

Abstract
Background
Bread wheat is not only an important crop, but its large (17 Gb), highly repetitive, and hexaploid genome makes it a good model to study the organization and evolution of complex genomes. Recently, we produced a high quality reference sequence of wheat chromosome 3B (774 Mb), which provides an excellent opportunity to study the evolutionary dynamics of a large and polyploid genome, specifically the impact of single gene duplications.
Results
We find that 27 % of the 3B predicted genes are non-syntenic with the orthologous chromosomes of Brachypodium distachyon, Oryza sativa, and Sorghum bicolor, whereas, by applying the same criteria, non-syntenic genes represent on average only 10 % of the predicted genes in these three model grasses. These non-syntenic genes on 3B have high sequence similarity to at least one other gene in the wheat genome, indicating that hexaploid wheat has undergone massive small-scale interchromosomal gene duplications compared to other grasses. Insertions of non-syntenic genes occurred at a similar rate along the chromosome, but these genes tend to be retained at a higher frequency in the distal, recombinogenic regions. The ratio of non-synonymous to synonymous substitution rates showed a more relaxed selection pressure for non-syntenic genes compared to syntenic genes, and gene ontology analysis indicated that non-syntenic genes may be enriched in functions involved in disease resistance.
Conclusion
Our results highlight the major impact of single gene duplications on the wheat gene complement and confirm the accelerated evolution of the Triticeae lineage among grasses.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0754-6) contains supplementary material, which is available to authorized users.
Background
Gene duplication is a major source of species adaptation, providing raw genetic material for functional diversification. The duplication of genes, along with alternative splicing, exon shuffling, and epigenetic regulation, has been shown to contribute to the vast complexity observed among eukaryotic genome architectures [1–4]. There are several types of gene duplication: large-scale, such as whole-genome duplication, and small-scale, where only one or a few genes are duplicated. Numerous marker-based comparative studies have demonstrated that grass genomes have a high degree of conserved synteny (homologous genes located on syntenic blocks between species) and collinearity (conserved gene order within syntenic blocks) [5–8]. Furthermore, access to the sequences of the rice, sorghum, maize, and Brachypodium genomes has enabled comparative analyses at a higher resolution [9–12], revealing that although synteny is well-conserved between orthologous grass chromosomes, many micro-rearrangements (including single gene duplications, insertions, and deletions) have disrupted the collinearity.
Hexaploid bread wheat (Triticum aestivum L.; 2n = 6x = 42; AABBDD) originated from two recent hybridizations between three diploid progenitors, donors of the A, B, and D subgenomes, which diverged an estimated 6.5 MYA [13]. The first hybridization occurred <0.8 MYA between the diploid donors of the A and B genomes, whose closest extant representatives are Triticum urartu (A genome) and Aegilops speltoides (S genome related to the B genome). It formed the allotetraploid Triticum turgidum that hybridized <0.4 MYA with the ancestor of Aegilops tauschii (D genome). Given its hexaploid composition, size of 17 Gb, and a percentage of transposable elements close to 90 % [14], the bread wheat genome is an interesting model to study the evolution of complex genomes and the impact of allopolyploidy on genome structure evolution and the fate of duplicated genes.
Several previous studies have estimated the proportion of non-syntenic genes in the wheat genome with model grass species to range from one-third to two-thirds of the genes. However, without access to a complete genome sequence, these analyses were based on ESTs mapped to genetic bins [15–17] or a subset of genomic sequence data from the wheat physical map [18]. In the closely related barley (Hordeum vulgare; wheat/barley divergence estimated at approximately 11.6 MYA [19]), a higher degree of synteny with wheat has been described, with less than one-third of the genes estimated as non-syntenic [18, 20, 21]. Additionally, comparative analyses using the physical map of the D genome progenitor, Ae. tauschii, led to the estimation that 26 % of the genes are non-collinear with Brachypodium [22]. Finally, in a study based on sequencing 2 % of the wheat 3B chromosome, Choulet et al. showed that 48 % of the genes are non-collinear with rice, Brachypodium, and sorghum [23]. Thus, there is convergent evidence to suggest that the Triticeae lineage, and wheat in particular, underwent accelerated evolution via gene duplication and movement. This is further evidenced by the higher number of inversions and translocations observed in Ae. tauschii compared to Brachypodium, sorghum and rice [22, 24], and by the elevated rate of alternative splicing and codon substitution observed in wheat [25].
Recently, we produced a high quality reference sequence of the wheat 3B chromosome [14]. This reference sequence was assembled into a pseudomolecule of 774 Mb, with 7,264 predicted protein-coding genes. In this study, we used the chromosome 3B reference sequence and the chromosome survey sequence assemblies of the bread wheat genome [26] to conduct a deeper analysis on the origin and fate of non-syntenic genes at the whole genome level as well as to study their distribution along a chromosome. Our results indicate that wheat has a higher duplication/fixation rate of non-syntenic genes compared to other grass species, and a selection for non-syntenic genes insertions in distal regions of the chromosomes. Compared to syntenic genes, non-syntenic genes have a more specific expression pattern, more relaxed selection pressure, and are enriched in functions that may provide adaptive advantages.
Results
Conserved genes in wheat and related model grasses
In this study, we compared the wheat 3B chromosome reference sequence with the genomes of three related species: Brachypodium distachyon, Oryza sativa (rice), and Sorghum bicolor (sorghum), representing the Pooideae, Ehrhartoideae, and Panicoideae clades, respectively (Fig. 1). These species were chosen to explore the evolutionary dynamics of the highly complex and polyploid wheat genome compared to smaller, more compact model grass genomes. We verified the syntenic relationships between wheat chromosome 3 (Ta3B), rice chromosome 1 (Os1), sorghum chromosome 3 (Sb3), and the distal regions of Brachypodium chromosome 2 (Bd2) [12, 15, 27–29] and delineated their exact borders using EnsemblPlants Synteny viewer [30] (Additional file 1: Figures S1-S4).
Fig. 1.
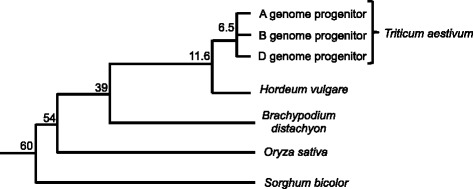
Phylogeny of the model grass species used in this study. Dating information (in MYA) was taken from [12, 13, 19]
Since different methods of genome annotation can result in spurious gene predictions [31], we applied a filtration process to define a gene set that could be compared between species (for a flow chart of the methodology, see Fig. 2). We first discarded alternative splice variants in each genome, taking the longest as the representative. Second, we removed transposable element (TE)-related genes from our dataset. For rice, Brachypodium, and sorghum, this was done based on the annotation summary files downloaded for each genome (genes either already classified as TEs, or having ‘transposon’ in their description). For wheat 3B, we used the TE annotation performed by Daron et al. [32]. Third, we removed potentially mispredicted genes by only including genes for which we could find homology in at least one of the other species used in the study. Finally, in order to focus on functional genes, we removed predictions annotated as pseudogenes.
Fig. 2.
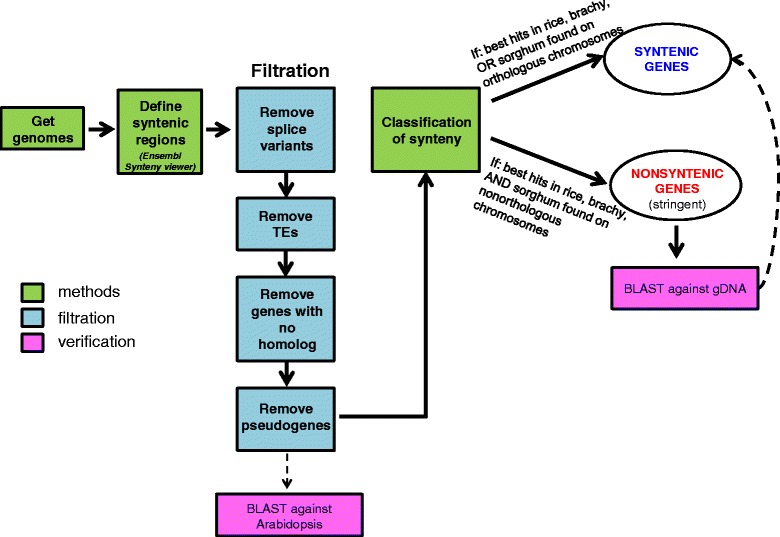
Methodology applied for classifying syntenic and non-syntenic genes
This filtration process allowed us to work on a ‘core’ gene set while removing mispredictions and/or potential lineage-specific genes. The core gene set consisted of 5,125, 3,804, 3,582, and 4,023 genes on the orthologous chromosomes of wheat 3B, Brachypodium 2, rice 1, and sorghum 3, respectively (Table 1). These results indicate that Brachypodium, rice, and sorghum have a similar number of genes in their core gene set (mean 3,803; Table 1) whereas wheat chromosome 3B carries 35 % more genes (5,125) than would be expected by comparison with the other grasses used in this study. We are confident that these additional approximately 1,000 genes in wheat are likely functional protein-coding genes because of the rigorous annotation process that was used for chromosome 3B based on [14, 33]. This includes: (1) training ab initio predictors based on thousands of wheat genes in order to improve the accuracy; (2) combining evidence from different methods of prediction and selecting the best gene model at a given locus based on a scoring system; (3) validation of 59 % of gene predicted splice sites based on transcript evidence (RNAseq, ESTs, mRNA); and (4) manual curation of 48 % of the 3B gene predictions. Moreover, 95 % of the 5,125 wheat core genes have significant sequence similarity to genes in the well-curated Arabidopsis thaliana genome, indicating these are likely to be real genes rather than mispredictions (Additional file 1: Table S1).
Table 1.
Filtration results for the four species compared in this study
| Species | Chr. | Number of genes (no ASVs or TEs) | Number of genes with at least 1 homolog | Number of genes after removing pseudogenes |
|---|---|---|---|---|
| Triticum aestivum | 3B | 7,703 | 6,254 | 5,125 |
| Brachypodium distachyon | 2 | 4,293 | 3,804 | 3,804 |
| Oryza sativa | 1 | 5,070 | 3,882 | 3,582 |
| Sorghum bicolor | 3 | 4,555 | 4,023 | 4,023 |
Sequential process of filtration, starting with the total numbers of genes in the syntenic regions of each chromosome after removing transposon related genes and alternative splice variants (ASVs). The numbers of genes with at least 1 homolog are those with a significant BLAST hit (>35 % amino acid identity and >35 % gene overlap) in at least one of the other species
These results demonstrate that wheat chromosome 3B has an increased number of genes in the core gene set compared to other species, and thus provide a first indication for an accelerated evolution via gene duplication of the gene repertoire in the wheat lineage.
High rate of inter-chromosomally duplicated (non-syntenic) genes in wheat
We examined synteny along chromosome 3B and the orthologous regions in the other grasses. Orthologous gene pairs found in syntenic counterparts between at least two species were classified as syntenic genes (Fig. 2). For example, for each of the filtered genes of wheat, if the best BLAST hits in rice, Brachypodium, or sorghum were found on orthologous chromosomes, the wheat gene was considered syntenic. In contrast, non-syntenic genes were defined as genes having their best BLAST hits located on non-syntenic chromosomes in all the species compared (that is, if the best blast hits in rice, Brachypodium, and sorghum were found on non-orthologous chromosomes, the wheat gene was considered non-syntenic) [14].
Thus, non-syntenic genes originate from duplication and/or translocation having occurred in a lineage-specific manner. Finally, in order to be certain that we were not overestimating the number of non-syntenic genes simply due to a lack of a predicted ortholog in the gene annotation, we searched for sequence similarity of the non-syntenic genes of each species on the genomic sequence (pseudomolecules) of orthologous chromosomes in the other species. This verification step allowed us to reclassify on average 2.25 % of the core genes of each species from non-syntenic to syntenic, and to confirm that the remaining non-syntenic genes are indeed not present in orthologous locations rather than simply not predicted (Additional file 1: Table S2).
While approximately 10 % of the conserved genes in Bd2, Os1, and Sb3 were found to be non-syntenic, 27 % (1,397 genes) of the filtered Ta3B genes were classified as non-syntenic (Table 2). Based on recent divergence time estimates between the three last common ancestors of the species studied here, non-syntenic genes have been inserted in the past 39, 54, and 60 MY for wheat/Brachypodium, rice, and sorghum, respectively (Fig. 1) [12]. Taking these divergence times into consideration, we calculated the rates of non-syntenic genes insertion/fixation and found that for Os1, Bd2, and Sb3, they were highly similar, ranging from 1.7 × 10−3 to 2.3 × 10−3 locus−1 MY−1. In contrast, non-syntenic gene fixation rate was approximately 3.5 times higher for wheat 3B with a rate of 7.0 × 10−3 locus−1 MY−1 (Table 2). Although only partially sequenced, we found barley chromosome 3H to have a similar percentage of non-syntenic genes (31 %) and fixation rate (8.1 × 10−3 locus−1 MY−1) (Additional file 1: Table S3) as wheat 3B, suggesting that the high rate of interchromosomal duplication is a feature of the Triticeae lineage.
Table 2.
Proportion of non-syntenic genes and their fixation rate for each species
| Species | No. syntenic genes (%) | No. non-syntenic genes (%) | Divergence time (MYA) | non-syntenic gene fixation rate (locus−1 MY−1) |
|---|---|---|---|---|
| Ta3B | 3,728 (72.7 %) | 1,397(27.3 %) | 39 | 7.0 × 10−3 |
| Bd2 | 3,509 (92.2 %) | 295 (7.8 %) | 39 | 2.0 × 10−3 |
| Os1 | 3,257 (90.9 %) | 325 (9.1 %) | 54 | 1.7 × 10−3 |
| Sb3 | 3,472 (86.3 %) | 551 (13.7 %) | 60 | 2.3 × 10−3 |
Percentages are out of the total number of genes. Divergence times were taken from [12]
The contrasting levels of synteny between wheat and the other species studied here may reflect a major difference in evolutionary dynamics between small and large genomes. We performed the same synteny analysis with the regions of maize chromosomes 3 and 8 that are syntenic to wheat 3B. We found 8 % of the maize filtered genes to be non-syntenic (Additional file 1: Table S3), far from the 27 % in wheat. This gives further evidence that the high percentage of interchromosomal duplications is specific to the Triticeae lineage and not just a feature of large genomes.
We then compared the sequence of wheat chromosome 3B with the chromosome survey sequence assemblies of all chromosomes produced by the International Wheat Genome Sequencing Consortium [26], excluding 3A, 3B, and 3D. We found that 52 % of the Ta3B non-syntenic genes have at least one copy on a non-homeologous chromosome elsewhere in the wheat genome (BLASTN hit at ≥80 % identity and ≥50 % overlap). This is likely an underestimate due to the fact that the survey sequence dataset is not an exhaustive representation of the wheat gene set, and genes might be partially assembled or unannotated. Therefore, there is evidence that more than half of the non-syntenic genes originated from lineage-specific interchromosomal duplications. The remaining non-syntenic genes may be the result of translocations, missing sequence data, or duplications followed by loss of the ancestral locus. Thus, we conclude that interchromosomal duplications which occurred in the past 39 MY have contributed to the increased number of genes on wheat chromosome 3B compared to other grass species and that this pattern is likely a feature of the Triticeae lineage.
Contrasted distribution of non-syntenic genes along the chromosome
The proportion of non-syntenic genes is positively correlated with the distance to the centromere (r = 0.6807) on wheat chromosome 3B. Interestingly, this pattern is opposite in the genomes of rice and sorghum in which a negative correlation is observed (r = −0.7279, r = −0.7797, respectively; Fig. 3). (Brachypodium was not used because the region syntenic to Ta3B only covers about half of the chromosome.) For barley 3H, we observed a similar pattern of non-syntenic gene distribution to that of wheat 3B (Additional file 1: Figure S5), suggesting that evolutionary forces governing such genome plasticity predate the divergence of the Triticeae lineage.
Fig. 3.
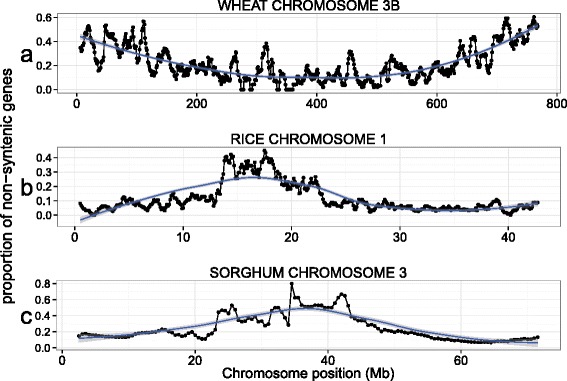
Chromosomal distributions of non-syntenic genes. Proportion of non-syntenic genes per total number of genes for (a) wheat chromosome 3B, (b) rice chromosome 1, and (c) sorghum chromosome 3. For wheat, the proportion of non-syntenic genes per total gene count was plotted in a sliding window of 10 Mb with a step of 1 Mb; in rice, 1 Mb sliding window with a step of 0.1 Mb, and for sorghum a 5 Mb sliding window with a step of 0.5 Mb. Pseudogenes were removed
In this study, we define 3B non-syntenic genes as those genes originating from duplication or translocation events that occurred specifically in the wheat lineage after the divergence with Brachypodium approximately 39 MYA. In order to gain deeper insights into the more recent evolution history, we used the partially sequenced genomes of barley (common ancestor: approximately 11.6 MYA) and wheat homeologous chromosomes 3A and 3D from the chromosome survey sequence (common ancestor: approximately 6.5 MYA; Fig. 1). We distinguished a set of 3B-specific genes, representing 3B non-syntenic genes for which no ortholog was found on barley 3H and no homeolog on wheat chromosomes 3A or 3D. We compared these 3B-specific genes to the non-syntenic genes that were conserved with either 3A, 3D, or 3H. These two subclasses allowed us to distinguish ‘old’ and ‘recent’ non-syntenic genes. Old non-syntenic genes relocated before the divergence of the A/B/D progenitor species more than 6.5 MYA. We know from previous studies that intrachromosomal duplicates have a higher proportion at the distal regions of chromosome 3B [14], so we chose one random representative per duplicate family to avoid biasing the chromosomal distribution plot. In total, we found 161 recent (duplicated <6.5 MYA) and 738 old (duplicated between 6.5 and 39 MYA) non-syntenic gene representatives on the 3B pseudomolecule. Their distribution pattern revealed that the recent interchromosomal gene duplication and/or translocation events occurred on average at a similar rate along the chromosome (Fig. 4a). Interestingly, there is a spike in the centromeric region, suggesting that there may be a slight preferential insertion of non-syntenic genes into this region, as in rice and sorghum (Fig. 3). For older non-syntenic genes, the proportion increases towards the distal ends of the chromosome (Fig. 4b), corresponding to the regions where meiotic recombination mainly occurs [14].
Fig. 4.
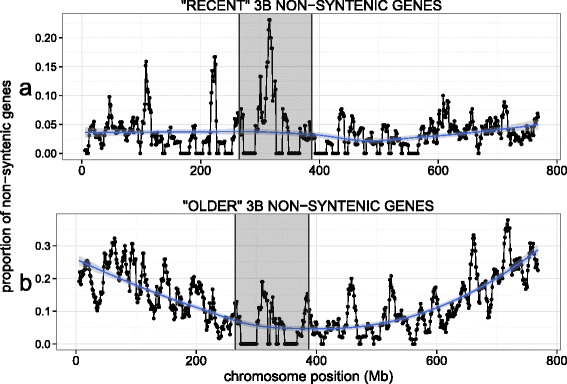
Comparison of the distributions of older versus more recent wheat non-syntenic genes along chromosome 3B. a “Recent” non-syntenic genes that moved after the divergence of the wheat progenitors (carried by 3B but without homolog on 3A, 3D, or 3H). b “Older” non-syntenic genes that have moved in the Triticeae lineage after divergence with Brachypodium and before the divergence of the wheat progenitors. These are 3B non-syntenic genes with a homolog on 3A, 3D, or 3H. The proportion of older and more recent non-syntenic genes out of the total number of genes was calculated within a 10 Mb sliding window with a step of 1 Mb. The centromeric/pericentromeric region is highlighted in the gray box
Non-syntenic genes are functional and may provide adaptive advantages
Non-syntenic genes appear to be significantly shorter in size than syntenic genes when considering gene length (introns + exons), coding sequence (CDS) length, and number of exons (Table 3). However, although the distribution of the syntenic gene CDS size has a higher upper tail (Additional file 1: Figure S6), the median CDS for syntenic and non-syntenic genes is nearly the same. Using deep transcriptome sequencing data from 15 different conditions (see Materials and Methods, [34]), we observed that 74 % of the non-syntenic genes are expressed in at least one condition (Table 3). Although this proportion is significantly lower than the 83 % of expressed syntenic genes, this indicates that the majority of non-syntenic genes are still functional and not only remnants of intense duplication activity. Interestingly, expressed non-syntenic genes are transcribed in fewer conditions on average (9 vs. 12 conditions) and at a lower intensity than syntenic genes (142 vs. 261 mean FPKM; Table 3 and Additional file 1: Figure S6). Forty-two percent of the genes expressed specifically in one condition are non-syntenic, whereas 84 % of those expressed constitutively are syntenic. Furthermore, non-syntenic genes have fewer numbers of alternative splice variants compared to syntenic genes (3.6 vs. 5.3) (Table 3). These results suggest that either non-syntenic genes have acquired a tissue-specific expression pattern through processes like subfunctionalization, or that genes expressed in a tissue-specific manner are more likely to be duplicated.
Table 3.
Structural and functional features of the 3B syntenic and non-syntenic genes
| Syntenic | Non-syntenic | Significance | |
|---|---|---|---|
| Mean genomic size | 3,299 bp | 3,008 bp | P valuea = 1.1e-4 |
| Median genomic size | 2,259 bp | 1,907 bp | |
| Mean CDS size | 1,252 bp | 1,150 bp | P valuea = 8.3e-3 |
| Median CDS size | 1,070 bp | 1,044 bp | |
| Mean number exons | 4.8 | 3.6 | P valuea = 3.2e-16 |
| Median number exons | 3 | 2 | |
| Expressed | 83 % | 74 % | P valueb = 1.8e-13 |
| Mean number conditions expressedc | 12.0 | 9.2 | P valuea <2.2e-16 |
| Median number conditions expressedc | 15 | 10 | |
| Mean fpkmc | 260.8 | 142.0 | P valuea <2.2e-16 |
| Median fpkmc | 130.8 | 46.3 | |
| Mean number alternative splice variants | 5.3 | 3.6 | P valuec <2.2e-16 |
| Median number alternative splice variants | 2 | 1 |
Percentages are out of the total filtered gene count
a Mann–Whitney-Wilcoxon test
b Chi-squared test
c Of those genes expressed
We then investigated the selection pressure on non-syntenic versus syntenic genes by aligning their sequence with their closest homolog in Brachypodium to determine their synonymous and non-synonymous substitution rates since these species have diverged. We observed that non-syntenic genes have a significantly higher Ka/Ks compared to syntenic genes, indicating that non-syntenic genes are under a more relaxed selection pressure (Fig. 5).
Fig. 5.
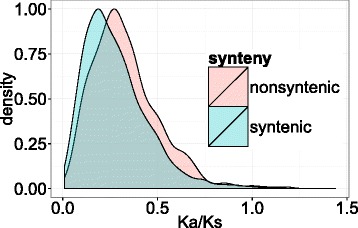
Frequency distributions of Ka/Ks rates of syntenic (blue) and non-syntenic genes (red). Coding sequences of 3,877 3B genes were aligned with their closest homolog in Brachypodium
Finally, we investigated the functions of non-syntenic genes in order to analyze the potential overrepresentation of specific categories. Gene ontology (GO) enrichment was analyzed for both syntenic genes and non-syntenic genes, and several GO terms were found to be significantly overrepresented in both types of genes. Syntenic genes were enriched in biological processes that are essential, metabolic functions, particularly: regulation of primary metabolic process, regulation of gene expression, regulation of cellular biosynthetic process, nucleobase-containing compound metabolic process, gene expression, among others (Table 4). In contrast, non-syntenic genes were enriched in far fewer categories: programmed cell death and macromolecule modification (Table 4) thereby suggesting that non-syntenic genes may provide some adaptive advantages to biotic or abiotic factors.
Table 4.
GO enrichment analysis of Biological Process terms for syntenic genes and non-syntenic genes
| Syntenic genes | ||||||
| GO ID | Term | Annotated | Significant | Expected | P value | Adjusted P value |
| GO:0019219 | Regulation of nucleobase-containing compound metabolic process | 372 | 229 | 176.14 | 4.3e-09 | 8.2e-07 |
| GO:0019222 | Regulation of metabolic process | 437 | 263 | 206.92 | 6.2e-09 | 9.9e-07 |
| GO:0031323 | Regulation of cellular metabolic process | 382 | 232 | 180.88 | 1.9e-08 | 2.7e-06 |
| GO:0060255 | Regulation of macromolecule metabolic process | 404 | 243 | 191.3 | 2.9e-08 | 3.67e-06 |
| GO:0010468 | Regulation of gene expression | 378 | 228 | 178.98 | 6.0e-08 | 5.97e-06 |
| GO:0009889 | Regulation of biosynthetic process | 373 | 225 | 176.62 | 7.4e-08 | 5.97e-06 |
| GO:0010556 | Regulation of macromolecule biosynthetic process | 373 | 225 | 176.62 | 7.4e-08 | 5.97e-06 |
| GO:0031326 | Regulation of cellular biosynthetic process | 373 | 225 | 176.62 | 7.4e-08 | 5.97e-06 |
| GO:0006139 | Nucleobase-containing compound metabolic process | 695 | 391 | 329.09 | 9.7e-08 | 6.8e-06 |
| GO:0010467 | Gene expression | 679 | 378 | 321.51 | 8.5e-07 | 5.0e-05 |
| GO:0044248 | Cellular catabolic process | 163 | 100 | 77.18 | 0.00017 | 0.0090 |
| GO:1901576 | Organic substance catabolic process | 178 | 106 | 84.28 | 0.00054 | 2.1639e-02 |
| GO:0044249 | Cellular biosynthetic process | 742 | 393 | 351.34 | 0.00033 | 1.5428e-02 |
| GO:0016485 | Protein processing | 100 | 64 | 47.35 | 0.00050 | 2.0778e-02 |
| GO:0071704 | Organic substance metabolic process | 2000 | 998 | 947.01 | 0.00029 | 1.4790e-02 |
| GO:0009058 | Biosynthetic process | 750 | 396 | 355.13 | 0.00044 | 1.9747e-02 |
| GO:0015672 | Monovalent inorganic cation transport | 41 | 30 | 19.41 | 0.00067 | 2.3120e-02 |
| GO:0009057 | Macromolecule catabolic process | 146 | 87 | 69.13 | 0.00163 | 4.6894e-02 |
| GO:0006812 | Cation transport | 67 | 44 | 31.72 | 0.00175 | 4.9087e-02 |
| GO:0019941 | Modification-dependent protein catabolic process | 97 | 62 | 45.93 | 0.00064 | 2.3120e-02 |
| GO:0070647 | Protein modification by small protein conjugation or removal | 99 | 62 | 46.88 | 0.00139 | 4.1042e-02 |
| Non-syntenic genes | ||||||
| GO ID | Term | Annotated | Significant | Expected | P value | Adjusted P value |
| GO:0012501 | Programmed cell death | 172 | 53 | 30.12 | 8.5e-06 | 0.00374 |
| GO:0043412 | Macromolecule modification | 663 | 150 | 116.1 | 0.00011 | 0.030855 |
Discussion
Our comparative analyses of a stringently filtered core set of genes reveal that wheat chromosome 3B carries a significantly higher number of genes than was previously expected based on the number of genes in the model grass species rice, Brachypodium, and sorghum. We are confident that the 5,125 genes filtered from the wheat chromosome 3B sequence correspond to real genes rather than transposable elements, mispredicted genes, or pseudogenes and therefore, that the observation is robust. This is because of four reasons: (1) TEs were previously annotated on 3B and removed from the wheat gene set [14]; (2) all of these genes show similarity to at least one other gene in rice, Brachypodium, or sorghum; (3) 95 % of the genes show similarity to A. thaliana (Additional file 1: Table S1), in accordance with the expected 11 % of genes to be monocot-specific [35]; and (4) pseudogenes (genes with nonsense mutations or large deletions) were removed from the dataset. This unusually high amount of genes indicates major single gene duplication activity during the evolution of the wheat genome over the past 39 MY.
The higher number of genes in the wheat 3B core gene set compared to other grass species most likely holds true at the whole genome level, as confirmed by the prediction of 44,523 high confidence genes in the B subgenome chromosome survey sequence assemblies [26]. This is the highest number of protein-coding genes observed for a diploid grass genome (Brachypodium: 26,552 genes; sorghum: 34,496; rice: 39,045; maize: 39,389). These small scale interchromosomal duplications, together with an increased proportion of intrachromosomal duplications [14] and two rounds of polyploidization have led to a highly redundant genome, providing a rich arsenal of raw genetic material and potential means of adaptation.
The wheat genome has undergone more interchromosomal duplications than related grasses
The definitions of synteny and collinearity have become blurred in recent years. In this paper, we make the distinction as follows: two genes in different species are syntenic if they are conserved on their corresponding orthologous chromosome, and collinear if they are on their corresponding chromosome and with a preserved gene order. Thus, collinearity is a more stringent form of synteny, and detects gene duplication and movement on the same chromosome. This is consistent with the original definition of synteny [36, 37]. Because we were only concerned with studying the extent of interchromosomal duplication (genes which have moved to a different chromosome in their specific lineage), we only considered synteny and conservation of overall orthologous chromosomal location.
We quantified the proportion of non-syntenic genes (27 %), and determined that most of these originated from interchromosomal duplications. Previous estimates using partial sequence information were in the range of 41–48 % for wheat chromosome 3B [18, 23]. Our estimation of 27 % is likely to be an underestimate due to our stringent filtration process in order to focus only on a high confidence core gene set. Even when considering only non-syntenic genes defined by our stringent criteria, wheat has more than double the proportion of non-syntenic genes (about 10 % vs. 27 %), and a three-fold duplication/retention rate (2 × 10−3 vs. 7.0 × 10−3) than the three other grasses analyzed here. In addition, the rate in sorghum is potentially overestimated since it is the outgroup of the four species used in this study. Thus we cannot distinguish between genes that relocated in the sorghum lineage and genes which were lost in the common ancestor of rice/Brachypodium/wheat.
This ‘accelerated evolution’ in the wheat lineage was previously observed in the D genome progenitor, Ae. tauschii, through the analysis of large scale genomic rearrangements [24]. As similar results were found for barley chromosome 3H, there is evidence that the increased rate of gene duplication is a feature of the Triticeae lineage. It will be interesting to perform similar analyses in rye to confirm when single gene duplications have started to increase the gene number in the Triticeae lineage. This increased rate of duplication supports the previously described notion of accelerated evolution in the wheat genome based on the increased number of alternative splicing events, non-synonymous substitution rates, and gene exon rearrangements compared to other grass lineages [25].
Potential mechanisms of interchromosomal gene movement
Single gene duplication can originate from different mechanisms. Retroposition, where genes are reverse-transcribed and reinserted back into the genome, has been shown to play a role in gene creation in plants [38, 39]. Alternatively, there has been a growing amount of evidence that long-distance gene movement may be the result of ectopic recombination during the process of double strand break (DSB) repair [40]. In grasses, it has been suggested that gene movement is most likely due to synthesis-dependent strand annealing upon DSB repair [41]. In addition, genes can be also be created and rearranged in genomes by TE-mediated transposon capture or exon shuffling [42]. Indeed, several studies have demonstrated that genes can be captured in TEs and moved throughout plant genomes [43–46]. This phenomenon has been demonstrated on wheat chromosome 3B, where CACTA transposons were shown to have captured and moved 140 of the non-syntenic genes [32]. However, this only explains a small percentage of the 5,125 non-syntenic genes. Upon duplication, one copy of the gene may turn into a pseudogene [47], or the duplicated gene may be retained, possibly undergoing sub- or neo-functionalization of one or both copies [48]. More studies are needed to precisely investigate which one of the potential mechanisms of interchromosomal gene movement led to the increased proportions we observe on wheat 3B.
Chromosomal distribution patterns of non-syntenic genes
Our finding of an increased proportion of retained non-syntenic genes in the distal regions of wheat chromosome 3B (Fig. 4) and barley chromosome 3H confirms previous studies [22, 23, 49]. When looking at the most recently duplicated genes (3B-specific non-syntenic genes), we found no such gradient, suggesting that non-syntenic genes are not preferentially inserted at the distal regions of the chromosome but that their elimination is biased. The spike in the proportion of 3B-specific non-syntenic genes observed in the centromeric region of chromosome 3B is similar to what is found in rice and sorghum chromosomes and support previous observations that synteny correlates with recombination [50]. As discussed in [51], a potential explanation for the difference in the distribution of syntenic vs. non-syntenic genes is that for species with a lower degree of synteny at their centromeric region, there may be more loss of single-gene duplications in euchromatin regions compared to heterochromatic regions due to the lack of recombination near the centromere. This in turn may preserve duplicated genes in the pericentromeric regions [50]. In addition, non-syntenic genes may not be counter-selected in the pericentromeric regions because of the low gene density in these areas (insertions being less deleterious than in areas of high gene density) [51].
Interestingly, this pattern contrasts with the distribution of the ‘older’ Ta3B non-syntenic genes (>6.5 MY), that increase in proportion towards the ends of the chromosome. So, which evolutionary forces shaped this pattern? Recombination as well as gene density increases with relative distance from the centromere, and intrachromosomally duplicated genes are found more frequently at the distal regions of the chromosome [14, 28]. The higher recombination rate at the distal regions could in turn promote rapid evolution [49]. On wheat 3B, DNA transposons represent 18 % of the sequence and tend to be located near the distal regions of the chromosome [32]. These small, repetitive TEs could serve as homologous sequences for unequal recombination, which could in turn can generate duplicated genes [2]. Previous studies found that, in Triticeae, the level of gene sequence polymorphism increases towards the distal regions [52]. This is also supported in Drosophila where beneficial mutations are fixed more efficiently in the high-recombination regions [53]. Thus, in wheat, the probability for a new gene to be retained seems to be higher in distal regions, and this may be due to an interplay of a number of factors, including gene density, TE content, and recombination.
Fate of non-syntenic genes
A previous study using draft sequences of individual chromosomes from wheat group 1 [54] observed a high amount of non-syntenic genes and concluded that many of these are pseudogenes. Here, we show that on chromosome 3B, even upon removing pseudogenes in the filtered set, the majority of the non-syntenic genes are expressed (74 %), indicating that most of these non-syntenic genes are functional. We also observed that older, syntenic, more conserved genes are more constitutively expressed and at a higher level than younger, non-syntenic genes. This supports other findings showing that recently evolved genes have a narrower expression range and level [55] than older and more conserved genes. Genes expressed in a broad range of tissues may evolve slower because the sequence divergence is restricted due the pleiotropic effects of many proteins interacting with each other [56].
Previous studies have shown that alternative splicing is reduced in duplicated genes shortly after they have been duplicated [57, 58]. The authors postulate that since duplicated genes are known to diverge in expression pattern, the reduction of alternative splicing capabilities, and thus, protein functional diversity, in young duplicates is compensated for by subfunctionalization. We also found that non-syntenic genes have fewer isoforms than syntenic genes, which supports this hypothesis. Additionally, duplicated genes in particular are known to evolve much faster than singletons due to sub- or neo-functionalization [59] and may provide adaptive advantages [60].
Although using GO for gene enrichment analysis has some inherent limitations and common drawbacks, it can also be a powerful tool for functional profiling [61]. We found that 3B syntenic genes tend to be enriched in biological processes that are essential, metabolic functions, whereas non-syntenic genes were enriched in processes that could provide some sort of adaptive advantages against biotic or abiotic factors, such as disease resistance. In this study we only investigated syntenic and non-syntenic genes on chromosome 3B, thus the enrichment was normalized by using only 3B as a reference, rather than the entire genome. The question still remains if non-syntenic and syntenic genes will be enriched for the same functions on the remaining chromosomes in the genome. Furthermore, the GO annotations for 3B were inferred computationally, so further experimental evidence will be necessary to confirm these functions.
Nevertheless, the most significantly enriched GO term for non-syntenic genes was programmed cell death (Table 4), which is a known plant defense strategy by which specific cells are destroyed, keeping neighboring cells intact. Programmed cell death can be induced by drought, salt, high temperatures, and other abiotic stresses [62], and is a common mechanism of pathogen resistance in plants [63]. Interestingly, wheat disease resistance genes have been shown to be located near the distal regions of the chromosomes [64]. Additionally, there is evidence that many cereal disease resistance genes are likely non-syntenic [65]. A recent study of mammalian genomes found that the proportion of genes having undergone small scale duplication was correlated with habitat variability, suggesting that species in variable habitats maintain small scale duplications as a way to adapt to their environment [66].
Conclusions
This in-depth analysis of interchromosomal duplications on the reference sequence of chromosome 3B enabled us to study non-syntenic genes between wheat, rice, Brachypodium, and sorghum at the highest resolution to date. The 3B pseudomolecule provides a valuable resource for studies of duplicated genes-- the BAC-by-BAC sequencing and assembly technique allows for fewer collapsed duplicated genes (assemblies with several paralogs merged into chimeric sequences). Thus we have obtained a more accurate picture of interchromosomal gene duplication in the wheat genome.
We performed a stringent analysis with many filtering steps in order to avoid spurious gene annotations. For the first time, we show that even without pseudogenes, the wheat genome has a higher proportion of non-syntenic genes compared to rice, Brachypodium, and sorghum. These non-syntenic genes have nearly the same coding sequence length as the syntenic genes, and the majority are expressed, showing that these are not ‘dead on arrival’, but functional (albeit with a more relaxed selection pressure). We show that non-syntenic genes are not preferentially inserted at the distal regions, but rather are selected for there.
This study provides another piece evidence for accelerated evolution in the Triticeae lineage. This accelerated evolution is in the form of massive interchromosomal duplications, and resulted in a higher number of genes than other grass species and an increased potential for gene adaption. Although polyploidization is one route towards adaptation, single gene duplications that have been occurring before and after the hybridization events have also greatly contributed to inflating the wheat gene repertoire. The structural and functional redundancy provided by the high duplication activity in the wheat genome has likely provided a selective advantage to wheat for adapting to a large range of environments, making it one of the most successful crops.
Methods
Species used and gene filtration
We downloaded the Brachypodium MIPS v1.2 (available at [67]), rice MSU v7 (available at ftp.plantbiology.msu.edu/), and sorghum v8.0 (available at ftp.jgi-psf.org) protein annotations for sequence comparisons. We delineated the regions of synteny between species by visualizing it with Ensembl Plants release 24 Synteny viewer [30]. The entire chromosomes of wheat 3B, rice 1, sorghum 3, and the distal regions of Brachypodium chromosome 2 are syntenic (Additional file 1: Figures S1-S4). For Brachypodium chromosome 2, we determined the borders of the syntenic distal regions: from the telomere of short arm to position 12,348,272 bp and from position 40,348,989 to the telomere of the long arm. In addition, we used the recently released Hordeum vulgare (barley) draft genome sequence [68] to compare to wheat as another representative of the Triticeae lineage. However, we did not use this draft for classifying Ta3B syntenic vs. non-syntenic genes because of the partial representation of the genome, with 2478 genes (less than half expected based on comparison with the other grasses) anchored onto chromosome 3H (syntenic with Ta3B).
The construction of a pseudomolecule for the wheat chromosome 3B was previously described in [14] and the chromosome survey sequence assemblies of each of the 20 other chromosomes were described in [26]. Genes from wheat 3B, rice 1, Brachypodium 2, sorghum 3, and barley 3H were subject to two rounds of filtration: we removed alternative splice variants (taking one representative model for each locus), all genes annotated as related to transposons, and, finally, all genes from the dataset that did not have a significant BLASTP hit (e-value ≤ 1e-5 with ≥35 % amino acid identity and ≥35 % sequence overlap).
Pseudogenes were removed from the dataset as follows: genes were defined as pseudogenes if their model contained internal stop codons, frame shift mutations, or deletions (leaving less than 70 % of the length of a complete homolog) within the CDS.
Classification of syntenic vs. non-syntenic genes
In order to identify syntenic and non-syntenic genes, amino acid sequences of the entire gene set of all species considered were compared using all-by-all BLAST. The best BLAST hit (e-value cutoff 1e-5) in each species was identified for each gene. We used best BLAST hit rather than reciprocal best hits to infer orthologs because in highly duplicated genomes such as wheat, methods based on reciprocal best hits have a high rate of false negatives and will miss many of the true orthologs [69]. If a gene had at least one best hit in another species on a syntenic counterpart, it was classified as syntenic. If the gene had all best BLAST hits carried by non-syntenic chromosomes, it was classified as non-syntenic. The non-syntenic gene fixation rate for each species was calculated by the number of non-syntenic genes/total number of genes in the genome/millions of years since the divergence from the last common ancestor. Non-syntenic genes were determined to have originated by duplication events if a nucleotide BLAST hit (at least 80 % identity and 50 % overlap) was found on a non-homeologous chromosome (not 3A, 3B, or 3D) of the chromosome survey sequence.
Distributions along the chromosomes
For wheat, the chromosome distribution of non-syntenic genes was performed by calculating the proportion of non-syntenic genes/total number of genes within a window of 10 Mb sliding at 1 Mb along the chromosome. For rice, this was a window of 1 Mb sliding at 0.1 Mb, and for sorghum 5 Mb window, sliding at 0.5 Mb. Centromere locations were estimated based on [10, 12, 14, 70]. R was used for calculation of the Pearson correlation coefficients. The loess function in ggplot2 was used to draw the curve.
Calculation of non-synonymous (Ka) and synonymous (Ks) substitution rates
We used the coding sequences of non-syntenic genes and their best BLASTP hit in Brachypodium distachyon to make an alignment with TranslatorX [71] for calculating Ka and Ks. Rates were calculated by the Nei and Gojobori method using codeml (part of the PAML package; [72]). This resulted in Ka and Ks rates for 3,962 pairs after removing those with a frameshift or stop codon mutation. To calculate the Ka/Ks ratio, we removed the samples that had Ks of 0, leaving 3,866 pairs.
Gene expression analysis
RNA extraction, library construction and sequencing was described in [14]. Briefly, total RNAs were extracted in duplicates from the wheat cv. Chinese Spring using five organs (root, leaf, stem, spike, and grain) at three developmental stages each. IlluminaTruSeqTM RNA sample preparation Kit (Illumina, USA) was used to create non-oriented RNA-seq libraries (4 μg of total RNA used with a library insert fragmentation time of 12 min). Libraries were sequenced on an Illumina HiSeq2000 with 2 × 100-bp paired-end reads. Read quality was verified using FastQC v0.10.0 [73]. Illumina reads were mapped to chromosome 3B using Tophat2 v2.0.8 [74] and bowtie2 [75] with the default parameters except: 0 mismatch, 0 splice-mismatch. PCR duplicates were removed with Samtools [76] with the rmdup option and an annotation-guided read alignment was performed with Cufflinks v2.1.1 [77] to reassemble and quantify transcripts in units of fragments per kb of exon per million mapped reads (FPKM). Expressed regions were considered as those with an FPKM higher than zero. Hierarchical clustering was performed using the Hierarchical Clustering Explorer 3.5 software [78] with the complete linkage method and the Pearson correlation coefficient. FPKM values were log2 transformed (log2(FPKM + 1)). The minimal similarity to establish the clusters was set to 0.641 which is the Pearson correlation significant at the P value threshold of 0.01.
Gene Ontology term enrichment analysis
Gene ontology (GO) term enrichment was described in [14]. Briefly, similarity searches using BLASTP (e-value < 1e-05) were performed for each amino acid sequence, against the PLAZA 2.5 protein database [79]. Consensus functional information was assigned to the 3B gene products based on homolog GO or InterPro information. Only the five best homologs with more than 50 % coverage were used for the analysis. The topGO R package was used for enrichment calculations [80]. The full set of 3B gene products was used as the reference comparison set against non-syntenic and syntenic gene sets. P values were calculated using Fisher’s exact test and they were corrected for multiple testing with FDR method using the R module called ‘p-adjust’. Finally, the redundancy from the list of enriched GO terms was removed using the program GO Trimming [81] using default parameters.
Acknowledgements
The authors would like to thank the International Wheat Genome Sequencing Consortium for early access to the chromosome survey sequence data, collaboration and guidance. We would also like to thank Philippe Leroy and Sebastien Theil for providing early access to the 3B gene model predictions. This work was supported by a grant of the French National Research Agency (ANR-09-GENM-025 3BSEQ), and a grant from France Agrimer. NG was funded by a grant of the European Commission research training program Marie-Curie Actions (FP7-MC-IIF-NoncollinearGenes). JD was funded by a grant from the French Ministry of Research. LP was funded by a grant from the Region Auvergne. KV is supported by the Ghent University Multidisciplinary Research Partnership (‘Bioinformatics: from nucleotides to networks’ (Project 01MR0310W)).
Abbreviations
- ASV
alternative splice variant
- BAC
bacteria artificial chromosome
- Bd2
Brachypodium chromosome 2
- CDS
coding sequence
- DSB
double-strand break
- EST
expressed sequence tag
- FPKM
Fragments Per Kilobase of transcript per Million mapped reads
- GO
gene ontology
- Ka
non-synonymous substitution rate
- Ks
synonymous substitution rate
- MYA
million years ago
- Os1
rice chromosome 1
- Sb3
sorghum chromosome 3
- Ta3B
wheat chromosome 3B
- TE
transposable element
Additional file
Comparison of wheat filtered gene set to Arabidopsis gene set. Table S2. Genes reclassified from non-syntenic to syntenic genes by BLAST against genomic DNA of orthologous chromosomes. Table S3 Synteny analysis of barley 3H and maize chromosomes 3 and 8 (regions syntenic to wheat 3B). Figures S1-S4. Rice, Brachypodium, sorghum, and wheat syntenic chromosomes, respectively. Figure S5. Proportion of non-syntenic genes per total genes for barley chromosome 3H. (PDF 759 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NG performed the gene filtration and analyses of: interchromosomal duplication, chromosomal distribution, gene characterization, and gene ontology, as well as drafted the manuscript. JD analyzed the Ka and Ks rates of syntenic vs. non-syntenic genes. LP collected and analyzed RNAseq expression data and carried out the cluster analysis. KV contributed to the gene ontology enrichment analysis and revision of the manuscript. EP participated in the design of the study and revision of the manuscript. FC originally planned the study, participated in its design, played a major role in data interpretation, and helped to draft and revise the manuscript. CF participated in the design of the project, data interpretation, coordination of the project, and critical revision of the manuscript. All authors read and approved the final manuscript.
References
- 1.Ohno S. Evolution by gene duplication. Berlin, Heidelberg: Springer Berlin Heidelberg; 1970. [Google Scholar]
- 2.Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–8. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- 3.Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11:345–55. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- 4.Patthy L. Genome evolution and the evolution of exon-shuffling – a review. Gene. 1999;238:103–14. doi: 10.1016/S0378-1119(99)00228-0. [DOI] [PubMed] [Google Scholar]
- 5.Gale MD, Devos KM. Comparative genetics in the grasses. Proc Natl Acad Sci. 1998;95:1971–4. doi: 10.1073/pnas.95.5.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore G, Devos KM, Wang Z, Gale MD. Cereal genome evolution: grasses, line up and form a circle. Curr Biol. 1995;5:737–9. doi: 10.1016/S0960-9822(95)00148-5. [DOI] [PubMed] [Google Scholar]
- 7.Sorrells ME, Rota ML, Bermudez-Kandianis CE, Greene RA, Kantety R, Munkvold JD, et al. Comparative DNA sequence analysis of wheat and rice genomes. Genome Res. 2003;13:1818–27. doi: 10.1101/gr.1113003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miftahudin, Ross K, Ma X-F, Mahmoud AA, Layton J, Milla MAR, et al. Analysis of expressed sequence tag loci on wheat chromosome group 4. Genetics. 2004;168:651–63. doi: 10.1534/genetics.104.034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 10.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–6. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 11.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 12.Vogel JP, Garvin DF, Mockler TC, Schmutz J, Rokhsar D, Bevan MW, et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–8. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 13.Marcussen T, Sandve SR, Heier L, Spannagl M, Pfeifer M, Jakobsen KS, et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science. 2014;345:1250092. doi: 10.1126/science.1250092. [DOI] [PubMed] [Google Scholar]
- 14.Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, et al. Structural and functional partitioning of bread wheat chromosome 3B. Science. 2014;345:1249721. doi: 10.1126/science.1249721. [DOI] [PubMed] [Google Scholar]
- 15.Singh NK, Dalal V, Batra K, Singh BK, Chitra G, Singh A, et al. Single-copy genes define a conserved order between rice and wheat for understanding differences caused by duplication, deletion, and transposition of genes. Funct Integr Genomics. 2006;7:17–35. doi: 10.1007/s10142-006-0033-4. [DOI] [PubMed] [Google Scholar]
- 16.Singh NK, Raghuvanshi S, Srivastava SK, Gaur A, Pal AK, Dalal V, et al. Sequence analysis of the long arm of rice chromosome 11 for rice-wheat synteny. Funct Integr Genomics. 2004;4:102–17. doi: 10.1007/s10142-004-0109-y. [DOI] [PubMed] [Google Scholar]
- 17.Munkvold JD, Greene RA, Bermudez-Kandianis CE, Rota CML, Edwards H, Sorrells SF, et al. Group 3 chromosome bin maps of wheat and their relationship to rice chromosome 1. Genetics. 2004;168:639–50. doi: 10.1534/genetics.104.034819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rustenholz C, Hedley PE, Morris J, Choulet F, Feuillet C, Waugh R, et al. Specific patterns of gene space organisation revealed in wheat by using the combination of barley and wheat genomic resources. BMC Genomics. 2010;11:714. doi: 10.1186/1471-2164-11-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalupska D, Lee HY, Faris JD, Evrard A, Chalhoub B, Haselkorn R, et al. Acc homoeoloci and the evolution of wheat genomes. Proc Natl Acad Sci. 2008;105:9691–6. doi: 10.1073/pnas.0803981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayer KFX, Martis M, Hedley PE, Simková H, Liu H, Morris JA, et al. Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell. 2011;23:1249–63. doi: 10.1105/tpc.110.082537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho S, Garvin DF, Muehlbauer GJ. Transcriptome analysis and physical mapping of barley genes in wheat–barley chromosome addition lines. Genetics. 2006;172:1277–85. doi: 10.1534/genetics.105.049908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo M-C, Gu YQ, You FM, Deal KR, Ma Y, Hu Y, et al. A 4-gigabase physical map unlocks the structure and evolution of the complex genome of Aegilops tauschii, the wheat D-genome progenitor. Proc Natl Acad Sci U S A. 2013;110:7940–5. doi: 10.1073/pnas.1219082110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choulet F, Wicker T, Rustenholz C, Paux E, Salse J, Leroy P, et al. Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell. 2010;22:1686–701. doi: 10.1105/tpc.110.074187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo MC, Deal KR, Akhunov ED, Akhunova AR, Anderson OD, Anderson JA, et al. Genome comparisons reveal a dominant mechanism of chromosome number reduction in grasses and accelerated genome evolution in Triticeae. Proc Natl Acad Sci U S A. 2009;106:15780–5. doi: 10.1073/pnas.0908195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akhunov ED, Sehgal S, Liang H, Wang S, Akhunova AR, Kaur G, et al. Comparative analysis of syntenic genes in grass genomes reveals accelerated rates of gene structure and coding sequence evolution in polyploid wheat. Plant Physiol. 2013;161:252–65. doi: 10.1104/pp.112.205161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International Wheat Genome Sequencing Consortium A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 27.Schnable JC, Freeling M, Lyons E. Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol Evol. 2012;4:265–77. doi: 10.1093/gbe/evs009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Quraishi UM, et al. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell. 2008;20:11–24. doi: 10.1105/tpc.107.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491:705–10. doi: 10.1038/nature11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monaco MK, Stein J, Naithani S, Wei S, Dharmawardhana P, Kumari S, et al. Gramene 2013: comparative plant genomics resources. Nucleic Acids Res. 2014;42:D1193–9. doi: 10.1093/nar/gkt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet. 2012;13:329–42. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
- 32.Daron J, Glover N, Pingault L, Theil S, Jamilloux V, Paux E, et al. Organization and evolution of transposable elements along the bread wheat chromosome 3B. Genome Biol. 2014;15:546. doi: 10.1186/s13059-014-0546-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leroy P, Guilhot N, Sakai H, Bernard A, Choulet F, Theil S, et al. TriAnnot: a versatile and high performance pipeline for the automated annotation of plant genomes. Front Plant Sci. 2012;3:5. doi: 10.3389/fpls.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pingault L, Choulet F, Alberti A, Glover N, Wincker P, Feuillet C, et al. Deep transcriptome sequencing provides new insights into the structural and functional organization of the wheat genome. Genome Biol. 2015;16:29. doi: 10.1186/s13059-015-0601-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paterson AH, Bowers JE, Chapman BA, Peterson DG, Rong J, Wicker TM. Comparative genome analysis of monocots and dicots, toward characterization of angiosperm diversity. Curr Opin Biotechnol. 2004;15:120–5. doi: 10.1016/j.copbio.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. Synteny and collinearity in plant genomes. Science. 2008;320:486–8. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 37.Passarge E, Horsthemke B, Farber RA. Incorrect use of the term synteny. Nat Genet. 1999;23:387–7. doi: 10.1038/70486. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Zheng H, Fan C, Li J, Shi J, Cai Z, et al. High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell Online. 2006;18:1791–802. doi: 10.1105/tpc.106.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Z, Zhang Y, Long M. Extensive structural renovation of retrogenes in the evolution of the populus genome. Plant Physiol. 2009;151:1943–51. doi: 10.1104/pp.109.142984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puchta H. The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot. 2005;56:1–14. doi: 10.1093/jxb/eri123. [DOI] [PubMed] [Google Scholar]
- 41.Wicker T, Buchmann JP, Keller B. Patching gaps in plant genomes results in gene movement and erosion of colinearity. Genome Res. 2010;20:1229–37. doi: 10.1101/gr.107284.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennetzen JL. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev. 2005;15:621–7. doi: 10.1016/j.gde.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–73. doi: 10.1038/nature02953. [DOI] [PubMed] [Google Scholar]
- 44.Lai J, Li Y, Messing J, Dooner HK. Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci U S A. 2005;102:9068–73. doi: 10.1073/pnas.0502923102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wicker T, Guyot R, Yahiaoui N, Keller B. CACTA transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiol. 2003;132:52–63. doi: 10.1104/pp.102.015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin YK, Bennetzen JL. Integration and nonrandom mutation of a plasma membrane proton ATPase gene fragment within the Bs1 retroelement of maize. Plant Cell. 1994;6:1177–86. doi: 10.1105/tpc.6.8.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurles M. Gene duplication: The genomic trade in spare parts. PLoS Biol. 2004;2:e206. doi: 10.1371/journal.pbio.0020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conant GC, Wolfe KH. Turning a hobby into a job: How duplicated genes find new functions. Nat Rev Genet. 2008;9:938–50. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 49.Akhunov ED, Goodyear AW, Geng S, Qi L-L, Echalier B, Gill BS, et al. The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms. Genome Res. 2003;13:753–63. doi: 10.1101/gr.808603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bowers JE, Arias MA, Asher R, Avise JA, Ball RT, Brewer GA, et al. Comparative physical mapping links conservation of microsynteny to chromosome structure and recombination in grasses. Proc Natl Acad Sci U S A. 2005;102:13206–11. doi: 10.1073/pnas.0502365102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK. Recombination: an underappreciated factor in the evolution of plant genomes. Nat Rev Genet. 2007;8:77–84. doi: 10.1038/nrg1970. [DOI] [PubMed] [Google Scholar]
- 52.Dvorák J, Luo MC, Yang ZL. Restriction fragment length polymorphism and divergence in the genomic regions of high and low recombination in self-fertilizing and cross-fertilizing aegilops species. Genetics. 1998;148:423–34. doi: 10.1093/genetics/148.1.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Presgraves DC. Recombination enhances protein adaptation in Drosophila melanogaster. Curr Biol CB. 2005;15:1651–6. doi: 10.1016/j.cub.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 54.Wicker T, Mayer KFX, Gundlach H, Martis M, Steuernagel B, Scholz U, et al. Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell. 2011;23:1706–18. doi: 10.1105/tpc.111.086629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang YE, Landback P, Vibranovski M, Long M. New genes expressed in human brains: Implications for annotating evolving genomes. BioEssays. 2012;34:982–91. doi: 10.1002/bies.201200008. [DOI] [PubMed] [Google Scholar]
- 56.Pál C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–31. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhan Z, Ren J, Zhang Y, Zhao R, Yang S, Wang W. Evolution of alternative splicing in newly evolved genes of Drosophila. Gene. 2011;470:1–6. doi: 10.1016/j.gene.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Su Z, Wang J, Yu J, Huang X, Gu X. Evolution of alternative splicing after gene duplication. Genome Res. 2006;16:182–9. doi: 10.1101/gr.4197006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellegren H. Comparative genomics and the study of evolution by natural selection. Mol Ecol. 2008;17:4586–96. doi: 10.1111/j.1365-294X.2008.03954.x. [DOI] [PubMed] [Google Scholar]
- 60.Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci. 2012;279:5048–57. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yon Rhee S, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nat Rev Genet. 2008;9:509–15. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- 62.Petrov V, Hille J, Mueller-Roeber B, Gechev TS. ROS-mediated abiotic stress-induced programmed cell death in plants. Plant Physiol. 2015;6:69. doi: 10.3389/fpls.2015.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greenberg JT. Programmed cell death in plant-pathogen interactions. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:525–45. doi: 10.1146/annurev.arplant.48.1.525. [DOI] [PubMed] [Google Scholar]
- 64.Dilbirligi M, Erayman M, Sandhu D, Sidhu D, Gill KS. Identification of wheat chromosomal regions containing expressed resistance genes. Genetics. 2004;166:461–81. doi: 10.1534/genetics.166.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leister D, Kurth J, Laurie DA, Yano M, Sasaki T, Devos K, et al. Rapid reorganization of resistance gene homologues in cereal genomes. Proc Natl Acad Sci. 1998;95:370–5. doi: 10.1073/pnas.95.1.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tamate SC, Kawata M, Makino T. Contribution of nonohnologous duplicated genes to high habitat variability in mammals. Mol Biol Evol. 2014;31:1779–86. doi: 10.1093/molbev/msu128. [DOI] [PubMed] [Google Scholar]
- 67.Phytozome. Available at: http://phytozome.jgi.doe.gov.
- 68.International Barley Genome Sequencing Consortium. Mayer KFX, Waugh R, Brown JWS, Schulman A, Langridge P, et al. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–6. doi: 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
- 69.Dalquen DA, Dessimoz C. Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol Evol. 2013;5:1800–6. doi: 10.1093/gbe/evt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan H, Talbert PB, Lee H-R, Jett J, Henikoff S, Chen F, et al. Intergenic locations of rice centromeric chromatin. PLoS Biol. 2008;6:e286. doi: 10.1371/journal.pbio.0060286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 73.Babraham Bioinformatics. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 74.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hierarchical Clustering Explorer. Available at: http://www.cs.umd.edu/hcil/hce/.
- 79.Van Bel M, Proost S, Wischnitzki E, Movahedi S, Scheerlinck C, Van de Peer Y, et al. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 2012;158:590–600. doi: 10.1104/pp.111.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alexa A, Rahnenfuhrer J. topGO: topGO: Enrichment analysis for Gene Ontology. Available at: http://www.bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf.
- 81.Jantzen SG, Sutherland BJ, Minkley DR, Koop BF. GO Trimming: Systematically reducing redundancy in large Gene Ontology datasets. BMC Res Notes. 2011;4:267. doi: 10.1186/1756-0500-4-267. [DOI] [PMC free article] [PubMed] [Google Scholar]