

Abstract
A palladium-catalyzed three-component coupling of α-olefins, aryldiazonium salts, and bis-(pinacolato)diboron affords direct access to chiral benzylic boronic esters. This process is rendered highly enantioselective using an unprecedented example of cooperative chiral anion phase transfer and transition-metal catalysis.
Benzylic boronic esters are attractive building blocks for complex biologically active natural products and pharmaceuticals as they participate in several well-established stereospecific transformations that forge C–O, C–N, or C–C bonds.1 Accordingly, there has been significant interest in developing new enantioselective methods that afford these motifs. Examples of catalytic enantioselective hydroboration,2 allylic borylation,3 conjugate borylation,4 reductive transformations of vinyl boronates5 and 1,2-diborylation of aromatic imines and styrenes have been reported.6 Furthermore, several enantioselective borylation reactions utilizing stoichiometric amounts of chiral auxiliaries have been disclosed.7 Recently, several methods have been reported that rely on transformations of 1,1-diborylated alkanes. For example, Hall8 and Yun9 have reported the preparation of enantioenriched 1,1-diborylated alkanes that undergo chemoselective coupling to provide highly enantioenriched benzylic organoboronates (Figures 1a and 1b). In a related contribution, Morken and co-workers have disclosed an enantioselective Suzuki reaction of 1,1-diboronates to provide highly enriched boronic esters (Figure 1c).10 While these methods ultimately access valuable enantioenriched benzylic boronates, they require a multistep synthetic sequence. From the perspective of synthetic divergence and step-economy, an ideal transformation would involve one-step installation of both the boronate and aryl functional groups (Figure 1d).
Figure 1.

Recent examples of multistep enantioselective 1,1-arylborylation by (a) Hall and co-workers, (b) Yun and co-workers, (c) Morken and co-workers, and (d) the single-step 1,1-arylborylation reported herein. dan = 1,8-diaminonaphthalene; pin = pinacolato.
Inspired by seminal examples of Pd-catalyzed 1,1-diarylation from Sigman and co-workers,11,12 we envisioned direct formation of 1,1-arylborylated products (9) from unactivated olefins (7) via a three-component Heck–Matsuda arylation/Miyuara borylation cascade reaction (Figure 1d). In these initial reports, efficient 1,1-difunctionalization required “ligand-free” conditions, as efforts to induce enantioselectivity through application of chiral ligands attenuated the desired three-component reaction pathway.12 Cognizant of this limitation, we were intrigued by the possibility of providing enantioinduction via a chiral ion-pairing strategy. While the intermediacy of non-cationic Pd(0) species presented a challenge due to the lack of continuous Coulombic attraction between the chiral anion and a cationic metal center, we hypothesized that a chiral anion phase-transfer (CAPT) strategy13-15 could enable association of a chiral anion with the key cationic Pd(II) intermediates involved in the enantiodetermining step. In this scenario, an insoluble aryldiazonium salt (11, Figure 2) would undergo phase transfer to provide soluble, chiral ion pair 12.16,17 Oxidative addition of the Pd(0) species followed by migratory insertion of the olefinic coupling partner would yield chiral Pd complex 14. Sequential β-hydride elimination and reinsertion steps would yield enantioenriched benzylic Pd complex 15 that is poised to undergo transmetalation and subsequent reductive elimination to provide the desired benzylic boronic ester. Importantly, the non-cationic Pd(0) species generated in this process can be reoxidized by soluble diazonium chiral ion pair 12 to regenerate the chiral Pd(II) species 13. In this Communication we report the successful execution of this methodological hypothesis through the single-step 1,1-arylbor-ylation of α-olefins, with high levels of enantioselectivity achieved via implementation of a CAPT strategy.
Figure 2.

Proposed dual catalytic cycle for Pd-CAPT 1,1-arylborylation.
Given the lack of precedent for this transformation, we sought to provide proof-of-principle with the development of a non-stereoselective 1,1-arylborylation of α-olefins. We were pleased to find that a room-temperature THF suspension of catalytic Pd2(dba)3, phenyldiazonium tetrafluoroborate, allyl methyl carbonate, and bis(pinacolato)diboron smoothly provided racemic boronate 16 in 72% yield (Table 1). Both electron-rich (17) and electron-poor aryldiazonium salts (18) proved competent in the transformation of allyl methyl carbonate to provide boronic esters 17 and 18 in 90% and 71% yield, respectively. Use of ortho-substituted aryldiazonium cations also provided the product in good yield (19). Notably, utilization of homoallylic carbonates (20), as well as protected allylic amines, furnished the borylated products in good to excellent yields (21,22). Further, a nitrile-substituted substrate underwent smooth arylborylation in 99% yield, demonstrating that substrates containing chelating groups did not impede the reaction (23). As an important demonstration of scope, electron poor alkenes were also effective under our reaction conditions, providing the arylborylated products 24–26, albeit in diminished yields.18
Table 1. Non-enantioselective Scopea,b.
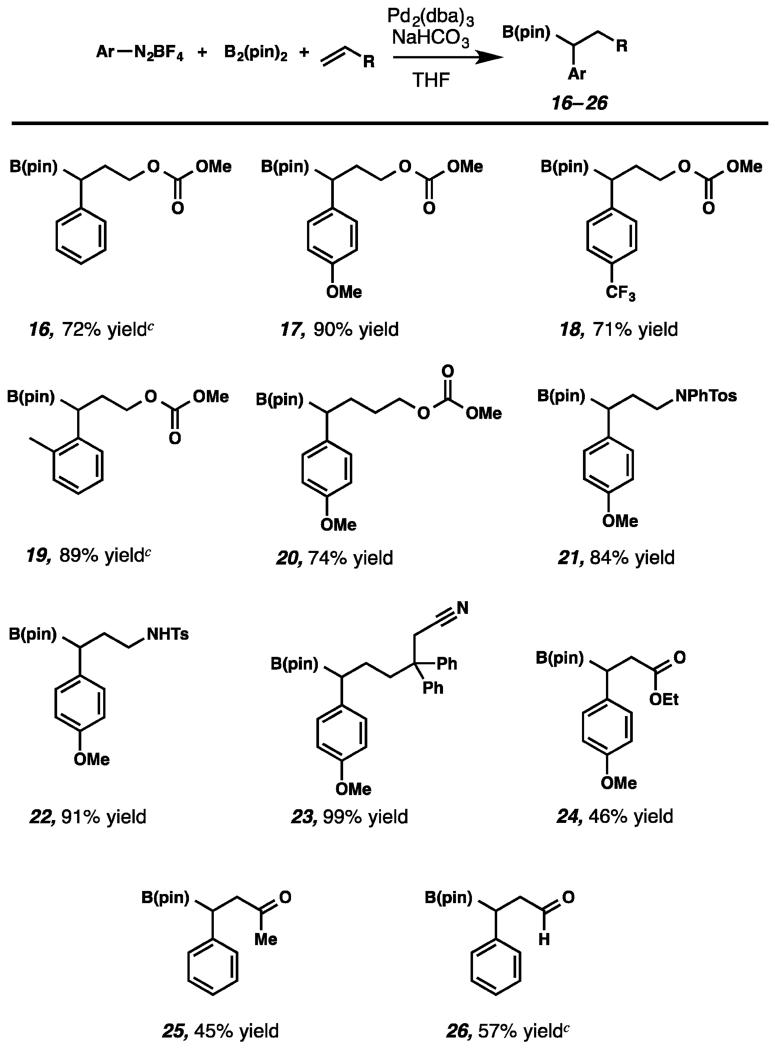
|
Conditions: ArN2BF4 (1 equiv), B2(pin)2 (1.2 equiv), Pd2(dba)3 (0.025 equiv), alkene (1 equiv), THF, 25 °C, 2–8 h.
Isolated yields.
NMR yield.
Having provided proof-of-principle for a single-step 1,1-arylborylation reaction, we began work toward rendering the process enantioselective via CAPT. Utilizing allyl methyl carbonate and phenyldiazonium tetrafluoroborate, an extensive study of reaction conditions was undertaken. In initial experiments, nonpolar solvents such as hexanes provided poor conversion (Table 2, entry 1), while THF allowed for excellent conversion albeit with poor enantioselectivity (entry 2). Et2O was identified as optimal, furnishing the product in 33% ee and 45% yield (entry 3).19 Examination of commonly employed CAPT catalysts (27–29) identified TCyP (27) as superior in terms of enantioselectivity, providing the desired product in 88% ee (entry 5). Further, Na3PO4 provided the highest yield of the bases examined, furnishing the product in 36% yield (entry 7). Finally, addition of exogenous dibenzylideneacetone (dba) derivatives20 (30) improved the efficiency of the reaction, with m-CF3-dba (30b) allowing for retention of enantioselectivity, providing the product in 39% yield and 90% ee (entry 9).21
Table 2. Optimization of Enantioselective 1,1-Arylborylationa,b.
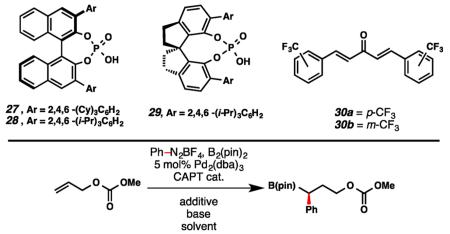
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | cat. | solvent | base | additive | ee (%) | yield (%) |
| 1 | 28 | hexanes | NaHCO3 | – | – | <5 |
| 2 | 28 | THF | NaHCO3 | – | <5 | 72 |
| 3 | 28 | Et2O | NaHCO3 | – | 33 | 45 |
| 4 | 29 | Et2O | NaHCO3 | – | <5 | 14 |
| 5 | 27 | Et2O | NaHCO3 | – | 88 | 25 |
| 6 | 27 | Et2O | K2CO3 | – | 94 | 25 |
| 7 | 27 | Et2O | Na3PO4 | – | 93 | 26 |
| 8 | 27 | Et2O | Na3PO4 | 30a | 70 | 40 |
| 9 | 27 | Et2O | Na3PO4 | 30b | 90 | 39 |
Enantiomeric excess determined by chiral phase HPLC.
Yield determined by 1H NMR utilizing dimethyl sulfone as an internal standard.
With optimized conditions in hand, we examined the aryldiazonium scope. Utilizing allyl methyl carbonate we found that aryldiazonium salts functionalized with a methoxy group at the meta- or para-positions provided products in synthetically useful yields and good to excellent enantioselectivities (Table 3a, 32 and 33). Alkyl substitution of the para- or meta-positions also provided highly enantioenriched benzylic boronic ester products (34–37). Finally, substitution with a vinyl group at the para-position afforded enriched boronic ester (38) in 40% yield and 98% ee.
Table 3. Enantioselective Scopea,b.
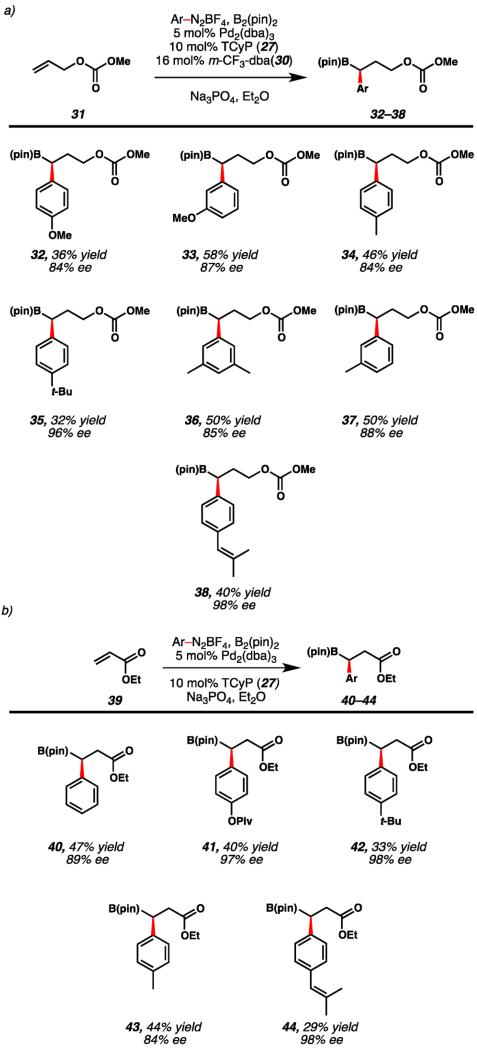
|
Enantiomeric excess determined by chiral phase HPLC.
Yield determined by 1H NMR utilizing dimethyl sulfone as an internal standard.
In addition to protected allylic alcohols, α,β-unsaturated esters were utilized as an alternative olefinic precursor for the enantioselective transformation (Table 3b). Notably, use of m-CF3-dba (30b) was not required to achieve useful yields and high enantioselectivities with acrylate coupling partners. Utilization of phenyldiazonium tetrafluoroborate allowed for formation of β,β-arylborylated ester 40 in 47% yield and 89% ee. The aryl moiety of the diazonium salt was tolerant of substitution of the para-position with heteroatom or alkyl functional groups, providing a variety of β,β-arylborylated esters in synthetically useful yields and excellent enantioselectivities (41–44).
In conclusion, we have disclosed a modular and step-economical method for the direct preparation of chiral benzylic boronates from terminal alkenes. Furthermore, this process was rendered enantioselective through the use of a chiral anion phase-transfer strategy. We expect the coupling of CAPT and Pd catalysis to have broad implications, as it provides an alternative strategy for achieving enantioinduction in “ligandless” reaction manifolds where chiral ancillary ligands have a deleterious effect.22
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the NIGMS (R01 GM104534) for financial support. We acknowledge Prof. Matthew Sigman for useful discussions and experimental advice. H.M.N. also acknowledges the UNCF and Merck for generous funding. J.M. thanks the University of Valencia for a predoctoral fellowship.
Footnotes
ASSOCIATED CONTENT
Procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Hall DG. Boronic Acids. 2nd ed. Wiley-VCH; Weinheim: 2011. [Google Scholar]; (b) Crudden CM, Edwards D. Eur. J. Org. Chem. 2003:4695. [Google Scholar]
- (2).(a) Carroll AM, O’Sullivan TP, Guiry PJ. Adv. Synth. Catal. 2005;347:609. [Google Scholar]; (b) Smith SM, Takacs JM. J. Am. Chem. Soc. 2010;132:1740. doi: 10.1021/ja908257x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Corberań R, Mszar NW, Hoveyda AH. Angew. Chem., Int. Ed. 2011;50:7079. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]
- (3).(a) Guzman-Martinez A, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10634. doi: 10.1021/ja104254d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park JK, Lackey HH, Ondrusek BA, McQuade DT. J. Am. Chem. Soc. 2011;133:2410. doi: 10.1021/ja1112518. [DOI] [PubMed] [Google Scholar]
- (4).Calow AD, Whiting A. Org. Biomol. Chem. 2012;10:5485. doi: 10.1039/c2ob25908g. [DOI] [PubMed] [Google Scholar]
- (5).(a) Ding J, Lee JCH, Hall DG. Org. Lett. 2012;14:4462. doi: 10.1021/ol301958x. [DOI] [PubMed] [Google Scholar]; (b) Morgan JB, Morken JP. J. Am. Chem. Soc. 2004;126:15338. doi: 10.1021/ja044396g. [DOI] [PubMed] [Google Scholar]; (c) Lee JCH, Hall DG. J. Am. Chem. Soc. 2010;132:5544. doi: 10.1021/ja9104057. [DOI] [PubMed] [Google Scholar]
- (6).(a) Kliman LT, Mlynarski SN, Morken JP. J. Am. Chem. Soc. 2009;131:13210. doi: 10.1021/ja9047762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong K, Morken JP. J. Am. Chem. Soc. 2013;135:9252. doi: 10.1021/ja402569j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Matteeon DS, Sadhu KM, Ray R, Peterson ML, Majumdar D, Hunt GD, Jemthi PK, Teai DJS, Erdik B. Pure Appl. Chem. 1988;57:1741. [Google Scholar]; (b) Beckmann E, Desai V, Hoppe D. Synlett. 2004;13:2275. [Google Scholar]; (c) Beenen MA, An C, Ellman JA. J. Am. Chem. Soc. 2008;130:6910. doi: 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]; (d) Scott HK, Aggarwal VK. Chem.—Eur. J. 2011;17:13124. doi: 10.1002/chem.201102581. [DOI] [PubMed] [Google Scholar]
- (8).Lee JCH, McDonald R, Hall DG. Nat. Chem. 2011;3:894. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- (9).Feng X, Jeon H, Yun J. Angew. Chem., Int. Ed. 2013;52:3989. doi: 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]
- (10).Sun C, Potter B, Morken JP. J. Am. Chem. Soc. 2014;136:6534. doi: 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Saini V, Sigman MS. J. Am. Chem. Soc. 2012;134:11372. doi: 10.1021/ja304344h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liao L, Jana R, Urkalan KB, Sigman MS. J. Am. Chem. Soc. 2011;133:5784. doi: 10.1021/ja201358b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Saini V, Liao L, Wang Q, Jana R, Sigman MS. Org. Lett. 2013;15:5008. doi: 10.1021/ol4023358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]; (b) Lackner A, Samant A, Toste FD. J. Am. Chem. Soc. 2013;135:14090. doi: 10.1021/ja4082827. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neel AJ, Hehn JP, Tripet PF, Toste FD. J. Am. Chem. Soc. 2013;135:14044. doi: 10.1021/ja407410b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang X, Phipps RJ, Toste FD. J. Am. Chem. Soc. 2014;136:5225. doi: 10.1021/ja500882x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zi W, Wang Y-M, Toste FD. J. Am. Chem. Soc. 2014;136:12864. doi: 10.1021/ja507468u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:406. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S, List B. J. Am. Chem. Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]; (c) Rauniyar V, Wang ZJ, Burks HE, Toste FD. J. Am. Chem. Soc. 2011;133:8486. doi: 10.1021/ja202959n. [DOI] [PubMed] [Google Scholar]; (d) Jiang G, Halder R, Fang Y, List B. Angew. Chem., Int. Ed. 2011;50:9752. doi: 10.1002/anie.201103843. [DOI] [PubMed] [Google Scholar]
- (15).(a) Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]; (b) Brak K, Jacobsen EN. Angew. Chem., Int. Ed. 2013;52:534. doi: 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mahlau M, List B. Angew. Chem., Int. Ed. 2013;52:518. doi: 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- (16).Nelson HM, Reisberg SH, Shunatona HP, Patel JS, Toste FD. Angew. Chem., Int. Ed. 2014;53:5600. doi: 10.1002/anie.201310905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nelson HM, Patel JS, Shunatona HP, Toste FD. Chem. Sci. 2015;6:170. doi: 10.1039/c4sc02494j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Unfunctionalized alkenes afforded the product in lower yields. For example, 1-octene underwent arylborylation in 37% yield. In the enantioselective reaction, the desired product was obtained in trace amounts, and the corresponding sytrene constituted the primary product.
- (19).The major byproduct in the enantioselective arylborylation reaction of both the allylic alcohol and acrylate substrates was the corresponding styrene. Unlike the non-enantioselective reaction in THF solvent, use of Et2O solvent and phase-transfer catalyst results in significant termination via β-hydride elimination.
- (20).The dba derivatives utilized in this study were prepared according to the procedure outlined in the following report: Kapdi AR, Whitwood AC, Williamson DC, Lynam JM, Burns MJ, Williams TJ, Reay AJ, Holmes J, Fairlamb IJS. J. Am. Chem. Soc. 2013;135:8388. doi: 10.1021/ja403259c.
- (21).While the role of dba derivatives in improving the efficiency of this process has not been rigorously investigated, and direct ligand exchange between Pd2(dba)3 and m-CF3-dba has not been observed under the reaction conditions, we attribute this effect to stabilization of unobservable Pd species along the catalytic cycle.
- (22).For an alternative approach to the use of chiral anions in palladium catalysis with achiral cationic ancillary ligands, see: Ohmatsu K, Ito M, Kunieda T, Ooi T. J. Am. Chem. Soc. 2013;135:590. doi: 10.1021/ja312125a. Ohmatsu K, Yoshiyuki H, Ooi T. Chem. Sci. 2014;5:3645. Ohmatsu K, Ito M, Kunieda T, Ooi T. Nat. Chem. 2012;4:473. doi: 10.1038/nchem.1311.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.