

Abstract
Severe asthma (SA) is a challenge to control, as patients are not responsive to high doses of systemic corticosteroids (CS). In contrast, mild-moderate asthma (MMA) is responsive to low doses of inhaled CS, indicating that Th2 cells, which are dominant in MMA, do not solely orchestrate SA development. Here, we analyzed broncholalveolar lavage cells isolated from MMA and SA patients and determined that IFN-γ (Th1) immune responses are exacerbated in the airways of individuals with SA, with reduced Th2 and IL-17 responses. We developed a protocol that recapitulates the complex immune response of human SA, including the poor response to CS, in a murine model. Compared with WT animals, Ifng–/– mice subjected to this SA model failed to mount airway hyperresponsiveness (AHR) without appreciable effect on airway inflammation. Conversely, AHR was not reduced in Il17ra–/– mice, although airway inflammation was lower. Computer-assisted pathway analysis tools linked IFN-γ to secretory leukocyte protease inhibitor (SLPI), which is expressed by airway epithelial cells, and IFN-γ inversely correlated with SLPI expression in SA patients and the mouse model. In mice subjected to our SA model, forced SLPI expression decreased AHR in the absence of CS, and it was further reduced when SLPI was combined with CS. Our study identifies a distinct immune response in SA characterized by a dysregulated IFN-γ/SLPI axis that affects lung function.
Introduction
Severe asthma (SA) is classified as a separate category of asthma in which disease symptoms are poorly controlled by corticosteroids (CS), the mainstay of asthma therapy (1–3). Although only 5% to 10% of asthmatics have disease that belongs to this category, because of frequent hospitalizations and need for emergency care, up to 50% of health care costs for all asthma is spent on SA patients in the US and in Europe (4, 5). In humans, airway inflammation is a feature of both mild-moderate asthma (MMA) and SA (6). However, while in MMA, airway inflammation is usually eosinophil dominated, in the more severe form of the disease, mixed granulocytic inflammation comprising both neutrophils and eosinophils is more common (6). Our recent study that employed unsupervised phenotyping of asthma characteristics using machine-learning approaches showed that asthma subjects with the lowest lung function exhibited persistent airway inflammation and were all on systemic CS use (7). Despite these basic understandings of disease characteristics, there is still a gap in our knowledge of the immune response in the airways of SA patients, whose disease, unlike MMA, is not adequately managed by CS. Because of this deficiency, sufficient advancement has not been made in the development of better therapy for SA.
The present study reports that the immune response in the airways of SA patients is distinct from that in subjects with MMA, with dominance of a Th1 immune response in the former despite ongoing treatment with high doses of CS. This information allowed us to establish a new animal model of SA in which immune response and poor control by CS resembled that observed in human disease. Using genetically engineered mice subjected to the SA model, IFN-γ but not IL-17 was found to be responsible for heightened airway hyperresponsiveness (AHR), which was in line with findings in a recent human study that failed to associate IL-17 with worse lung function in SA patients (8). In contrast, mice subjected to a Th2-dominated model displayed lower AHR and complete resolution by CS. Computer-assisted analysis of molecules linked to IFN-γ and AHR allowed us to focus on the anti-protease secretory leukocyte protease inhibitor (SLPI). SLPI expression in the airway epithelial cells of SA patients negatively correlated with IFN-γ expression in bronchoalveolar lavage (BAL) cells. Overexpression of SLPI in mice attenuated AHR without concomitant use of CS and did so to a greater degree with CS. Thus, our bedside-bench approach establishes that the immune response in SA is distinct from that in MMA with a dysregulated IFN-γ–SLPI immune-epithelial axis.
Results
SA subjects harbor more IFN-γ+CD4+ T cells in their airways compared with MMA subjects.
A total of 66 subjects, 33 classified with MMA and 33 classified with SA, were included in this study; details of patient characteristics are included in Table 1. Of note, biological samples, such as cells in BAL fluid used for differential cell counts and cytokine expression, were analyzed from a subset of these subjects based on availability, as described in each figure legend. Since the recovery of BAL fluid from different individuals is variable, especially from SA subjects, percentage of cell type in BAL fluid is shown rather than absolute numbers. Mean percentages of lymphocytes, neutrophils, and eosinophils were higher in the airways of SA as compared with MMA subjects (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI80911DS1). The mean baseline forced expiratory volume in 1 second (FEV1) % predicted was 55.9 in the SA group and 87.5 in the MMA group (Supplemental Figure 1B and Table 1). The dominant T cell population in cells collected by BAL was CD4+ T cells (Supplemental Figure 1C). BAL cells from 9 MMA and 11 SA subjects were available for cytokine analysis by intracellular cytokine staining (ICS), and representative flow plots of cytokine+CD4+ T cells in the subjects are shown in Figure 1, A and B. The percentage of IFN-γ+CD4+ T cells (Th1 cells) was significantly greater in SA subjects, averaging 20% versus 5% in MMA (Figure 1A). The percentage of Th2 cytokine+ cells detected by ICS was low in each category (not shown). The percentage of IL-17+CD4+ T cells was overall lower in both groups and was marginally higher in SA as compared with MMA, which reached statistical significance (Figure 1B). Total BAL cells from 9 MMA and 14 SA subjects were cultured overnight, and cytokines released were assayed by ELISA. The levels of secreted IFN-γ were also higher in SA compared with those in MMA (Figure 1C). The mean levels of IL-5 and IL-17 secreted by BAL cells were comparable between the 2 groups, although the levels of secreted IL-13 were greater in the SA group (Figure 1C). Secreted levels of IL-4, IL-9, and IL-12p40 were also assayed. Overall, the levels of all of these cytokines were low, possibly due to their rapid utilization by T cells (Figure 1C). There was a trend toward increased IL-12p40 protein from SA BAL cells. IL-12p70 was below the level of detection. Since more samples were available for mRNA analysis, we also examined expression of IFNG and IL12P40 mRNA in the total BAL cells. As shown in Figure 1D, the expression of both was higher in the SA group as compared with that in the MMA group, which reached statistical significance. We further analyzed the data derived from the MMA cohort based on whether or not the subjects were on inhaled CS (ICS). As shown in Supplemental Figure 2A, the production of all of these cytokines was lower when the subjects were on ICS, with the difference reaching statistical significance in the case of IL-9 and IL-13. A comparison of cytokine levels from BAL cells of MMA subjects on ICS versus those from SA subjects revealed higher levels of all in SA, with the difference in IFN-γ and IL-13 levels being statistically significant (Supplemental Figure 2B).
Table 1. Characteristics of MMA and SA subjects evaluated in the study.
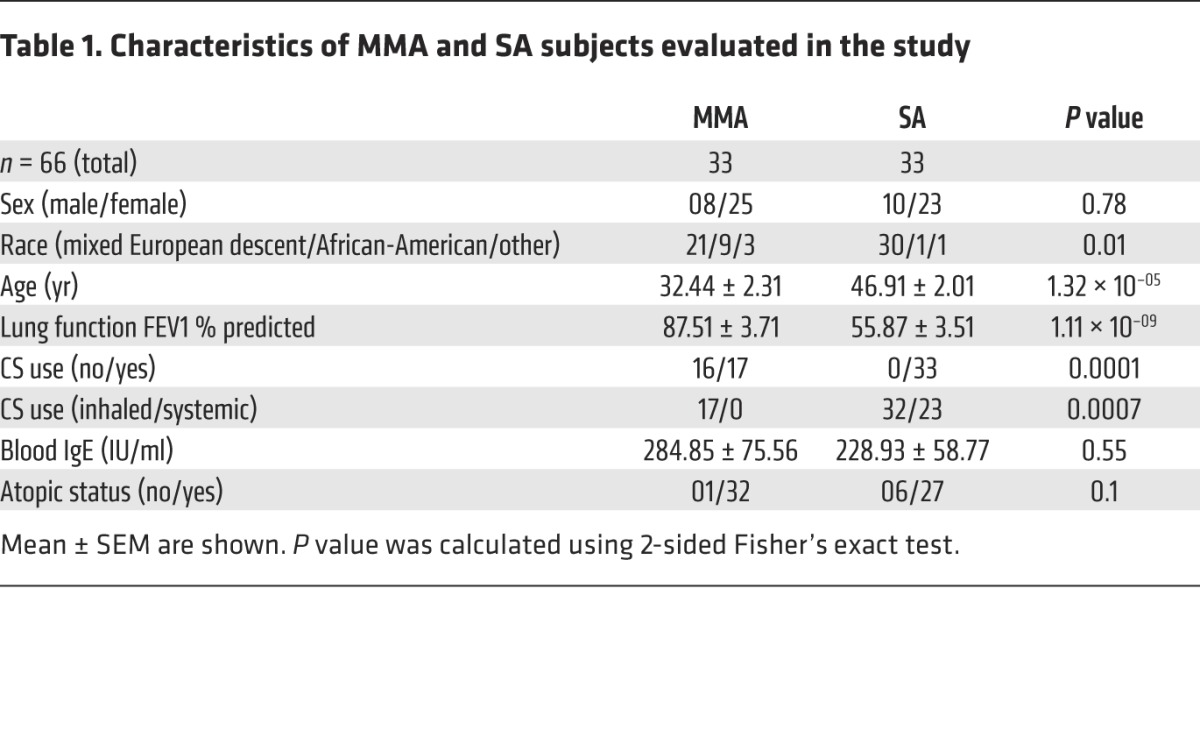
Figure 1. SA subjects harbor more IFN-γ+ and IL-17A+CD4+ T cells in their airways compared with MMA subjects.

Representative flow plots (upper panels) of (A) IFN-γ+ and (B) IL17A+ CD4+ T cells in total BAL cells. Lower panels are graphical representations of percentages of (A) IFN-γ– and (B) IL-17A–expressing CD4+ T cells in total BAL cells obtained from the airways of SA (n = 11) and MMA (n = 9) subjects studied. (C) Concentration of indicated cytokines in culture supernatants of total unfractionated BAL cells from MMA and SA groups briefly cultured ex vivo. For IL-5 and IL-13 estimation, n = 9 and n = 14 for MMA and SA subjects, respectively; for IFN-γ and IL-17A estimation, n = 14 in each category. For IL-12p40, IL-4, and IL-9 estimation, n = 9 and n = 8 for MMA and SA subjects, respectively. (D) IFNG and IL12P40 mRNA expression in total BAL cells from MMA and SA subjects; n = 8 and n = 10 for MMA and SA, respectively. *P ≤ 0.05; ***P ≤ 0.001; NS, nonsignificant, Student’s unpaired t test or Mann-Whitney U test based on normality of data.
Establishment of a mouse model of SA.
Our next goal was to establish a mouse model of SA that would (a) display the complex immune response identified in the airways of human SA, (b) demonstrate mixed granulocytic infiltrates in the airways, and (c) demonstrate resistance to CS therapy, as observed in the SA subjects. To model this complex immune response in rodents, we took into consideration the current concept that pathogen infections play an important role in asthma pathogenesis (9, 10). In this regard, bacterial infections have been associated with asthma exacerbations. Intracellular bacteria such as Chlamydia pneumoniae have been linked to fixed airflow limitation (11–13), and Haemophilus influenzae has been detected in the sputum of SA patients (14). In a more recent study, SA patients were found to harbor neutrophilic airway inflammation and infection with either Moraxella catarrhalis or members of the Haemophilus or Streptococcus genera (15). All of these bacterial species are capable of intracellular growth and hence can produce the second messenger cyclic-di-GMP (c-di-GMP) (16). c-di-GMP has been shown to be a potent mucosal adjuvant that induces a Th1-Th17 response accompanied by a low Th2 response (17). Thus, we hypothesized that this adjuvant may be useful in modeling the complex immune response observed in human SA. We sensitized mice with house dust mite (HDM) allergen and c-di-GMP and subsequently challenged the mice with HDM and a lower dose of c-di-GMP (Figure 2A). A second group of mice was sensitized and challenged with HDM alone following the same regimen. AHR of mice immunized with both HDM and c-di-GMP increased with increasing doses of methacholine (MCh) and was higher than that induced using HDM alone (Figure 2B). Upon treatment with the CS dexamethasone (Dex), which was initiated during challenge and maintained until harvest, AHR in the SA model was reduced only to the response induced by HDM alone (note the almost complete overlap of the black and blue lines in Figure 2B). In mice that were sensitized and challenged with HDM alone, AHR was considerably lower than that induced by HDM plus c-di-GMP (Figure 2B). Also, Dex treatment reduced AHR further, but not down to the level measured in MCh-challenged naive mice (Figure 2B). Differential cell counts in the BAL fluid recovered from mice subjected to the 2 models showed greater neutrophil recruitment in the SA model, but more eosinophil infiltration in the HDM-only model (Figure 2C). Dex treatment substantially reduced airway inflammation in the mice that received HDM alone, but was only partly effective in reducing airway inflammation in the SA mice (Figure 2C and Supplemental Figure 3A). Histological examination of lung tissue sections showed that the mice subjected to the SA model harbored more intense inflammation and mucus staining compared with those sensitized and challenged with HDM alone (Figure 2D). The increase in lung CD4+ T cell numbers was greater in mice subjected to the SA model compared with that in mice that received HDM alone, and the cells were reduced by 50% upon Dex treatment (Supplemental Figure 3B). When the models were assessed for cytokine levels, we observed a significantly higher level of IFN-γ in the lungs of SA mice compared with the mild asthma (MA) mice (Figure 3A). In contrast, the type 2 cytokines IL-4, IL-5, and IL-13 were detected at higher levels in the MA mice compared with the SA mice (Figure 3A). We have hereafter referred to the model established with HDM plus c-di-GMP as SA and that in which HDM alone was instilled following the same regimen as the MA Th1loTh2 model. IL-9, another cytokine associated with a type 2 response, was not detected in either mouse model. IL-17A was detected only in the SA mice (Figure 3A). Dex treatment only partially reduced lung IFN-γ levels in the SA mice, and IFN-γ was also not completely suppressed in the Dex-treated MA mice (Figure 3A). The levels of Th2 cytokines also partially decreased upon Dex treatment in both models, as did the levels of IL-17 in the SA model (Figure 3A). Of note, the IL-17 levels in the presence of Dex were reduced but relatively high in the mouse model compared with what is typically observed in our analysis of human subjects (Figure 1B). The cytokines detected at the whole lung level in SA mice were detected in CD4+ T cells by ICS (Figure 3B). Also, Dex treatment reduced the frequency of IL-13+CD4+ T cells (Figure 3B). However, since CS treatment inhibits the expression of a wide range of molecules, such as cell adhesion molecules (18), Dex treatment also led to lower T cell accumulation in the tissue (Supplemental Figure 3B). Thus, based on the reduced CD4+ T cell numbers in Dex-treated mice (Supplemental Figure 3B), the total number of each of the cytokine-expressing T cells was lower upon Dex treatment at the whole lung level.
Figure 2. Establishment of a mouse model of SA.

(A) Schematics of the models of asthma used in the study. Mice were sensitized and challenged as shown either with or without Dex treatment, and 24 hours after the last allergen challenge, mice were euthanized and analyzed for different end points. (B) Assessment of central airway resistance (Newtonian resistance [Rn]) as a marker of AHR after challenge with different doses of MCh. ****P ≤ 0.0001, versus naive mice; ††††P < 0.0001, SA versus MA Th1loTh2; ###P < 0.001, SA versus SA plus Dex; #P < 0.05, MA Th1loTh2 versus MA Th1loTh2 plus Dex using 1-way ANOVA with Tukey’s post hoc test. (C) Differential cell counts in BAL fluid cytospins showing total cells, eosinophils (Eos), macrophages (Macs), neutrophils (Neu), and lymphocytes (Lymph) of mice sensitized and challenged in SA and MA Th1loTh2 models ± Dex. **P ≤ 0.01; ***P ≤ 0.001, Student’s unpaired t test. (D) PAS staining of lung sections. Scale bar: 200 mm. Data shown are mean ± SEM and representative of 2 to 3 independent experiments. n = 8–16 mice (B); n = 3–4 mice (C and D).
Figure 3. Lung cytokine levels in mice subjected to the asthma models.

(A) Levels of cytokine proteins in whole lung homogenates of mice subjected to the SA or MA Th1loTh2 model. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001, 1-way ANOVA with Tukey’s post-hoc test. (B) Percentages of cytokine+CD4+ T cells in the lungs of mice subjected to the SA model. *P ≤ 0.05, Student’s unpaired t test. Data shown are mean ± SEM and representative of 2 to 3 independent experiments. n = 3–4 mice.
Airway inflammation and AHR in a second MA model, MA Th2.
As described above, Dex treatment of the MA Th1loTh2 mice significantly attenuated airway inflammation, mucus production, and AHR (Figure 2, B–D, Figure 3A, and Supplemental Figure 3, A and C). However, AHR in these mice was still not completely reduced to the level induced in naive MCh-challenged mice (Figure 2B). We next examined another mouse model in which Th2 cytokines were exclusively induced in response to HDM, which we named MA Th2. This was achieved by instilling HDM on a daily basis initially for 10 days and subsequently challenging the mice with HDM after a period of rest (Figure 4A). As shown in Figure 4, B and C, repetitive HDM instillation induced a Th2 response and eosinophilic airway inflammation with barely detectable IFN-γ or IL-17 induction and low neutrophil infiltration in the airways. Levels of Th2/type 2 cytokines produced in the lungs were comparable between the MA Th1loTh2 and MA Th2 models (Figure 3A and Figure 4B). The key difference in cytokine response between the MA Th2 model and the MA Th1loTh2 model shown in Figures 2 and 3 was that IFN-γ was induced only in the latter, albeit at levels below that induced in the SA model. No IL-17 was detected in either model. Dex treatment completely ablated airway inflammation and mucus production in this model (Figure 4D) and completely suppressed AHR induced by MCh challenge (Figure 4E). The level of AHR induced in this Th2-driven model was lower than that observed in the SA model, but was equivalent to that induced in the MA Th1loTh2 model (Figure 2). Dex partially reduced tissue Th2 cytokine levels, as observed in the other models (Figure 4B).
Figure 4. Airway inflammation, cytokine expression, and AHR in MA Th2 mouse model.

(A) Schematic of MA Th2 model. (B–E) Mice were sensitized and challenged according to the scheme shown in A either with or without Dex treatment, and different end points were analyzed 24 hours after the last allergen challenge. (B) Cytokine levels in whole lungs. *P ≤ 0.05, Student’s unpaired t test. (C) Differential cell counts in BAL fluid cytospins showing total cells, eosinophils, macrophages, neutrophils, and lymphocytes. *P ≤ 0.05; **P ≤ 0.01, Student’s unpaired t test. (D) PAS staining of lung sections. Scale bar: 200 mm. (E) AHR (Rn) assessment. ****P ≤ 0.0001, using 1-way ANOVA with Tukey’s post-hoc test. Data shown are mean ± SEM and are representative of 2 to 3 independent experiments. n = 3–4 mice (B–D); n = 8–12 mice (E).
IFN-γ, but not IL-17, controls AHR in the SA mouse model.
The data from the 3 models suggested that the presence of IFN-γ prevents complete attenuation of AHR by Dex. This is because, among the 3 models, in the MA Th2 model only, no IFN-γ was induced and Dex completely suppressed AHR. Our next question, therefore, was the relative contribution of IL-17 versus IFN-γ to increased AHR in the SA mice. Toward this end, we used Il17ra–/– mice, which are nonresponsive to multiple IL-17 family members, including IL-17A and IL-17F, and Ifng–/– mice. We studied the consequence of selective deletion of either the IL-17 response or of IFN-γ on AHR, airway inflammation, and cytokine response in WT or gene-deficient mice subjected to the SA model. Surprisingly, AHR in Il17ra–/– mice subjected to the SA model remained similar to that measured in WT mice (Figure 5A). However, as expected, neutrophil recruitment to the airways and tissue inflammation were lower in the Il17ra–/– mice compared with their WT counterparts (Figure 5, B and C). Addition of Dex did not mitigate the inflammation. These results suggest that neutrophils and IL-17A do not contribute to AHR in this model of allergic airway disease. At the whole lung level, the levels of secreted Th1/Th2/Th17 cytokines were largely comparable between WT and Il17ra–/– mice subjected to the SA model (Figure 5D). Cytokine mRNA expression was similar between WT and Il17ra–/– mice except for a reduction in IL-17A in the Il17ra–/– mice (Supplemental Figure 4A).
Figure 5. IL-17 controls neutrophil infiltration but not AHR in the SA mouse model.

(A–D) WT and Il17ra–/– mice were subjected to the SA model, and different end points were analyzed 24 hours after the final allergen challenge. (A) AHR in WT and Il17ra–/– mice after MCh challenge. 1-way ANOVA with Tukey’s post-hoc test. (B) Differential cell counts in BAL fluid cytospins showing total cells, eosinophils, macrophages, neutrophils, and lymphocytes. ***P ≤ 0.001, using Student’s unpaired t test. (C) PAS staining of lung sections and (D) cytokine levels in total lungs. ND, not detected. ***P ≤ 0.001, Student’s unpaired t test. Scale bar: 200 mm (C). Data shown are mean ± SEM and representative of 2 to 3 independent experiments. n = 8–16 mice (A); n = 3–4 mice (B–D).
We next focused on the contribution of IFN-γ to AHR and airway inflammation in the SA model. While the WT mice showed increased AHR (as shown in Figure 2), the Ifng–/– mice failed to develop AHR (Figure 6A). Again, Dex treatment only partially reduced AHR in the WT mice (Figure 6A). Despite the inability of Ifng–/– mice to mount appreciable AHR upon MCh challenge, airway inflammation was unabated in these mice with or without Dex treatment (Figure 6, B and C). The Ifng–/– mice showed slightly greater neutrophil and eosinophil infiltration in their airways (Figure 6B). These mice also displayed a significantly higher level of IL-17A protein and mRNA in the lungs compared with the WT mice and yet failed to mount AHR (Figure 6D and Supplemental Figure 4B). The increase in IL-17 levels in the absence of IFN-γ was expected because of crossregulation between IFN-γ and IL-17 (19). Taken together, these data show that IFN-γ, and not IL-17, contributes to AHR in this model of asthma. However, IL-17 contributes to neutrophil infiltration in the airways, as previously shown (20–23).
Figure 6. IFN-γ controls AHR in the SA mouse model.

(A–D) WT and Ifng–/– mice were subjected to the SA model, and different end points were analyzed 24 hours after the final allergen challenge. (A) AHR in WT and Ifng–/– mice after MCh challenge. ****P ≤ 0.0001, 1-way ANOVA with Tukey’s post-hoc test. (B) Differential cell counts in BAL fluid cytospins showing total cells, eosinophils, macrophages, neutrophils, and lymphocytes. *P ≤ 0.05; **P ≤ 0.01, Student’s unpaired t test. (C) PAS staining of lung sections and (D) cytokine levels in total lungs. ***P ≤ 0.001, Student’s unpaired t test. Scale bar: 200 mm (C). Data shown are mean ± SEM and representative of 2 to 3 independent experiments. n = 8–16 mice (A); n = 3–4 mice (B–D).
We next asked how IFN-γ might associate with increased AHR. Using the Ingenuity Knowledge Base (IKB), we identified multiple genes that have been associated with AHR in humans or mice (Supplemental Table 1). We then further narrowed down the output by examining molecules modulated by IFN-γ and also expressed in either epithelial cells or smooth muscle cells in the lungs (Supplemental Figure 5). The expression of all of these genes except for CCL3L3 was analyzed by quantitative reverse-transcriptase PCR (qRT-PCR) in the lungs of WT and Ifng–/– mice subjected to the SA model (Figure 7). qRT-PCR analysis of 2 epithelial-restricted genes and 2 nonepithelial genes confirmed the enrichment of airway epithelium using the brush technique (Supplemental Figure 6). As already discussed, levels of Il17a mRNA and protein were increased several-fold in the lungs of Ifng–/– mice compared with WT mice, and yet the mice did not develop AHR, which ruled out a role for IL-17 in promotion of AHR in our model. The expression of 2 other differentially expressed genes, slpi and Ccl22, was of interest based on available information on their role in asthma.
Figure 7. Comparison of AHR-linked IFN-γ–regulated genes in WT and Ifng–/– mice subjected to the SA model.

qRT-PCR analysis of the indicated genes in WT and Ifng–/– mice subjected to the SA model. **P ≤ 0.01, Student’s unpaired t test. Data shown are mean ± SEM and representative of 3 independent experiments.
Inverse correlation between IFN-γ and SLPI expression in human SA.
SLPI is expressed by the airway epithelium and was previously associated with suppression of asthma features in mouse models (24, 25). Indeed, Ifng–/– mice with greater SLPI expression were resistant to development of AHR (Figure 6A). There is no report in the literature on the relative expression of SLPI in SA versus MA, which we then examined. As shown in Figure 8A, SLPI mRNA expression was lower in the bronchial brushings of SA subjects compared with MMA subjects. We next explored the relationship between epithelial SLPI expression and IFNG expression in BAL cells in the same subjects. All MMA subjects and most SA subjects (except for 2) used for this analysis were new subjects (not included in Figure 1, but included in Table 1). MMA subjects displayed high epithelial SLPI mRNA expression, but again, low IFNG mRNA expression in their BAL cells (Figure 8B). In the majority of SA subjects, the opposite was true, and epithelial cell–expressed SLPI showed a significant negative correlation with IFNG expressed by the corresponding BAL cells (Figure 8B). Given that Ccl22 expression was also higher in Ifng–/– mice compared with WT mice, we assayed CCL22 mRNA expression in bronchial brushings of MMA and SA subjects. CCL22 mRNA expression was comparable between the 2 cohorts (Supplemental Figure 7). Importantly, both MA models (MA Th1loTh2 and MA Th2) displayed higher Slpi mRNA levels compared with the SA model (Supplemental Figure 8A). Also, Slpi mRNA expression was similar in the lungs of WT and Il17ra–/– mice, suggesting that IL-17 signaling is not involved in Slpi regulation (Supplemental Figure 8B).
Figure 8. Inverse correlation between IFN-γ and SLPI expression in human SA.

(A) SLPI mRNA expression analyzed by qRT-PCR in epithelial brushings of MMA and SA subjects. n = 17 and n = 20 for MMA and SA, respectively. **P ≤ 0.01, Student’s unpaired t test. (B) Correlation analysis between BAL cell IFNG and airway epithelial cell–expressed SLPI mRNA in MMA (top) and SA (bottom) subjects. Spearman’s rank correlation test was used to calculate the correlation coefficient (rs) using GraphPad Prism software. Regression line for the SA cohorts is shown in the right panel. n = 9 and n = 13 for MMA and SA, respectively. (C) Primary airway (bronchial) epithelial cells from nonasthmatic human subjects were stimulated with rhIFN-γ ± anti–IFN-γ or left untreated for 8 hours. SLPI mRNA expression was analyzed by qRT-PCR, and fold change over untreated was calculated using HPRT as internal reference control. n = 5. ***P ≤ 0.001, 1-way ANOVA with Tukey’s post-hoc test. (D) Detection of hSLPI protein in sera of mice that received hSLPI expression plasmid by tail vein injection. *P ≤ 0.05, 1-way ANOVA with Tukey’s post-hoc test. (E) AHR measurement in mice subjected to the SA model with or without hSLPI introduced in an expression plasmid via hydrodynamic tail vein injection. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001, 1-way ANOVA with Tukey’s post-hoc test. (F) Cytokine concentrations in whole lungs of mice treated as in E. **P ≤ 0.01, 1-way ANOVA with Tukey’s post-hoc test. For D–F, data shown represent mean ± SEM and representative of 2 independent experiments.
We next treated primary human bronchial epithelial cells with recombinant IFN-γ, causing suppression of SLPI mRNA levels that was reversed with neutralizing anti–IFN-γ antibody (Figure 8C). We then asked whether overexpression of SLPI in mice subjected to the SA model would attenuate AHR. An expression plasmid for human SLPI (hSLPI), which was shown to be functional in mice (25), was introduced by hydrodynamic tail vein injection into mice. hSLPI was detectable in the sera of mice 4 days after the last tail vein injection of hSLPI expression plasmid, with or without Dex treatment, but not in control mice, which received PBS only (Figure 8D). As compared with control mice, those that received the hSLPI expression plasmid (but no Dex treatment) mounted lower AHR. Mice that received the hSLPI expression plasmid along with Dex had even lower AHR at the highest dose of MCh than those that received the expression plasmid alone (Figure 8E). IFN-γ concentration in lung homogenates of mice that received the hSLPI expression plasmid was lower than in mice that received PBS, suggesting a feedback regulation between SLPI and IFN-γ. SLPI overexpression did not alter the level of IL-17A (Figure 8F).
Regulation of IFN-γ production by c-di-GMP via IL-12 induction is dependent on STAT1.
STAT1 is a well-studied molecule involved in IFN-γ signaling (26). To study the effect of c-di-GMP signaling on IFN-γ induction, bone marrow–derived macrophages (BMDMs) were used, since macrophages can express both IL-12 (27) and IFN-γ (28) and IFN-γ can upregulate its own expression (29). Also, c-di-GMP, via cytosolic recognition, has been shown to induce inflammatory cytokine production in BMDMs (30). BMDMs generated from WT and Stat1–/– mice were transfected with c-di-GMP. c-di-GMP–induced IL-12p40 production was significantly lower in Stat1–/– BMDMs compared with that in WT BMDMs (Supplemental Figure 9A). c-di-GMP transfection did not induce IFN-γ production in either WT or Stat1–/– BMDMs (Supplemental Figure 9B), suggesting that c-di-GMP promotes IFN-γ production from CD4+ T cells via stimulation of IL-12 production in APCs, which is partly dependent on STAT1. We also observed reduced expression of Il12p35 and Il12p40 mRNA expression in the lung-draining lymph nodes of sensitized Stat1–/– mice as compared with WT mice in the SA model (Supplemental Figure 9C). Ifng mRNA expression in the lungs of Stat1–/– mice subjected to the SA model was significantly lower compared with that in WT mice (Supplemental Figure 9D), which resulted in higher Slpi mRNA expression in the airway epithelium (Supplemental Figure 9E).
Discussion
Our analysis of human disease has revealed a dysregulated Th1/IFN-γ immune response in SA. Use of an animal model that resembles the characteristics of human disease shows an important role for IFN-γ, but not for IL-17, in promoting AHR. The combined human-animal data in the present study also suggest a role for lower SLPI levels in the context of high IFN-γ levels contributing to increased AHR in SA subjects. While a relationship between IL-17 levels and neutrophil recruitment in the airways is evident in both humans and mice (20–23), neutrophilic inflammation per se may not portend poor lung function. That airway inflammation and AHR may not always be linked is being increasingly appreciated (31, 32). This focuses our attention on other effector functions of the T cell cytokines in asthma pathophysiology.
We have recently shown synergistic interactions between IFN-γ and low levels of IL-13 in induction of nitro-oxidative stress in primary airway epithelial cells (33). Despite the use of high doses of oral CS, IL-13 was still detectable at low levels in the BAL cell culture supernatants of SA subjects in our study (Figure 1C). It will be interesting to determine whether, in addition to CD4+ T cells, the recently identified IL-25–induced IL-17RB+ cells in the blood of SA patients that is associated with steroid-resistant asthma (34) is a source of increased IL-13 from BAL cells isolated from SA patients. In the mouse model as well, despite Dex treatment, a low level of IL-13 along with IFN-γ and IL-17 was maintained in the lungs of the mice. Whether the combination of different levels of Th cytokines causes different degrees of disease severity will be important to investigate in future studies with large patient cohorts. However, our analysis has shown that among the 3 Th subsets, Th1 is dominant in approximately 70% of SA subjects.
Although a previous study showed the ability of IL-17 to cause airway smooth muscle (ASM) contraction using tracheal rings from humans and mice (35), a recent study failed to associate an IL-17 sputum signature with worse lung function in SA subjects (36). The animal model in the former study employed i.p. sensitization with OVA and alum followed by i.n. challenge with OVA, and thus, the primary site of T cell priming was not the lung. When mice with CD11c+ cell–specific deletion of αvβ8 were subjected to this model, the partial reduction in AHR was attributed to a deficiency of Th17 cells (35). In all of the animal models used in our study, mice were sensitized and challenged i.n. to induce T cell priming in the lung-draining lymph nodes. In our SA model, in which both IFN-γ and IL-17 were induced, failure to respond to IL-17 in Il17ra–/– mice did not reduce AHR, while lack of IFN-γ in Ifng–/– prevented AHR. It is not known whether any IFN-γ was induced in the OVA model, since Th17 cells can also produce IFN-γ (37). In our analysis of cytokine production in human asthmatics, IFN-γ was the dominant cytokine detectable in appreciable amounts in SA patients despite use of high doses of CS, and the amount of IL-17 secreted by BAL cells from SA or MMA patients was much lower and essentially similar (Figure 1C). Our cytokine analysis of BAL cell culture supernatants revealed a mean concentration of 0.29 ng/ml of IFN-γ in cultures established from MMA subjects. Among SA subjects, BAL cells from 10/14 (70%) produced more than this level while only 3/14 (20%) of MMA subjects were found to produce at a level higher than average. Of the 3 animal models used we used, in 2 models (SA and MA Th1loTh2) in which IFN-γ was induced, AHR was only incompletely suppressed by Dex. In contrast, in the MA Th2 model, which did not produce any detectable IFN-γ protein, AHR was completely suppressed by Dex. Most remarkably, AHR in the Il17ra–/– mice was almost identical to that induced in WT mice, although inflammation was substantially reduced. It is important to note that, among the 4 SA subjects whose BAL cells generated below average IFN-γ, IL-17 concentrations were less than 0.050 ng/ml and IL-13 levels were below the mean level detected in the SA group (~40 pg/ml). Thus, in some subjects, other mechanisms, which may involve tissue-resident cells such as ILC2s (38), may drive the SA phenotype. Collectively, these data show that in the context of a complex immune response, as observed in 70% of human SA patients and also modeled in mice, IFN-γ plays an important role in AHR while IL-17 promotes neutrophilic inflammation in the airways.
An inverse relationship between IFN-γ expression and SLPI, as observed in the SA subjects and in the mouse SA model, has been also observed previously in entirely different contexts. For example, overexpression of IFN-γ in the airways of transgenic mice was shown to selectively inhibit expression of SLPI, but not of other protease inhibitors (39). How might low SLPI contribute to the SA disease state? After its cloning in 1986 and the description of its presence in various body fluids, including nasal and bronchial mucosal secretions, SLPI was described as an epithelial product. Later, it was also shown to be a product of macrophages hyporesponsive to LPS (40). IFN-γ was shown to inhibit SLPI expression, restoring LPS responsiveness to macrophages (40). Most interestingly, relevant to SA, overexpression of SLPI in macrophages inhibited nitric oxide production (40). It is important to note that in humans, airway epithelial cells rather than alveolar macrophages express iNOS and that reduced SLPI expression in these cells in SA patients, as observed in our study, may contribute to increased exhaled nitric oxide (FeNO) production. In our recent cluster analysis, subjects in cluster 6 with mixed granulocytic airway inflammation, who required the highest health care utilization and were on high doses of oral CS, also expressed the highest levels of FeNO (7). It is possible, therefore, that lower SLPI expression in SA due to high IFN-γ expression contributes to increased FeNO levels.
SLPI broadly inhibits multiple leukocyte serine proteases including chymase and tryptase produced by mast cells. Tryptase was shown to promote AHR by activating protease-activated receptor 2 (PAR-2), which rendered PAR-2 responsive to peptide agonists (41). Stimulation of activated PAR-2 caused AHR by release of neurokinins from afferent neurons in the bronchial tissue (41). It is interesting that in another OVA-based model of chronic asthma in which mice were sensitized i.p. with OVA (without alum) and then challenged i.n. with OVA for 9 weeks, a mast cell–dependent role for IFN-γ on airway remodeling, AHR, and airway inflammation was found (42). It is possible that in this chronic model as well, IFN-γ–mediated decrease in SLPI expression in airway epithelial cells with derepression of mast cell protease activity is involved in increased AHR in mice. Although in the chronic OVA-induced model, a role for IFN-γ in airway inflammation was also observed, in our SA model, a role for IFN-γ in airway inflammation was not evident, but instead, IL-17 was involved in promoting airway inflammation. In a study of wound healing in skin, Slpi-deficient mice were found to generate active TGF-β, which played a role in wound healing (43). Since TGF-β is a central mediator of airway remodeling (44), SLPI deficiency may also contribute to airway remodeling in asthma, another hallmark of SA and chronic asthma, which is believed to be responsible for persistent AHR (45). It is interesting to note that multiple allergens have protease activities as do different cell types, such as neutrophils and mast cells. Thus, it is possible that SLPI may play a fundamental role in inhibiting both allergen- and cell-associated proteases such that its downregulation promotes the SA phenotype via effects on AHR, FeNO levels, and airway remodeling. It will be useful to develop an inducible airway–specific SLPI transgenic mouse system to study effects of SLPI expression on allergic airway disease — this approach may inhibit AHR to a greater degree due to local overexpression in airway epithelial cells where SLPI is normally expressed.
In summary, our study was initiated with the analysis of immune response in human subjects who were clinically diagnosed with SA or MMA. This analysis highlighted a complex Th1-dominated immune response in the majority of SA subjects with concurrent low but detectable Th17 and Th2 responses, all of these subjects being maintained on CS therapy, with the majority being on systemic CS. The SA immune response, including its limited response to CS, was modeled in mice to determine the functional significance of the higher Th1 response in SA. We report that IFN-γ, but not IL-17, plays an important role in mediating AHR in the SA model. However, IL-17 influences neutrophil accumulation in the airways of these mice. Both in humans and in mice, expression of SLPI is downregulated in the context of severe disease, which provides an explanation for poor lung function in SA, SLPI being an inhibitor of AHR (25). Increasing SLPI levels in the SA mice reduced AHR, which was more evident in the presence of Dex. Taken together, we show that an augmented Th1 immune response in the airways with reduced SLPI expression in airway epithelial cells differentiates SA from milder asthma in both humans and mice.
Methods
Human subjects.
Male and female nonsmoking (<10 pack year and no smoking in the previous year) subjects meeting the American Thoracic Society (ATS) 2000 definition of SA (46) and MMA subjects on no or lower doses of inhaled CS (FEV1) > 60% predicted with no history of frequent or severe exacerbations of asthma in previous year) of diverse racial/ethnic backgrounds and between the ages of 18 and 60 years old were recruited. Subjects underwent extensive baseline characterization, including physiologic (spirometry, diffusing capacity, and PC20), allergic, and clinical characterization (47). Subjects also underwent bronchoscopy per published protocols and consistent with procedures practiced in the SA research program (48).
Human BAL fluid processing and flow cytometry.
Red blood cells in BAL fluid were lysed using ammonium chloride solution. After washing, total cell numbers were determined. One million cells/ml were cultured ex vivo with antibody-coated (cocktail of anti-CD3/CD28/CD2) beads from a T cell activation/expansion kit (Miltenyi Biotec). After 48 hours, cell culture supernatants were collected, centrifuged, and stored at –80°C until analysis. A separate group of cells was stimulated with phorbol myristate acetate (PMA) (50 ng/ml) plus ionomycin (500 ng/ml) (Sigma-Aldrich) for 6 hours, with GolgiStop (BD) added for the last 3 hours. Cells were washed and processed for staining with fluorochrome-conjugated antibodies. Isotype control antibodies and cytokine-specific antibodies were used to assess cytokine production by flow cytometry. Briefly, after cells were stained for CD4+ and the relevant isotype controls, a lymphocyte gate was first applied based upon forward versus 90 degree light scatter of the whole population upon which a CD4+ gate was subsequently drawn. On the selected CD4+ population, the fluorescence intensity that represents the minimal cut-off/threshold was determined for each fluorescence channel using the relevant isotype control for each cytokine. Finally, this was then applied to test samples stained with antibodies against the cytokines.
Mice.
BALBc/ByJ, C57BL/6J, Ifng–/– mice on a BALB/c background (catalog 002286), BALB/cJ mice (catalog 000651; WT control for Ifng–/– mice), and Stat1–/– mice on a C57BL/6J background (catalog 012606) were purchased from The Jackson Laboratory. Il17ra–/– mice on a BALBc/ByJ background were obtained through Taconic Farms. Stat1–/–, Ifng–/–, and Il17ra –/– mouse strains were bred at the Department of Laboratory Resources (DLAR) at the University of Pittsburgh. Mice were housed under pathogen-free conditions and used at between 8 to 10 weeks of age. Age-matched male mice were used in all experiments.
Asthma models.
For the SA model, mice were sensitized with 25 μg HDM (low-endotoxin, Greer Laboratories) and 5 μg c-di-GMP (Biolog Inc.) i.n. on days 1, 3, and 5. Mice were rested for 5 days and then subjected to 3 challenge sets involving 3 consecutive challenges with HDM and c-di-GMP with a rest of 4 days in between challenge sets. 0.5-μg c-di-GMP was instilled with HDM on the first day of each challenge set, followed by 25 μg HDM only in the next 2 challenges. The MA model followed the same sensitization and challenge scheme except that c-di-GMP was not used in this model. For the MA Th2 model, 25 μg HDM was instilled i.n. on days 1 to 10. Mice were then rested for 5 days and then subjected to 2 HDM challenge sets, each involving 25 μg HDM i.n. instillation on 3 consecutive days. For all 3 models outlined above, Dex (Baxter Inc.) at a concentration of 4 mg/kg was given i.p. starting on the first day of the challenge and then repeated every third day. Mice were sacrificed 24 hours after the last challenge. Schematics of these models are shown in Figure 2A and Figure 4A.
BAL and differential cell counts.
Twenty-four hours after the final allergen challenge, mice were anesthetized and BAL was performed. The left bronchus in each mouse was tied off, and the right lobe was lavaged with 0.7 ml of sterile PBS to obtain BAL fluid. Lavaged lungs were stored in Safefix II (Fisher Scientific) for 48 hours for fixation and then transferred to 70% ethanol until paraffin embedding for periodic acid–Schiff (PAS) staining. Total BAL cell numbers were determined using trypan blue staining and standard light microscopy, and 750,00 BAL fluid cells were cytospun onto clean glass slides for differential cell counts using Giemsa staining as previously described (49).
Brushing method for harvesting murine airway epithelium.
Brushings were collected following a method described previously (50). Brushes were made from 60-grit sandpaper–polished polyethylene PE-10 tubing (BD). The tube was inserted into the right main and left main bronchus with gentle brushing and immediately placed in Buffer RLT (QIAGEN) for RNA preparation.
RNA isolation and cDNA preparation.
Lungs were dissociated in TRIzol (Life Technologies) using a high-speed homogenizer. RNA was isolated using RNeasy Kit (QIAGEN) and treated with RNase-free DNase (QIAGEN). cDNA was synthesized using a High Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions. Integrity of the RNA was analyzed with Agilent Bioanalyzer.
Real-time PCR.
Real-time qRT-PCR was performed using validated TaqMan Gene Expression primer and probe sets (Life Technologies) according to the manufacturer’s instructions: IFNG (Hs00989291_m1), SLPI (Hs00268204_m1), CCL22 (Hs01574247_m1), HPRT1 (Hs02800695_m1), Ifng (Mm01168134_m1), Il5 (Mm00439646_m1), Il13 (Mm00434204_m1), Il17a (Mm00439618_m1), hprt1 (Mm01545399_m1), Slpi (Mm00441530_g1), Il18 (Mm00434225_m1), Il27 (Mm00461162_m1), Tnf (Mm00443258_m1), Il1rn (Mm00446186_m1), Stat3 (Mm01219775_m1), Cxcl12 (Mm00445553_m1), Ccl2 (Mm00441242_m1), Ccl22 (Mm00436439_m1), Il12p35 (Mm00434165_m1), and Il12p40 (Mm00434174_m1). qRT-PCR reactions were carried out using the ABI PRISM 7700 Sequence System (Applied Biosystems) at the Genomics Research Core at the University of Pittsburgh. Results were analyzed using SDS 2.2.2 software. mRNA expression was calculated using the 2–ΔCt method with hprt1 as an internal reference control.
Assay of secreted cytokines.
Lungs were homogenized in a buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.02% Tween 20, and Complete Mini, EDTA-Free Protease Inhibitor (Roche Applied Science), and debris-free supernatant was used for cytokine measurement. Secreted cytokines in human cell culture supernatants and mouse lung extracts were measured by magnetic bead–based assays (Millipore Inc. and Bio-Rad Laboratories) according to the manufacturers’ instructions. Data were acquired and analyzed using the Luminex Automated System (Bio-Rad Laboratories). hSLPI protein was assayed using the Human SLPI Quantikine ELISA Kit (catalog DPI00) from R&D Systems. Levels of IL-12p40 and IFN-γ protein in supernatants from BMDM were measured using DuoSet Elisa Kits (R&D Systems) according to the manufacturer’s protocol.
Assessment of AHR.
Mice were anesthetized with 90 mg/kg pentobarbital sodium delivered i.p. and tracheostomized; airway mechanics was assessed using the forced oscillation technique. Data were fit to the constant-phase model using FlexiVent, a computer-controlled small animal ventilator (SCIREQ). Mice were mechanically ventilated at 200 breaths/min with positive end-expiratory pressure (PEEP) of 3 cm H2O and given approximately 10 minutes to acclimate. AHR was assessed by a MCh challenge test with increasing doses of aerosolized Mch (0, 3.125, 12.5, 25 mg/ml), as previously described (51). Briefly, a standard lung volume history was established by delivering 2 total lung capacity maneuvers followed by 2 measurements of respiratory input impedance (Zrs). The ascending doses of nebulized MCh were delivered for 10 seconds each, followed by 3 minutes of Zrs measurements being collected every 10 seconds, which was repeated for each dose.
Cell isolation and sorting.
Lungs of anesthetized mice were perfused with sterile PBS, removed, and digested as described previously (52). Briefly, lungs were dissociated in a collagenase-DNase suspension on a gentleMACS Dissociator (Miltenyi Biotec) according to the manufacturer’s protocol. Single-cell suspensions were obtained by passing the dissociated tissue through a 70-μm cell strainer (BD Falcon) and washed with PBS containing 2% FBS. Cells were then stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) for 6 hours, with monensin added for the last 4 hours. Cells were processed for staining with fluorochrome-labeled antibodies.
Antibodies and flow cytometry.
For staining of human single-cell suspensions, antibodies to CD4 (FITC; clone RPA-T4; catalog 555346), IL-17A (PerCpCy5.5; clone N49-653; catalog 560799), and IL-5 (APC; clone TRFK5; catalog 554396) were purchased from BD Biosciences. Anti–IFN-γ (PE; clone 4S.B3; catalog 502510) was purchased from BioLegend. For staining of mouse lung single-cell suspensions, antibodies to CD4 (PE-TxRed; clone RM4-5, catalog 562314; 1:500), IFN-γ (PerCpCy5.5; clone XMG1.2; catalog 560660; 1:200), and IL-17A (AF488; clone TC11-18H10; 1:200) were also purchased from BD Biosciences. Anti–IL-13 (PE; clone ebio13A; catalog 12-7133-81; 1:200) was purchased from eBiosciences. Single-cell suspensions stained with combinations of the above-mentioned antibodies were acquired on FACSCalibur or FACSAria flow cytometers (BD Immunocytometry Systems), and the data were analyzed using FlowJo software (Tree Star).
Human bronchial epithelial cell culture.
Primary human bronchial epithelial cells were cultured from explanted donor lungs under approved protocol at the University of Pittsburgh. Cells were enzymatically dissociated, expanded in growth media, and seeded onto Transwell inserts at air-liquid interface. Polarized and differentiated cells were stimulated with recombinant human IFN-γ with or without anti-human IFN-γ for 8 hours. Unstimulated cells were used as controls.
Plasmid isolation and hydrodynamic tail vein injection.
pCDNA3-SLPI was obtained from Addgene and the recombinant plasmid was originally a gift from Ronny Drapkin (Dana-Farber Cancer Institute, Boston, Massachusetts, USA) (Addgene plasmid no. 18103). Endotoxin-free plasmid was injected into mice; 25 μg plasmid DNA in sterile saline was injected via the tail vein in mice, and sterile saline was injected into the control group. Injections were performed 1 day prior to the first challenge in each challenge set.
IKB analysis.
The disease and functions search module of IKB (Ingenuity Systems) was used to identify genes involved in regulating AHR. This gene set was further narrowed down by selecting for genes that are expressed in the lung tissue, epithelial cells, and smooth muscle cells using the build function. From this reduced set, genes that are known to be regulated by IFN-γ in lung tissue and epithelial cells were identified by IKB analysis.
Generation of BMDMs and transfection with c-di-GMP.
Bone marrow cells from femurs and tibias of WT and Stat1–/– mice were cultured for 7 days in the presence of CSF. A media change with fresh addition of CSF was carried out on day 4 of culture. BMDMs were transfected using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. c-di-GMP was mixed with LF2000 at a ratio of 1 μl LF2000/1 μg c-di-GMP, incubated at room temperature for 15 minutes, and added to cells at a final concentration of 5 μg/ml in Opti-MEM Reduced Serum Medium (Life Technologies). Culture supernatants were collected 24 hours later and centrifuged; cell-free supernatants were stored at –80°C until further use.
Statistics.
Results shown are mean values ± SEM. After testing for normal distribution of the populations, 1-way ANOVA with Tukey’s post hoc test was used for multiple pairwise comparisons. Student’s unpaired, 2-tailed t test was used for comparisons involving 2 groups. Differences between groups were considered significant at P ≤ 0.05. All statistical analyses were performed using GraphPad Prism 5 software.
Study approval.
For human asthma samples, written informed consent was received from participants prior to inclusion in the study. Samples were collected with approval from the University of Pittsburgh Institutional Review Board. All animal-related protocols were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee (IACUC).
Supplementary Material
Acknowledgments
This work was supported by NIH grants HL113956 (to A. Ray and P. Ray), AI048927 (to A. Ray), AI100012 (to P. Ray), HL69174 (to S.E. Wenzel), PO1 AI106684 (to A. Ray and S.E. Wenzel), R37 HL079142 (to J. Kolls), and P30 DK072506 and the Cystic Fibrosis Foundation Research Development Program (to J. Pilewski).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(8):3037–3050. doi:10.1172/JCI80911.
References
- 1.Chung KF, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343–373. doi: 10.1183/09031936.00202013. [DOI] [PubMed] [Google Scholar]
- 2.Jarjour NN, et al. Severe asthma: lessons learned from the National Heart, Lung, and Blood Institute Severe Asthma Research Program. Am J Respir Crit Care Med. 2012;185(4):356–362. doi: 10.1164/rccm.201107-1317PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan SD, Rasouliyan L, Russo PA, Kamath T, Chipps BE. Extent, patterns, and burden of uncontrolled disease in severe or difficult-to-treat asthma. Allergy. 2007;62(2):126–133. doi: 10.1111/j.1398-9995.2006.01254.x. [DOI] [PubMed] [Google Scholar]
- 5.Fitzpatrick AM, Baena-Cagnani CE, Bacharier LB. Severe asthma in childhood: recent advances in phenotyping and pathogenesis. Curr Opin Allergy Clin Immunol. 2012;12(2):193–201. doi: 10.1097/ACI.0b013e32835090ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18(5):673–683. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 7.Wu W, et al. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J Allergy Clin Immunol. 2014;133(5):1280–1288. doi: 10.1016/j.jaci.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manni ML, et al. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol. 2014;7(5):1186–1198. doi: 10.1038/mi.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242(1):10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renz H, et al. Gene-environment interaction in chronic disease: a European Science Foundation Forward Look. J Allergy Clin Immunol. 2011;128(6):S27–S49. doi: 10.1016/j.jaci.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 11.Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowalski ML, Cieslak M, Perez-Novo CA, Makowska JS, Bachert C. Clinical and immunological determinants of severe/refractory asthma (SRA): association with Staphylococcal superantigen-specific IgE antibodies. Allergy. 2011;66(1):32–38. doi: 10.1111/j.1398-9995.2010.02379.x. [DOI] [PubMed] [Google Scholar]
- 13.ten Brinke A, Zwinderman AH, Sterk PJ, Rabe KF, Bel EH. Factors associated with persistent airflow limitation in severe asthma. Am J Respir Crit Care Med. 2001;164(5):744–748. doi: 10.1164/ajrccm.164.5.2011026. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, et al. Bacteria in sputum of stable severe asthma and increased airway wall thickness. Respir Res. 2012;13:35. doi: 10.1186/1465-9921-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green BJ, et al. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS One. 2014;9(6):e100645. doi: 10.1371/journal.pone.0100645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol. 2005;187(5):1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebensen T, Schulze K, Riese P, Link C, Morr M, Guzman CA. The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine. 2007;25(8):1464–1469. doi: 10.1016/j.vaccine.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 18.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A. 1992;89(21):9991–9995. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laan M, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162(4):2347–2352. [PubMed] [Google Scholar]
- 21.Bullens DM, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang SC, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179(11):7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 23.McKinley L, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marino R, et al. Secretory leukocyte protease inhibitor plays an important role in the regulation of allergic asthma in mice. J Immunol. 2011;186(7):4433–4442. doi: 10.4049/jimmunol.1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright CD, et al. Secretory leukocyte protease inhibitor prevents allergen-induced pulmonary responses in animal models of asthma. J Pharmacol Exp Ther. 1999;289(2):1007–1014. [PubMed] [Google Scholar]
- 26.McBride KM, Reich NC. The ins and outs of STAT1 nuclear transport. Sci STKE. 2003;2003(195):RE13. doi: 10.1126/stke.2003.195.re13. [DOI] [PubMed] [Google Scholar]
- 27.Trinchieri G. Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood. 1994;84(12):4008–4027. [PubMed] [Google Scholar]
- 28.Robinson BW, McLemore TL, Crystal RG. γ-Interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J Clin Invest. 1985;75(5):1488–1495. doi: 10.1172/JCI111852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2(12):933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 30.McWhirter SM, et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206(9):1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janssen-Heininger YM, Irvin CG, Scheller EV, Brown AL, Kolls JK, Alcorn JF.Airway hyperresponsiveness and inflammation: causation, correlation, or no relation? J Allergy Ther. 20122012suppl 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157(1):4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 33.Voraphani N, et al. An airway epithelial iNOS-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. Mucosal Immunol. 2014;7(5):1175–1185. doi: 10.1038/mi.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen BC, Budelsky AL, Baptist AP, Schaller MA, Lukacs NW. Interleukin-25 induces type 2 cytokine production in a steroid-resistant interleukin-17RB+ myeloid population that exacerbates asthmatic pathology. Nat Med. 2012;18(5):751–758. doi: 10.1038/nm.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudo M, et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012;18(4):547–554. doi: 10.1038/nm.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manni ML, et al. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol. 2014;7(5):1186–1198. doi: 10.1038/mi.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30(1):92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnig C, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5(174):174ra126. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, et al. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med. 2000;192(11):1587–1600. doi: 10.1084/jem.192.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88(3):417–426. doi: 10.1016/S0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- 41.Barrios VE, Jarosinski MA, Wright CD. Proteinase-activated receptor-2 mediates hyperresponsiveness in isolated guinea pig bronchi. Biochem Pharmacol. 2003;66(3):519–525. doi: 10.1016/S0006-2952(03)00292-2. [DOI] [PubMed] [Google Scholar]
- 42.Yu M, et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121(8):3133–3143. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashcroft GS, et al. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat Med. 2000;6(10):1147–1153. doi: 10.1038/80489. [DOI] [PubMed] [Google Scholar]
- 44.Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-β in airway remodeling in asthma. Immunol Cell Biol. 2007;85(5):348–356. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- 45.Cockcroft DW, Davis BE. Airway hyperresponsiveness as a determinant of the early asthmatic response to inhaled allergen. J Asthma. 2006;43(3):175–178. doi: 10.1080/02770900600566348. [DOI] [PubMed] [Google Scholar]
- 46.[No authors listed]. Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, unanswered questions. American Thoracic Society. Am J Respir Crit Care Med. 200016262341–2351. 10.1164/ajrccm.162.6.ats9-00 [DOI] [PubMed] [Google Scholar]
- 47.Moore WC, et al. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol. 2007;119(2):405–413. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balzar S, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183(3):299–309. doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang DH, et al. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 1999;11(4):473–482. doi: 10.1016/S1074-7613(00)80122-3. [DOI] [PubMed] [Google Scholar]
- 50.Sugimoto K, et al. The αvβ6 integrin modulates airway hyperresponsiveness in mice by regulating intraepithelial mast cells. J Clin Invest. 2012;122(2):748–758. doi: 10.1172/JCI58815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H, et al. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses allergic inflammation. Proc Natl Acad Sci U S A. 2008;105(18):6690–6695. doi: 10.1073/pnas.0708809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnamoorthy N, et al. Activation of c-Kit in dendritic cells regulates T helper cell differentiation and allergic asthma. Nat Med. 2008;14(5):565–573. doi: 10.1038/nm1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.