R., Mann M. (2012) Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 84, 2631–2637 [DOI] [PubMed] [Google Scholar]
- 24. Erde J., Loo R. R., Loo J. A. (2014) Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 13, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zielinska D. F., Gnad F., Wisniewski J. R., Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 26. Kulak N. A., Pichler G., Paron I., Nagaraj N., Mann M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 [DOI] [PubMed] [Google Scholar]
- 27. Weekes M. P., Antrobus R., Lill J. R., Duncan L. M., Hor S., Lehner P. J. (2010) Comparative analysis of techniques to purify plasma membrane proteins. J. Biomol. Tech. 21, 108–115 [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y., Zhang W., Kho Y., Zhao Y. (2004) Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 [DOI] [PubMed] [Google Scholar]
- 29. Hughes C. S., Foehr S., Garfield D. A., Furlong E. E., Steinmetz L. M., Krijgsveld J. (2014) Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Slysz G. W., Schriemer D. C. (2003) On-column digestion of proteins in aqueous-organic solvents. Rapid Commun. Mass Spectrom. 17, 1044–1050 [DOI] [PubMed] [Google Scholar]
- 31. Slysz G. W., Schriemer D. C. (2005) Blending protein separation and peptide analysis through real-time proteolytic digestion. Anal. Chem. 77, 1572–1579 [DOI] [PubMed] [Google Scholar]
- 32. Slysz G. W., Schriemer D. C. (2009) Integrating accelerated tryptic digestion into proteomics workflows. Methods Mol. Biol. 492, 241–254 [DOI] [PubMed] [Google Scholar]
- 33. Krenkova J., Lacher N. A., Svec F. (2009) Highly efficient enzyme reactors containing trypsin and endoproteinase LysC immobilized on porous polymer monolith coupled to MS suitable for analysis of antibodies. Anal. Chem. 81, 2004–2012 [DOI] [PubMed] [Google Scholar]
- 34. Wang L., Pan H., Smith D. L. (2002) Hydrogen exchange-mass spectrometry: optimization of digestion conditions. Mol. Cell. Proteomics 1, 132–138 [DOI] [PubMed] [Google Scholar]
- 35. Ahn J., Jung M. C., Wyndham K., Yu Y. Q., Engen J. R. (2012) Pepsin immobilized on high-strength hybrid particles for continuous flow online digestion at 10,000 psi. Anal. Chem. 84, 7256–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palm A. K., Novotny M. V. (2005) A monolithic PNGase F enzyme microreactor enabling glycan mass mapping of glycoproteins by mass spectrometry. Rapid Commun. Mass Spectrom. 19, 1730–1738 [DOI] [PubMed] [Google Scholar]
- 37. Bynum M. A., Yin H., Felts K., Lee Y. M., Monell C. R., Killeen K. (2009) Characterization of IgG N-glycans employing a microfluidic chip that integrates glycan cleavage, sample purification, LC separation, and MS detection. Anal. Chem. 81, 8818–8825 [DOI] [PubMed] [Google Scholar]
- 38. Krenkova J., Lacher N. A., Svec F. (2009) Multidimensional system enabling deglycosylation of proteins using a capillary reactor with peptide-N-glycosidase F immobilized on a porous polymer monolith and hydrophilic interaction liquid chromatography-mass spectrometry of glycans. J. Chromatogr. A 1216, 3252–3259 [DOI] [PubMed] [Google Scholar]
- 39. Wang Y., Kumar N., Solt L. A., Richardson T. I., Helvering L. M., Crumbley C., Garcia-Ordonez R. D., Stayrook K. R., Zhang X., Novick S., Chalmers M. J., Griffin P. R., Burris T. P. (2010) Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands. J. Biol. Chem. 285, 5013–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chalmers M. J., Busby S. A., Pascal B. D., West G. M., Griffin P. R. (2011) Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev. Proteomics 8, 43–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chung K. Y., Rasmussen S. G., Liu T., Li S., DeVree B. T., Chae P. S., Calinski D., Kobilka B. K., Woods V. L., Jr., Sunahara R. K. (2011) Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477, 611–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen E. I., Cociorva D., Norris J. L., Yates J. R., 3rd (2007) Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J. Proteome Res. 6, 2529–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saveliev S., Simpson D., Wood K. V. (2009) Cleavable surfactants. WO 2009048611 A2. Google Patents, USPTO. [Google Scholar]
- 44. Chang Y. H., Gregorich Z. R., Chen A. J., Hwang L., Guner H., Yu D., Zhang J., Ge Y. (2015) New mass-spectrometry-compatible degradable surfactant for tissue proteomics. J. Proteome Res. Epub Jan 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ning Z., Seebun D., Hawley B., Chiang C. K., Figeys D. (2013) From cells to peptides: “One-stop” integrated proteomic processing using amphipols. J. Proteome Res. 12, 1512–1519 [DOI] [PubMed] [Google Scholar]
- 46. Waas M., Bhattacharya S., Chuppa S., Wu X., Jensen D. R., Omasits U., Wollscheid B., Volkman B. F., Noon K. R., Gundry R. L. (2014) Combine and conquer: surfactants, solvents, and chaotropes for robust mass spectrometry based analyses of membrane proteins. Anal. Chem. 86, 1551–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richards A. L., Merrill A. E., Coon J. J. (2015) Proteome sequencing goes deep. Curr. Opin. Chem. Biol. 24, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christianson J. C., Olzmann J. A., Shaler T. A., Sowa M. E., Bennett E. J., Richter C. M., Tyler R. E., Greenblatt E. J., Harper J. W., Kopito R. R. (2012) Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Babu M., Vlasblom J., Pu S., Guo X., Graham C., Bean B. D., Burston H. E., Vizeacoumar F. J., Snider J., Phanse S., Fong V., Tam Y. Y., Davey M., Hnatshak O., Bajaj N., Chandran S., Punna T., Christopolous C., Wong V., Yu A., Zhong G., Li J., Stagljar I., Conibear E., Wodak S. J., Emili A., Greenblatt J. F. (2012) Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 489, 585–589 [DOI] [PubMed] [Google Scholar]
- 50. Garavito R. M., Ferguson-Miller S. (2001) Detergents as tools in membrane biochemistry. J. Biol. Chem. 276, 32403–32406 [DOI] [PubMed] [Google Scholar]
- 51. Rosevear P., VanAken T., Baxter J., Ferguson-Miller S. (1980) Alkyl glycoside detergents: a simpler synthesis and their effects on kinetic and physical properties of cytochrome c oxidase. Biochemistry 19, 4108–4115 [DOI] [PubMed] [Google Scholar]
- 52. Thompson D. A., Suarez-Villafane M., Ferguson-Miller S. (1982) The active form of cytochrome c oxidase: effects of detergent, the intact membrane, and radiation inactivation. Biophys. J. 37, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. VanAken T., Foxall-VanAken S., Castleman S., Ferguson-Miller S. (1986) Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol. 125, 27–35 [DOI] [PubMed] [Google Scholar]
- 54. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 57. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brohawn S. G., del Marmol J., MacKinnon R. (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qin L., Hiser C., Mulichak A., Garavito R. M., Ferguson-Miller S. (2006) Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 103, 16117–16122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chae P. S., Rasmussen S. G., Rana R. R., Gotfryd K., Chandra R., Goren M. A., Kruse A. C., Nurva S., Loland C. J., Pierre Y., Drew D., Popot J. L., Picot D., Fox B. G., Guan L., Gether U., Byrne B., Kobilka B., Gellman S. H. (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Norton W. T., Abe T., Poduslo S. E., DeVries G. H. (1975) The lipid composition of isolated brain cells and axons. J. Neurosci. Res. 1, 57–75 [DOI] [PubMed] [Google Scholar]
- 63. Calderon R. O., Attema B., DeVries G. H. (1995) Lipid Composition of Neuronal Cell Bodies and Neurites from Cultured Dorsal Root Ganglia. J. Neurochem. 64, 424–429 [DOI] [PubMed] [Google Scholar]
- 64. Scandroglio F., Venkata J. K., Loberto N., Prioni S., Schuchman E. H., Chigorno V., Prinetti A., Sonnino S. (2008) Lipid content of brain, brain membrane lipid domains, and neurons from acid sphingomyelinase deficient mice. J. Neurochem. 107, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thompson A. A., Liu J. J., Chun E., Wacker D., Wu H., Cherezov V., Stevens R. C. (2011) GPCR stabilization using the bicelle-like architecture of mixed sterol-detergent micelles. Methods 55, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang X., Miller K. W. (2015) Dodecyl maltopyranoside enabled purification of active human GABA type A receptors for deep and direct proteomic sequencing. Mol. Cell. Proteomics 14, 724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller P. S., Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee S. C., Bennett B. C., Hong W. X., Fu Y., Baker K. A., Marcoux J., Robinson C. V., Ward A. B., Halpert J. R., Stevens R. C., Stout C. D., Yeager M. J., Zhang Q. (2013) Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 110, E1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Popot J. L., Althoff T., Bagnard D., Baneres J. L., Bazzacco P., Billon-Denis E., Catoire L. J., Champeil P., Charvolin D., Cocco M. J., Cremel G., Dahmane T., de la Maza L. M., Ebel C., Gabel F., Giusti F., Gohon Y., Goormaghtigh E., Guittet E., Kleinschmidt J. H., Kuhlbrandt W., Le Bon C., Martinez K. L., Picard M., Pucci B., Sachs J. N., Tribet C., van Heijenoort C., Wien F., Zito F., Zoonens M. (2011) Amphipols from A to Z. Annu. Rev. Biophys. 40, 379–408 [DOI] [PubMed] [Google Scholar]
- 70. Tao H., Lee S. C., Moeller A., Roy R. S., Siu F. Y., Zimmermann J., Stevens R. C., Potter C. S., Carragher B., Zhang Q. (2013) Engineered nanostructured beta-sheet peptides protect membrane proteins. Nat. Methods 10, 759–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Loo R. R., Dales N., Andrews P. C. (1994) Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci. 3, 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hebling C. M., Morgan C. R., Stafford D. W., Jorgenson J. W., Rand K. D., Engen J. R. (2010) Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal. Chem. 82, 5415–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Parker C. H., Morgan C. R., Rand K. D., Engen J. R., Jorgenson J. W., Stafford D. W. (2014) A conformational investigation of propeptide binding to the integral membrane protein gamma-glutamyl carboxylase using nanodisc hydrogen exchange mass spectrometry. Biochemistry 53, 1511–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang X., Tamot B., Hiser C., Reid G. E., Benning C., Ferguson-Miller S. (2011) Cardiolipin deficiency in Rhodobacter sphaeroides alters the lipid profile of membranes and of crystallized cytochrome oxidase, but structure and function are maintained. Biochemistry 50, 3879–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang X., Hiser C., Tamot B., Benning C., Reid G. E., Ferguson-Miller S. M. (2011) Combined genetic and metabolic manipulation of lipids in Rhodobacter sphaeroides reveals non-phospholipid substitutions in fully active cytochrome c oxidase. Biochemistry 50, 3891–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Orban T., Jastrzebska B., Gupta S., Wang B., Miyagi M., Chance M. R., Palczewski K. (2012) Conformational dynamics of activation for the pentameric complex of dimeric G protein-coupled receptor and heterotrimeric G protein. Structure 20, 826–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 78. Athauda S. B., Matsumoto K., Rajapakshe S., Kuribayashi M., Kojima M., Kubomura-Yoshida N., Iwamatsu A., Shibata C., Inoue H., Takahashi K. (2004) Enzymic and structural characterization of nepenthesin, a unique member of a novel subfamily of aspartic proteinases. Biochem. J. 381, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kadek A., Mrazek H., Halada P., Rey M., Schriemer D. C., Man P. (2014) Aspartic protease nepenthesin-1 as a tool for digestion in hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 86, 4287–4294 [DOI] [PubMed] [Google Scholar]
- 80. Hamuro Y., Coales S. J., Molnar K. S., Tuske S. J., Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046 [DOI] [PubMed] [Google Scholar]
- 81. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hao P., Ren Y., Alpert A. J., Sze S. K. (2011) Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol. Cell. Proteomics 10, O111 009381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu F., Breukelen B., Heck A. J. (2014) Facilitating protein disulfide mapping by a combination of pepsin digestion, electron transfer higher energy dissociation (EThcD) and a dedicated search algorithm SlinkS. Mol. Cell. Proteomics 13, 2776–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.