

Abstract
The present study aimed to investigate the molecular targets for colorectal cancer (CRC). Differentially expressed genes (DEGs) were screened between CRC and matched adjacent noncancerous samples. GENETIC_ASSOIATION_DB_DISEASE analysis was performed to identify CRC genes from the identified DEGs using the Database for Annotation, Visualization and Integrated Discovery, followed by Gene Οntology (GO) and Kyoto Encyclopedia of Genes and Genomes analysis for the CRC genes. A protein-protein interaction (PPI) network was constructed for the CRC genes, followed by determination and analysis of the hub genes, in terms of the protein domains and spatial structure. In total, 35 CRC genes were determined, including 19 upregulated and 16 downregulated genes. Downregulated N-acetyltransferase (NAT)1 and NAT2 were enriched in the caffeine metabolism pathway. The down-regulated and upregulated genes were enriched in a number of GO terms and pathways, respectively. Cyclin D1 (CCND1) and proliferating cell nuclear antigen (PCNA) were identified as the hub genes in the PPI network. The C-terminal and N-terminal domains were similar in PCNA, but different in CCND1. The results suggested PCNA, CCND1, NAT1 and NAT2 for use as biomarkers to enable early diagnosis and monitoring of CRC. These results form a basis for developing therapies, which target the unique protein domains of PCNA and CCND1.
Keywords: colorectal cancer, signaling pathway, protein-protein network, differentially expressed genes, hub gene
Introduction
Colorectal cancer (CRC) is the third most common type of cancer, which accounts for ~10% of all cases of cancer with 1,400,000 new cases and a cancer-associated mortality rate of 694,000 in 2012 (1). The incidence of CRC is higher in developed countries, compared with that in developing countries (1). A number of risk factors of the disease have been defined to date, including the consumption of red and processed meat, obesity, smoking and lack of physical activity (2). Its symptoms and signs vary and dependent predominantly on the location and metastasis of the tumor. Localized cancer at an early stage can be cured with surgical resection, however, advanced stages of cancer, accompanied with presence of metastasis, is less likely to be cured by surgery and has a poor prognosis with a significantly lower 5-year survival rate (3).
CRC is a multistep-process resulting from accumulative mutations of tumor suppressor genes and oncogenes (4,5). Increasing studies have demonstrated that heterogeneous mutations of the Wnt pathway, which potentiate the pathway activity can contribute to the initiation and development of CRC (6,7). In addition, the important roles of adenomatous polyposis coli (APC) mutation and relevant β-catenin signaling pathway have implicated in CRC (8). A common consensus has been reached that APC mutation can lead to activation of the Wnt pathway by regulating the accumulation of β-catenin in cells. In addition to the Wnt pathway, a previous study suggested that APC mutations affect CRC development via regulation of the energetic metabolite pathways (9). Furthermore, a previous in vitro study revealed that tumor suppressor gene, mothers against decapentaplegic homolog 4 (SMAD4), if lost, triggers the tumor suppressive bone morphogenetic protein (BMP) signaling pathway to exert a metastasis-promoting effect on CRC (10). By contrast, the tumor suppressor gene, p53 can induce the expression of micro (mi)RNA-34a, which affects the interleukin (IL)-6R/signal transducer and activator of transcription 3/miR-34a feedback loop, and reduces the rate of CRC progression (11). Despite these considerable insights, the molecular mechanism of CRC remain to be fully elucidated.
The interest in examining biomarkers of diseases using bioinformatics approaches has increased. Scavenger receptor class A, member 5 (SCARA5) is found to be downregulated and is involved in CRC (12). Unlike the above-mentioned studies, the present study investigated the differentially expressed genes (DEGs) between CRC and control samples, and then aimed to identify the CRC genes from the DEGs obtained prior to the functional annotation of the identified CRC genes through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. The study aimed to identify potential molecular targets for CRC. The findings of this study may provide useful information regarding the pathogenesis of CRC and lay the foundation for developing novel effective therapeutic strategies for the management of CRC.
Materials and methods
Data preprocessing
As the present study did not involve any humans or animals, there was no requirement for ethical approval. For microarray data preprocessing and DEG identification, the gene expression dataset (GSE7621) was downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/), consisting of 17 CRC tissues samples and 17 matched adjacent noncancerous tissues samples from patients with CRC.
The probe-level data in the CEL files were converted into expression measures using Affy package in R software (http://www.r-project.org/), followed by data normalization using the robust multiarray average 1 algorithm (13). The data, prior to and following normalization were presented as respective box plots. Subsequently, the probe number was converted into corresponding gene names, according to the HG-U133_Plus_2 Affymetrix Human Genome U133 Plus 2.0 Array platform (Affymetrix, Inc., Santa Clara, CA, USA). Expression values of multiple probes targeting one gene were averaged, and this average expression value was selected for the gene.
Following data preprocessing, Significance Analysis of Microarrays (SAM) was performed to screen for the DEGs between the CRC samples and noncancerous samples using samr package in R software (http://cran.r-project.org/web/packages/samr/index.html) (14). Subsequently, multiple assessment correction was performed to adjust the P-values using the Benjamini-Hochberg (BH) method (15). Genes with a false discovery rate (FDR) <0.05 and fold-change >2 were selected.
Identification of CRC genes
The Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) provides a high-throughput and desirable data-mining environment and combines functional genomic annotations with intuitive graphical representations, facilitating the transition between genomic data and the biological function meaning (16). Using DAVID, GENETIC_ASSOIATION_DB_DISEASE analysis was performed to identify the CRC genes from the identified DEGs. The CRC genes were defined as DEGs that were significantly associated with CRC (P <0.05) (17).
GO and KEGG pathway enrichment analysis
GO (http://www.geneontology.org/) functions as a database to provide vocabularies and classifications associated with the molecular and cellular structures and functions for biological annotations of genes (18). GO terms consist of three categories: Biological process (BP), cellular component (CC) and molecular function (MF). The KEGG (http://www.genome.jp/kegg/ or http://www.kegg.jp/) database contains rich information pertaining to known metabolic pathways and regulatory pathways, and facilitates the mapping of genes to KEGG pathways for systemic analysis of gene functions (19).
To provide an insight into the precise biological function and signaling pathways involved with the CRC genes identified in the present study, GO and KEGG pathway enrichment analysis were performed for the upregulated and downregulated DEGs, respectively. GO terms with P<0.05 and gene count ≥5, and KEGG pathways with P<0.05 were screened out.
Construction of a PPI network and identification of hub CRC genes
The Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org/) is a useful tool, which is capable of providing a comprehensive view of all the known and predicted interactions and associations among proteins (20). In order to elucidate the interactions between the CRC genes in the present study, STRING online software was used to construct a PPI network using the CRC genes, and the network was visualized using Cytoscape open-source software (http://www.cytoscape.org/) (21). In the PPI network, the proteins in the network served as 'nodes' and the link connecting two nodes represent a pairwise protein interaction. The degree of a node corresponds to the number of interactions that the protein is in possession of. The nodes with the highest degree of connection were considered the 'hub' genes in the PPI network.
Predicting coding sequence (CDS) and protein domains of hub genes
GENSCAN (http://genes.mit.edu/GENSCAN.html) is a complex computer program, which applies a probabilistic model of human genome structure to predict complete gene structures in human genomic sequences. The probabilistic model is comprised of detailed information of exons, introns and intergenic regions of human genome (22–24). In the present study, to obtain a better understanding of the structure of the identified hub CRC genes, GENSCAN was used to predict the CDS of the hub genes. The corresponding amino acid (aa) sequence was then obtained.
The protein domain of the hub genes was predicted using the Pfam database (http://pfam.sanger.ac.uk/), which is a widely used database containing information of protein domains and families, and assists in determining the structure of proteins (25). Finally, the structure of the obtained protein domain was visualized using Cn3D software version 4.0 (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml;). The Cn3D ('see in 3D') is a molecular graphics program, which facilitates viewing of the 3D structure of a protein sequence. An interactive view of sequences and sequences alignments is also available with Cn3D (26). The structures of the proteins encoded by the hub genes were analyzed using Conserved Domain Database (CDD). It is a protein annotation database that provides rich information concerning protein structure and function (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml).
Results
Data preprocessing
The box plots in Figs. 1 and 2 show the microarray data priot to and following normalization, respectively. In Fig. 1, the horizontal black lines representing the median gene expression value for each sample were varied. By contrast, the median gene expression vales appear on a straight line in Fig. 2, suggesting that the data following normalization was suitable for further analysis.
Figure 1.

Box plot for the gene expression data prior to normalization. Expression values were determined using Affy package in R software. The black bar indicates the median value.
Figure 2.

Box plot for the gene expression data following normalization. Expression values were determined using Affy package in R software, followed by normalization using the robust multiarray average algorithm The black bar indicates the median value.
Identification of DEGs and CRC genes
Following data normalization, a total of 1,389 DEGs were screened between the CRC and noncancerous samples, from which 691 genes were upregulated and 698 genes were downregulated (Fig. 3A). Consequently, from the obtained DEGs, 35 CRC genes were identified, including 19 upregulated and 16 downregulated genes (Fig. 3B).
Figure 3.

Identification of critical genes. (A) Analysis of the DEGs identified between the CRC and normal samples. Horizontal axis, alterations of gene expression; vertical axis, number of genes. (B) Analysis of the 35 identified CRC genes. Horizontal axis, alterations of gene expression; vertical axis, number of genes. DEGs, differentially expressed genes. MF, molecular function; BP, biological process; CC. cellular component.
GO and KEGG pathway enrichment analysis
The result of GO enrichment analysis revealed that the upregulated CRC genes were enriched in a number of BP terms associated with cell proliferation and DNA metabolism (Fig. 4). By contrast, the downregulated genes were predominately associated with response to organic substance and regulation of cell proliferation (Fig. 5).
Figure 4.

GO enrichment analysis of the upregulated colorectal cancer genes. Diamond, categories of GO terms; square, GO terms. GO, Gene Οntology; BP, biological process; CC, cellular component. A blue line links a diamond (GO term) and a square (GO category). It means that the diamond (GO term) belongs a square (GO category).
Figure 5.
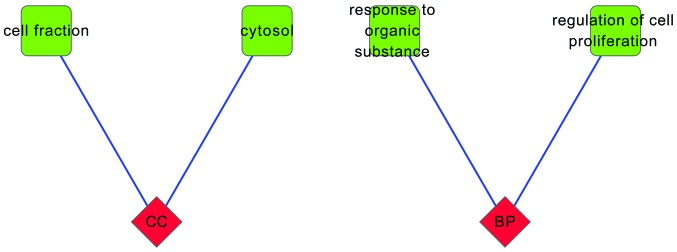
GO enrichment analysis of the downregulated colorectal cancer genes. Diamond, categories of GO terms; square, GO terms. GO, Gene Οntology. A blue line links a diamond (GO term) and a square (GO category). It means that the diamond (GO term) belongs a square (GO category).
As shown in Table I, the base excision repair, cell cycle, bladder cancer and p53 signaling pathways were critical for the upregulated CRC genes. The downregulated NAT1 and NAT2 genes were enriched in the caffeine metabolism pathway.
Table I.
KEGG pathways enriched with colorectal cancer genes.
| KEGG pathway | Gene | Change in expression |
|---|---|---|
| Caffeine metabolism | NAT1, NAT2 | Down |
| Base excision repair | PCNA, POLB, APEX1 | Up |
| Cell cycle | CCND1, PCNA, CHEK1, CHEK2 | Up |
| Bladder cancer | CCND1, IL8, MMP1 | Up |
| p53 signaling pathway | CCND1, CHEK1, CHEK2 | Up |
KEGG, Kyoto Encyclopedia of Genes and Genomes.
Identification of hub genes from the PPI network
A PPI network was constructed using the obtained CRC genes (Fig. 6). The two upregulated genes, CCND1 and (PCNA), were determined as the hub genes, as they were identified to exhibit the highest degrees of connection.
Figure 6.
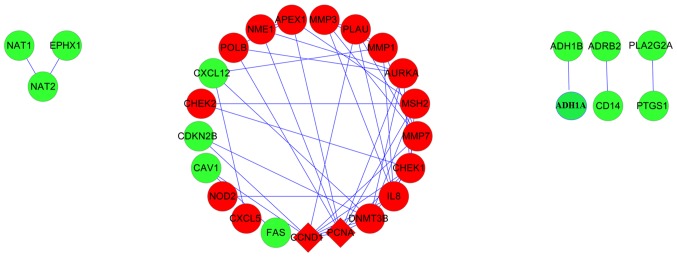
PPI network constructed using the colorectal cancer genes. Red round nodes, upregulated genes; green round nodes, downregulated genes; diamond nodes, hub genes. PPI, protein-protein interaction. A blue line indicates the interaction between two proteins that are linked by the blue line.
Analysis of CDS and protein domain of the hub genes
The CDSs and aa sequences of CCND1 and PCNA was obtained, following which the protein domains were retrieved for CCND1 and PCNA (Table II). According to the Conserved Domains database, the N-terminal and C-terminal domains of PCNA were topologically identical: Proliferating cell nuclear antigen, in which three PCNA molecules formed a closed ring zoning DNA duplex.
Table II.
Protein domains of CCND1 and PCNA.
| Coding-region | Family | Description | E-value |
|---|---|---|---|
| PCNA | PCNA_N | Proliferating cell nuclear antigen, N-terminal domain | 2.8e-63 |
| PCNA_C | Proliferating cell nuclear antigen, C-terminal domain | 1.5e-67 | |
| CCND1 | Cyclin_N | Cyclin, N-terminal domain | 1.8e-30 |
| Cyclin_C | Cyclin, C-terminal domain | 5.6e-22 |
CCND1, cyclin D1; PCNA, proliferating cell nuclear antigen.
However, the N-terminal and C-terminal domains of CCND1 were different. Cyclin_N: Cyclins modulated cyclin dependent kinases (CDKs) and human cyclin-O was a type of uracil-DNA glycosylase associated with other cyclins. Cyclins contain 2 domains of similar all-alpha fold, of which this family corresponds with the N-terminal domain. Cyclin_C: Cyclins regulated CDKs and human CCNO was a uracil-DNA glycosylase, linked to other cyclins. Cyclins contain 2 domains of similar all-alpha fold, of which this family corresponds with the C-terminal domain.
The spatial structures of the C-terminal and N-terminal domains of PCNA are shown in Figs. 7 and 8, respectively. For PCNA, the structure of the N-terminal domain was similar to that of the C-terminal domain. The C-terminal and N-terminal domains of CCND1 are shown in Figs. 9 and 10, respectively, in which the structure of the N-terminal domain of CCND1 differed from that of the C-terminal domain.
Figure 7.

Spatial structure of the C-terminal of proliferating cell nuclear antigen. The prominent colored frame represents the spatial structure of the protein domain corresponding to the coding region of the gene.
Figure 8.

Spatial structure of the N-terminal of proliferating cell nuclear antigen. The prominent colored frame represents the spatial structure of the protein domain corresponding to the coding region of the gene.
Figure 9.

Spatial structure of the C-terminal of cyclin D1. The prominent colored frame stands for the spatial structure of the protein domain corresponding to the coding region of the gene.
Figure 10.

Spatial structure of the N-terminal of cyclin D1. The prominent colored frame represents the spatial structure of the protein domain corresponding to the coding region of the gene.
Discussion
Although the mortality rates of CRC have reduced substantially, resulting from improved diagnosis and treatment of the disease, it remains a primary health concern, which is life threatening (27). The aim of the present study was to determine the key genes involved in CRC and their underlying mechanism. A total of 1,389 DEGs were screened out between the CRC and noncancerous samples. From the 1,389 identified DEGs, 35 CRC genes were further screened, which included 19 upregulated and 16 downregulated genes. GO enrichment analysis revealed that the downregulated CRC genes were predominately associated with response to organic substance and regulation of cell proliferation, whereas the upregulated CRC genes were predominantly involved in a number of biological functions pertaining to cell death and DNA repair. KEGG pathway analysis revealed that the downregulated CRC genes were enriched in the caffeine metabolism pathway, whereas the upregulated CRC genes were involved in base excision repair, cell cycle, bladder cancer and p53 signaling pathways. In the PPI network, CCND1 and PCNA were determined as hub genes, which had a number of interactions with other CRC genes. Subsequent structural analysis of the two hub genes revealed that the C-terminal and N-terminal domains were similar in PCNA, but different in CCND1.
It has been well-established that the primary function of N-acetyltransferase is in catalyzing the acetylation of arylamines and aromatic amines, as well as the detoxication of environmental toxins (28). NAT1 and NAT2 are two members of NAT family, of which NAT1 is distributed in almost all tissues, whereas NAT2 is detected predominantly in the liver. Although divergence exists in the distribution of NAT1 and NAT2, which locate on chromosome 8, the two are involved in metabolizing drugs and other xenobiotics, and are involved in folate catabolism and caffeine metabolism (29). In addition, the overexpression of NAT1 can result increase the survival rates of patients with cancer and has been recommended as a viable target for developing therapy against cancer (30). The association between NAT2 and CRC is a source of controversy. A meta-analysis, combining evidence from 40 studies, revealed that NAT2 phenotypes may not be linked to the progression of CRC (31). Another study demonstrated that people with fast NAT2 acetylator phenotypes are at increased risk of CRC (32). The present study found that NAT1 and NAT2 were critical downregulated genes for CRC and were predominantly enriched in the caffeine metabolism pathway. The conflicting results between these studies may be partly attributed to the specificity of population and inter-individual variations. A previous large retrospective study provided novel insight into the prevention of CRC, demonstrating that coffee intake appears to reduce the susceptibility to colon cancer, particularly in proximal cancer (33). These findings suggest that downregulated NAT1 and NAT2 may facilitate the initiation and development of CRC, through effects on the caffeine metabolism pathway.
Cyclin D1 is the protein encoded by CCND1 gene, belonging to the cyclin family that regulates CDKs. Cyclin D1 is involved in modulating cell cycle (34,35) and suggests the possibility that cyclin D1 may affect the development of CRC through modulation of the cancer cell cycle. Accumulating evidence has revealed that cyclin D1 polymorphisms may be associated with the prognosis of patients with advanced CRC, administrated with cetuximab, and confer susceptibility to CRC (36,37). However, a prospective, population-based study suggested a novel viewpoint that the expression of cyclin D1 is a preferred biomarker for predicting the prognosis of male patients with CRC, but not female patients (38). Furthermore, a previous bioinformatics study suggested that microRNA may exert an antitumor effect on colon cancer via targeting cyclin D1 (39). These studies demonstrated the importance of cyclin D1 in CRC. In agreement with the studies, the present study identified CCND1 as a hub gene in the PPI network, which exhibited numerous interactions with other genes, adding further support to suggestion that cyclin D1 is pivotal in CRC. The present study also demonstrated, through detailed analysis of the protein structure and spatial structure of the hub genes, that the N-terminal and C-terminal domains of CCND1 were topologically distinct. The protein domains of CCND1 may serve as targets for the development of novel therapies.
The PCNA protein, essential for DNA synthesis and DNA repair, resides in the nucleus and serves as a cofactor of DNA polymerase δ. Of particular concern is that PCNA is can cooperate with diverse partners in a coordinated manner, and is involved in several pathways, including DNA repair, DNA synthesis and cell cycle regulation (40). The expression of PCNA at the invasive tumor margin has been suggested to be positively correlated with the invasion and metastasis potential of CRC (41). In addition, it is also significantly associated with the liver metastasis of CRC (42). These evidence lead to a general acceptance that PCNA is implicated in CRC. Consistently, the present study provided evidence that PCNA was another hub gene of the PPI network, interacting with several other genes. Unlike CCND1, PCNA possessed almost identical N-terminal and C-terminal domains, allowing for the development of therapies that target the specific structure of PCNA.
In the present study, the upregulated PCNA and CCND1 were enriched in cell cycle pathway, consistent with the results of the GO enrichment analysis, suggesting that the upregulated genes were significantly linked to a variety of biological processes concerning cell and DNA metabolism, including cell cycle, cell cycle process, DNA repair and regulation of cell proliferation. In addition to the cell cycle pathway, CCND1 was also closely associated with bladder cancer and p53 signaling pathways. p53, a crucial tumor suppressor gene, acts as gatekeeper for cell growth and division, and the expression of p53 is associated with clinical outcomes and pathological characteristics of patients with CRC (43). Based on the findings of the present study, it was hypothesized that upregulated CCND1 may enhance the growth and proliferation of CRC cells through regulation of the cell cycle and p53 signaling pathways, and that upregulated PCNA promotes the progression of CRC via effects on the cell cycle pathway.
The present study had limitations due to the small sample size, therefore, further investigations with large sample sizes are warranted. Further in vivo and in vitro investigations are also required to validate the findings of the present study.
In conclusion, the present study found that PCNA, CCND1, NAT1 and NAT2 may be pivotal genes for CRC. The upregulation of PCNA and CCND1 may accentuate the development of CRC through regulation of the cell cycle and p53 signaling pathways, and downregulation of NAT1 and NAT2 may potentiate progression of the disease by the targeting caffeine metabolism pathway. In addition, the C-terminal and N-terminal domains were similar in PCNA, but different in CCND1. These findings suggested the potential use of these genes as molecular biomarkers in the early diagnosis and monitoring of CRC. They also offer potential in developing a range of potent therapies against CRC, which target the unique protein domains of PCNA and CCND1.
References
- 1.Stewart BW, Wild C. World Cancer Report 2014. World Health Organization; 2014. [Google Scholar]
- 2.Stegeman I, de Wijkerslooth TR, Stoop EM, van Leerdam ME, Dekker E, van Ballegooijen M, Kuipers EJ, Fockens P, Kraaijenhagen RA, Bossuyt PM. Colorectal cancer risk factors in the detection of advanced adenoma and colorectal cancer. Cancer Epidemiol. 2013;37:278–283. doi: 10.1016/j.canep.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Nelson H, Petrelli N, Carlin A, Couture J, Fleshman J, Guillem J, Miedema B, Ota D, Sargent D, National Cancer Institute Expert Panel Guidelines 2000 for colon and rectal cancer surgery. J Natl Cancer Inst. 2001;93:583–596. doi: 10.1093/jnci/93.8.583. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goyette MC, Cho K, Fasching CL, Levy DB, Kinzler KW, Paraskeva C, Vogelstein B, Stanbridge EJ. Progression of colorectal cancer is associated with multiple tumor suppressor gene defects but inhibition of tumorigenicity is accomplished by correction of any single defect via chromosome transfer. Mol Cell Biol. 1992;12:1387–1395. doi: 10.1128/mcb.12.3.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 7.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 8.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 9.Yoshie T, Nishiumi S, Izumi Y, Sakai A, Inoue J, Azuma T, Yoshida M. Regulation of the metabolite profile by an APC gene mutation in colorectal cancer. Cancer Sci. 2012;103:1010–1021. doi: 10.1111/j.1349-7006.2012.02262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voorneveld PW, Kodach LL, Jacobs RJ, Liv N, Zonnevylle AC, Hoogenboom JP, Biemond I, Verspaget HW, Hommes DW, de Rooij K, et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology. 2014;147:196–208. e13. doi: 10.1053/j.gastro.2014.03.052. [DOI] [PubMed] [Google Scholar]
- 11.Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–67. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khamas A, Ishikawa T, Shimokawa K, Mogushi K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H, Sugihara K. Screening for epigenetically masked genes in colorectal cancer Using 5-Aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genomics Proteomics. 2012;9:67–75. [PubMed] [Google Scholar]
- 13.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy - analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 14.Tibshirani R, Hastie T, Narasimhan B. samr: SAM: Significance Analysis of Microarrays. R package version 1.26. 2011 http://cran.r-project.org/web/packages/samr/index.html; Date accessed June 29, 2011.
- 15.Thissen D, Steinberg L, Kuang D. Quick and easy implementation of the Benjamini-Hochberg procedure for controlling the false positive rate in multiple comparisons. J Educ Behav Stat. 2002;27:77–83. doi: 10.3102/10769986027001077. [DOI] [Google Scholar]
- 16.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(3) [PubMed] [Google Scholar]
- 17.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. Gene Ontology Consortium: The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito R, Smoot ME, Ono K, Ruscheinski J, Wang P-L, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to Cytoscape plugins. Nat Methods. 2012;9:1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 23.Marasco E, Ross A, Dawson J, Moroose T, Ambrose T. Detecting STR peaks in degraded DNA samples; Proceedings of the 4th International Conference of Bioinformatics and Computational Biology; LasVegas, USA. 2012. pp. 1–8. [Google Scholar]
- 24.McEvoy CR, Seshadri R, Firgaira FA. Large DNA fragment sizing using native acrylamide gels on an automated DNA sequencer and GENESCAN software. Biotechniques. 1998;25:464–470. [PubMed] [Google Scholar]
- 25.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Geer LY, Chappey C, Kans JA, Bryant SH. Cn3D: sequence and structure views for Entrez. Trends Biochem Sci. 2000;25:300–302. doi: 10.1016/S0968-0004(00)01561-9. [DOI] [PubMed] [Google Scholar]
- 27.Jemal A, Ward E, Thun M. Declining death rates reflect progress against cancer. PLoS One. 2010;5:e9584. doi: 10.1371/journal.pone.0009584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans DA. N-acetyltransferase. Pharmacol Ther. 1989;42:157–234. doi: 10.1016/0163-7258(89)90036-3. [DOI] [PubMed] [Google Scholar]
- 29.Minchin RF, Hanna PE, Dupret J-M, Wagner CR, Rodrigues-Lima F, Butcher NJ. Arylamine N-acetyltransferase I. Int J Biochem Cell Biol. 2007;39:1999–2005. doi: 10.1016/j.biocel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Butcher NJ, Minchin RF. Arylamine N-acetyltransferase 1: A novel drug target in cancer development. Pharmacol Rev. 2012;64:147–165. doi: 10.1124/pr.110.004275. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Zhou J, Wang J, Liang G, Li J, Zhu Y, Su Y. Absence of association between N-acetyltransferase 2 acetylator status and colorectal cancer susceptibility: Based on evidence from 40 studies. PLoS One. 2012;7:e32425. doi: 10.1371/journal.pone.0032425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lilla C, Verla-Tebit E, Risch A, Jäger B, Hoffmeister M, Brenner H, Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- 33.Sinha R, Cross AJ, Daniel CR, Graubard BI, Wu JW, Hollenbeck AR, Gunter MJ, Park Y, Freedman ND. Caffeinated and decaffeinated coffee and tea intakes and risk of colorectal cancer in a large prospective study. Am J Clin Nutr. 2012;96:374–381. doi: 10.3945/ajcn.111.031328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 35.Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474:230–234. doi: 10.1038/nature10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Gordon M, Press OA, Rhodes K, Vallböhmer D, Yang DY, Park D, Fazzone W, Schultheis A, Sherrod AE, et al. Cyclin D1 and epidermal growth factor polymorphisms associated with survival in patients with advanced colorectal cancer treated with Cetuximab. Pharmacogenet Genomics. 2006;16:475–483. doi: 10.1097/01.fpc.0000220562.67595.a5. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y, Wang F, Shi C, Zou Y, Qin H, Ma Y. Cyclin D1 G870A polymorphism contributes to colorectal cancer susceptibility: Evidence from a systematic review of 22 case-control studies. PLoS One. 2012;7:e36813. doi: 10.1371/journal.pone.0036813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wangefjord S, Manjer J, Gaber A, Nodin B, Eberhard J, Jirström K. Cyclin D1 expression in colorectal cancer is a favorable prognostic factor in men but not in women in a prospective, population-based cohort study. Biol Sex Differ. 2011;2(10) doi: 10.1186/2042-6410-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nie J, Liu L, Zheng W, Chen L, Wu X, Xu Y, Du X, Han W. microRNA-365, downregulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis. 2012;33:220–225. doi: 10.1093/carcin/bgr245. [DOI] [PubMed] [Google Scholar]
- 40.Maga G, Hübscher U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 41.Teixeira CR, Tanaka S, Haruma K, Yoshihara M, Sumii K, Kajiyama G. Proliferating cell nuclear antigen expression at the invasive tumor margin predicts malignant potential of colorectal carcinomas. Cancer. 1994;73:575–579. doi: 10.1002/1097-0142(19940201)73:3<575::AID-CNCR2820730313>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 42.Sumiyoshi Y, Yamashita Y, Maekawa T, Sakai N, Shirakusa T, Kikuchi M. Expression of CD44, vascular endothelial growth factor, and proliferating cell nuclear antigen in severe venous invasional colorectal cancer and its ionship to liver metastasis. Surg Today. 2000;30:323–327. doi: 10.1007/s005950050594. [DOI] [PubMed] [Google Scholar]
- 43.Yu SJ, Yu JK, Ge WT, Hu HG, Yuan Y, Zheng S. SPARCL1, Shp2, MSH2, E-cadherin, p53, ADCY-2 and MAPK are prognosis-related in colorectal cancer. World J Gastroenterol. 2011;17:2028–2036. doi: 10.3748/wjg.v17.i15.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]