

Abstract
The localization of atherosclerotic lesion formation to regions of disturbed blood flow associated with certain arterial geometries, in humans and experimental animals, suggests an important role for hemodynamic forces in the pathobiology of atherosclerosis. There is increasing evidence that the vascular endothelium, which is directly exposed to various fluid mechanical forces generated by pulsatile blood flow, can discriminate among these different biomechanical stimuli and transduce them into genetic regulatory programs that modulate endothelial function. In this brief review, we discuss how biomechanical stimuli generated by blood flow can influence endothelial functional phenotypes, and explore the working hypothesis of “atheroprone” hemodynamic environments as “local risk factors” in atherogenesis. In addition, we consider the therapeutic implications of the activation of “atheroprotective genes” and their role as “critical regulatory nodes” in vascular homeostasis.
Keywords: Atherosclerosis, Endothelial pathobiology, Hemodynamic forces, Genomic analysis, Atheroprotective genes, Kruppel-like factor 2
1. Introduction
Atherosclerosis, the most common form of large vessel pathology responsible for syndromes of vital organ ischemic damage (e.g., myocardial infarction and stroke), remains a leading cause of mortality and morbidity in industrialized societies. Although its histopathologic features have been appreciated since the time of Virchow [1], mechanistic insights into its pathogenesis, at the cellular and molecular level, have been gained only relatively recently. While early pathogenetic theories (e.g., “lipid insudation” and “fibrin incrustation”) focused on the prominent microscopic features of advanced lesions, more recent working hypotheses have utilized the tools of modern cell biology and molecular genetics to probe the pathogenetic mechanisms of fibromuscular scarring and lipid accumulation that ultimately result in plaque instability and thrombotic sequelae [2–5].
An important organizing principle that has emerged is that the vascular endothelium is a dynamically mutable interface–whose structural and functional properties are responsive to a variety of stimuli, both local and systemic, and that its phenotypic modulation to a dysfunctional state can constitute a major risk factor for vascular diseases such as atherosclerosis [6]. Manifestations of endothelial dysfunction extend well beyond altered nitric oxide metabolism and vascular reactivity, and encompass increased lipoprotein permeability and oxidation, enhanced mononuclear leukocyte adhesion and intimal accumulation, altered extracellular matrix metabolism, and dysregulation of the hemostatic–thrombotic balance. Pathophysiologic stimuli of arterial endothelial dysfunction that are especially relevant to atherogenesis include proinflammatory cytokines, bacterial products and viruses, advanced glycation end products generated in diabetes and aging, hypercholesterolemia (per se), as well as oxidized lipoproteins and their components (e.g., lyso-phosphatidyl-choline) that accumulate within the arterial wall [7]. In addition to these biochemical stimuli, it is now clear that biomechanical forces, generated by flowing blood, can also influence the structure and function of endothelial cells and, remarkably, can act, at the level of complex transcriptional regulation, to orchestrate the pattern of expression of pathophysiologically relevant genes in atherogenesis [8].
The notion that hemodynamic forces can function as pathophysiologic stimuli for endothelial dysfunction provides a conceptual basis for the long-standing observation that the earliest lesions of atherosclerosis develop in a distinctive, nonrandom pattern, the geometry of which correlates with branch points and other regions of altered blood flow [9,10]. This strikingly localized pattern of lesion formation, across various species and in the face of systemic risk factors such as elevated plasma cholesterol, has intrigued experimental pathologists and fluid mechanical engineers alike, for decades, and has motivated the search for mechanistic links between hemodynamic forces and atherogenesis. In this brief review, we highlight our current understanding of the interrelationships of hemodynamics, endothelial pathobiology, and atherogenesis, with a view to future therapeutic implications.
2. Hemodynamics, endothelium, and vascular pathobiology
The pulsatile flow of blood through the branched tubular geometry of the arterial vasculature generates various types of hemodynamic forces–wall shear stresses, hydrostatic pressures, and cyclic strains–that can impact vessel wall biology [11]. As the cellular layer in intimate contact with blood, the endothelium in particular bears the frictional forces (wall shear stresses) imparted by the flow of this viscous fluid. Blood flow patterns can vary from the relatively uniform (time-averaged) well-developed laminar flows that occur in the unbranched portions of medium-sized arteries, to the complex disturbed laminar flow patterns (involving regions of flow separation, recirculation, and reattachment) that result in significant temporal and spatial gradients of wall shear stresses over relatively short distances [12]. The latter disturbed laminar flow patterns occur near branch points, bifurcations, and major curvatures–arterial geometries that are typically associated with the earliest appearance (and subsequent progression) of atherosclerotic lesions (Fig. 1A). In contrast, the unbranched, tubular portions of arteries that carry uniform laminar flow typically are relatively protected from atherogenesis (at least at the early stages of disease) [13,14]. For many years, the common wisdom therefore held that “low shear” areas (e.g., complex geometries in which the time-averaged fluctuations in wall shear stresses were small, due to forward–reverse flow cycles) were especially atherosclerosis susceptible, whereas “high shear” areas were relatively atherosclerosis resistant (Fig. 1B). Interestingly, this nonrandom pattern of lesion development is observed not only in various experimental animal models (dietary and/or genetic), across multiple animal species, but also is a feature of the natural history of atherosclerotic disease in humans, thus underscoring its potential pathogenetic significance [15].
Fig. 1.
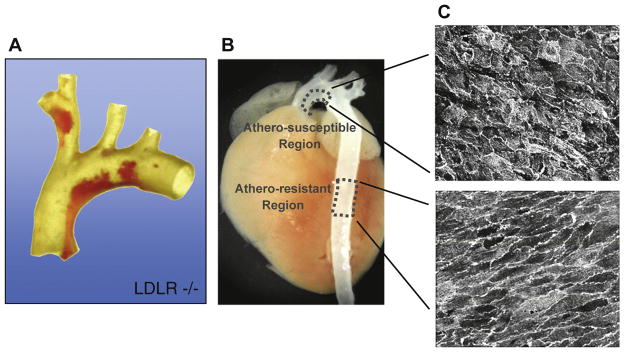
Nonrandom pattern of early atherosclerotic lesion development in mouse aorta. (A) Dissected aortic arch from a LDL receptor-deficient (LDLR−/−) mouse fed a cholesterol-rich diet stained with oil red-O to mark early atherosclerotic lesions (adapted from Ref. [65]). (B) Location of “atherosclerosis-resistant” and “atherosclerosis-susceptible” regions, indicated on an intact mouse aorta, corresponding to the descending thoracic aorta and the lesser curvature of the aortic arch, respectively. (C) En face confocal microscopy of the endothelium of these regions showing distinct cell shapes (cell junctions stained for CD31).
Various in vivo observations suggest that the structure and function of the endothelial lining is modulated by hemodynamic forces. Strikingly, endothelial cell morphology appears to reflect local flow conditions, with ellipsoidal cell (and nuclear) shape and coaxial alignment in the primary flow direction seen in laminar flow regions, and disruption of this orderly pattern in regions of disturbed flow [16,17] (Fig. 1C). Surgical manipulation of vascular architecture (e.g., the creation of coarctations or arteriovenous shunts) results in acute and chronic changes in the vessel wall that appear (at least in part) to be endothelial dependent. And, in the presence of hypercholesterolemia, these surgically modified vascular geometries can also develop atherosclerotic-like lesions [18]. While these in vivo observations are consistent with an effect of blood flow on arterial wall (patho)biology, evidence of the direct action of hemodynamic forces on endothelium has come primarily from in vitro studies. In the early 1980s, our group, in collaboration with colleagues in Fluid Mechanical Engineering at the Massachusetts Institute of Technology, utilized a modified cone-plate viscometer to subject cultured human and animal endothelial cells to defined fluid mechanical stimulation [19] and began to explore the resultant changes in endothelial structure and function [20]. Our early studies established that unidirectional, steady laminar shear stresses could induce time- and force-dependent cell-shape changes and alignment that mimicked those observed in the arterial vasculature in vivo [21]. Further studies by our group, and several others, went on to document that hemodynamic forces could significantly influence a spectrum of the vital properties of vascular endothelium that encompass its functions as a blood-compatible container, a selectively permeable barrier, and a metabolically active cellular component of the arterial wall [22]. This essentially established a new paradigm in vascular (patho)biology–biomechanical regulation of endothelial phenotype [23].
3. Endothelial gene regulation by biomechanical forces
In addition to eliciting immediate responses (e.g., the secretion of biologically active endothelial metabolites such as prostacyclin and nitric oxide via enzymatic regulation), in vitro applied shear stresses appeared to strongly influence endothelial cell gene expression [11]. Mechanistic analyses of this phenomenon revealed the existence of “shear-stress response elements” in the promoters of pathophysiologically relevant genes, such as the platelet-derived growth factor and vascular cell adhesion molecule-1 (VCAM-1), that acted to up- or down-regulate gene transcription [24,25]. Other studies revealed the presence of various “mechano-transducers” and downstream signaling pathways that link the externally applied mechanical stimuli to intracellular and, ultimately, intranuclear events [12,26,27]. Interestingly, individual genes as well as groups of multiple genes appeared to be differentially responsive to the spatial and temporal properties of applied shear stresses–suggesting a complex and dynamic system of biomechanical endothelial gene regulation.
A major logistical step forward was the move from single (candidate) gene analyses to high-throughput molecular biological techniques that could reveal patterns of coordinated gene regulation. Of particular interest were those genes whose level of expression was up-regulated in a sustained fashion by steady laminar shear stress stimulation. This approach indeed yielded a striking result–steady laminar shear stresses (but not turbulence) differentially up-regulated several endothelial genes with special relevance to atherogenesis, in particular eNOS (the endothelial isoform of nitric oxide synthase), COX-2 (the inducible isoform of cyclooxygenase), and manganese-dependent superoxide dismutase [28]. These endothelial genes encode enzymes that exert potent antithrombotic, antiadhesive, antiproliferative, anti-inflammatory, and antioxidant effects, both within the intimal lining and in interacting cells, such as platelets, leukocytes, and vascular muscle. Given the well-established observation that uniform laminar shear stresses are characteristically associated with atherosclerotic lesion-protected arterial geometries in vivo, this coordinated pattern of gene expression led us to hypothesize that this type of biomechanical stimulation acts to chronically up-regulate the expression of a set of “atheroprotective genes” in lesion-protected areas of the endothelium that then act locally to offset the effects of systemic risk factors, such as hypercholesterolemia, hyperglycemia, and hypertension.
The critical testing of this “atheroprotective gene hypothesis” required three significant refinements in our experimental strategy–first, a detailed characterization of the actual near-wall shear stress profiles in “atheroprone” and “atheroprotected” arterial geometries; second, the recreation of these complex flow regimes, with spatial and temporal fidelity, on the surface of cultured human endothelial monolayers; and third, a genome-wide comparative analysis of the resultant transcriptomes.
4. Atheroprone vs. atheroprotective endothelial phenotypes
In order to create this integrated experimental strategy, our laboratory first established platforms to perform genome-wide transcriptional profiling using microarrays coupled with several bioinformatics tools used for the systematic analyses of the resultant large datasets [29–31]. An in vitro model system was then created in which cultured human endothelial cells can be exposed to well-defined shear stress waveforms that accurately simulate those present in vivo [32]. Finally, the flow patterns in the carotid artery bifurcation of normal human subjects were analyzed using a 3D fluid dynamic analysis based on actual vascular geometries and measured blood flow profiles [33]. From this characterization, two prototypic arterial waveforms, atheroprone and atheroprotective, were defined as representative of the wall shear stresses in two distinct regions of the carotid artery (the carotid sinus and the distal internal carotid artery) that are typically “susceptible” or “resistant,” respectively, to atherosclerotic lesion development [33] (Fig. 2). With this novel biomechanical model system, human cultured endothelial cells exposed to wall shear stresses that mimic those present in atherosclerosis-susceptible arterial geometries in vivo acquired a pro-inflammatory phenotype, expressing several physiologically important chemokines and chemokine receptors. Exposure to this atheroprone waveform stimulation also induced a dysregulation of the expression and organization of cytoskeletal and junctional proteins, and the activation of the NF-κB signaling pathway [33]. These observations are in agreement with the atherosusceptible endothelial phenotype documented by other investigators using murine and swine models [34,35]. In contrast, preconditioning of human endothelial cells with atheroprotective waveform curtailed the IL-1β-inducible VCAM-1 expression [33]. Taken together, these data provided a link between certain forms of biomechanical stimulation, endothelial gene expression, and cellular and molecular changes associated with the pathogenesis of atherosclerosis. Moreover, analysis of transcriptional profiling data derived from the comparison between human endothelial cells exposed to these atheroprone vs. atheroprotective waveforms strongly indicated that a complex program of gene expression was being coordinately orchestrated, thus suggesting transcriptional activators/repressors in the endothelium that are biomechanically responsive. Using hierarchical clustering and other parallel analyses, we established that at least 48 distinct transcription factors were biomechanically regulated. Of these, the gene most highly differentially regulated between the two waveforms was the zinc finger transcription factor Kruppel-like factor 2 (KLF2) [33].
Fig. 2.

A novel in vitro biomechanical model system for the analysis of atheroprotective and atheroprone endothelial phenotypes in human endothelial cells. Computational fluid mechanical analysis of flow patterns of the normal human carotid artery yielded two prototypic arterial waveforms–“atheroprotective” and “atheroprone,” representative of the wall shear stresses of the carotid sinus and the distal internal carotid artery, which are typically “susceptible” or “resistant,” respectively, to atherosclerosis. These biomechanical stimuli were recreated on the surface of cultured human endothelial monolayers in a dynamic flow system, and the resultant transcriptomes were subjected to detailed bioinformatic analyses.
5. KLF2 as an integrator of the atheroprotective flow-mediated endothelial cell phenotype
Members of the Kruppel transcription factor family are characterized by a carboxy terminus containing three DNA-binding C2H2 zinc fingers. To date, 17 human members of this family have been identified, and several of them have been reported to regulate vascular functions [36]. In particular, KLF2 has been implicated in T-lymphocyte quiescence [37], lung development [38], and adipogenesis [39,40]. Within the murine vessel wall, KLF2 expression appears to be endothelial restricted, and generation of KLF2-null mouse embryos resulted in embryonic hemorrhaging and lethality at approximately E12.5–E14.5 [37,41]. The apparent cause of this phenotype was impairment in smooth muscle cell recruitment to nascent endothelial tubes, implying a possible paracrine effect of KLF2-dependent, endothelial-derived products. Deletion of KLF2 in the endothelial compartment in mice also resulted in embryonic lethality due to heart failure caused by high cardiac output [42]. Interestingly, in vitro studies have shown that KLF2 is one of a small number of genes up-regulated in human endothelial cells exposed to laminar shear stress for 7 days when compared to static (no flow) culture conditions [43]. Using in situ hybridization of adult human arteries, these authors demonstrated that KLF2 expression was also restricted to the endothelium within the human vessel wall [43]. The response of KLF2 expression to laminar shear stress in vitro has been confirmed by other groups, including our own [44,45]. Dissection of the basic biomechanical parameters relevant to KLF2 induction revealed an increased effect of pulsatile flow compared to non-pulsatile, steady laminar flow, and a lack of KLF2 response to cyclic stretch [46,47].
In vivo studies by our group using silent heart mutant zebrafish, which lack blood flow yet remain viable for several days, demonstrated the absence of vascular endothelial KLF2a (the homolog of the mammalian KLF2) expression in the aorta and cardinal vein [48]. These observations thus demonstrated that endothelial KLF2 expression is dependent on blood flow in vivo and that this mode of regulation may be evolutionarily conserved. Further in vitro analyses performed in our laboratory revealed that the mechanisms linking hemodynamic forces and KLF2 expression involve activation of a MEK5/ERK5/MEF2 pathway and that MEK5 activation is necessary and sufficient for the flow-mediated up-regulation of KLF2 in vascular endothelium. These results are consistent with a model in which flow activates MEK5, which in turn phosphorylates ERK5, resulting in the activation of the MEF2 family of transcription factors at the KLF2 promoter [48].
Collectively, these observations raised some important questions. For example, what are the flow-mediated downstream targets of KLF2 in the endothelium? And, how does expression of these genes impact endothelial phenotype? Experiments performed in our laboratory and others have documented that KLF2 regulates a vast array of genes of major functional importance in the endothelium, and it has become apparent that the sum of these complex actions confers a phenotype that promotes endothelial homeostasis and an anti-atherogenic phenotype [48,49]. Thus, for example, KLF2 overexpression in cultured human endothelial cells was shown to inhibit IL-1β-dependent induction of the pro-inflammatory adhesion molecules VCAM-1 and E-selectin, and to promote the expression of eNOS [45]. Subsequent studies have revealed an important role for KLF2 in regulating certain genes involved in thrombosis and hemostasis [50]. Moreover, using a systems biology approach combined with gain-and loss-of-function experiments, we showed that KLF2 acts as a critical integrator of the global transcriptional responses of endothelial cells to atheroprotecive flow. Specifically, we demonstrated that the atheroprotective flow-mediated induction of KLF2 results in the orchestrated regulation of endothelial transcriptional programs controlling inflammation, thrombosis/hemostasis, and vascular tone [48]. As a consequence of these coordinated transcriptional changes, we also demonstrated that KLF2 is necessary to evoke the endothelial functional phenotype triggered by atheroprotective shear stress. For example, silencing KLF2 results in an increase in leukocyte adhesion to endothelial monolayers and the loss of protection against oxidative stress in endothelial cells preconditioned with atheroprotective flow [48].
The identification of KLF2-dependent transcriptional programs involved in the regulation of several endothelial functions contributing to a vasoprotective phenotype indicates a critical role for this transcription factor in maintaining the functional integrity of the vascular endothelium (Fig. 3). Furthermore, the requirement for KLF2 expression in the atheroprotective flow-mediated phenotype, together with the selective expression of KLF2 in atheroprotective regions of human arteries, strongly suggests that the focal nature of atherosclerosis and its long-established correlation with particular hemodynamic environments might be explained in part by spatial patterns of KLF2 expression.
Fig. 3.

Critical regulatory nodes in vascular homeostasis. Induction of the transcription factors KLF2 and Nrf2 by atheroprotective flow orchestrates a multifunctional genetic program, the net effects of which contribute to the maintenance of endothelial vasoprotective phenotypes. Statins, and potentially other yet-to-be-defined agents, through their induction of KLF2 can function as pharmacomimetics of atheroprotective flow.
While the importance of KLF2 for endothelial integrity in vivo remains to be fully elucidated, several lines of evidence do suggest that KLF2 expression is linked to atheroprotection. First, ApoE-null mice bearing a hemizygous deficiency in KLF2 exhibited a 31–37% increase in atherosclerotic lesion area relative to littermate controls [51,52]; second, myeloid cell-specific KLF2 inactivation leads to an increase in atherosclerotic lesion formation in the LDLR−/− murine model [52]; and, third, suppressing the expression of KLF2 in T cells and monocytes leads to a pro-inflammatory phenotype of these cell types [53,54].
6. Other endothelial transcription factors involved in flow-mediated atheroprotection
To explore whether additional mechanisms are responsible for flow-mediated endothelial atheroprotection, we sought to further characterize the transcriptional activators responsible for flow-dependent resistance to oxidative challenges that we had previously observed [33]. To this end, we analyzed the gene expression programs of human endothelial cells exposed to atheroprotective or atheroprone waveforms and found that atheroprotective flow up-regulated certain antioxidant genes and strongly activated the transcription factor nuclear factor erythroid-2-related factor-2 (Nrf2). In particular, we identified several redox-related genes, including heme oxygenase 1, ferritin heavy chain, NAD(P)H dehydrogenase quinone 1, and thioredoxin reductase, as being dependent on Nrf2 in the context of atheroprotective flow (Fig. 3). In addition, we showed that atheroprotective flow activates Nrf2 via the PI3K/Akt pathway and that this activation occurs differentially in atherosclerosis-resistant and atherosclerosis-susceptible regions of the mouse aorta [55]. Recent work by other laboratories indicates that KLF2 and Nrf2 act in a synergistic manner to activate the expression of a substantial fraction of atheroprotective flow-dependent gene expression [56]. Mechanistically, this synergy is based on the effects of KLF2 expression on the activation of Nrf2 and its antioxidant downstream targets. Taken together, the documented effects of the expression of KLF2 and Nrf2 in the vascular endothelium strongly suggest that these transcription factors are acting as “critical regulatory nodes” in vascular homeostasis and, as such, may be relevant targets for pharmacological intervention [57]. This notion has been recently supported by experimental evidence indicating that expression of KLF2 in the vascular endothelium triggers the production of extracellular vesicles containing microRNAs (miRNAs). Notably, some of these miRNAs appear to act in a paracrine manner on adjacent smooth muscle cells and are capable of reducing atherosclerosis in the murine ApoE−/− model [58].
7. Statins exert endothelial atheroprotective effects via KLF2
Based on the similarity between KLF2-dependent transcriptional and functional effects observed in endothelial cells exposed to atheroprotective flow, and those previously reported to be exerted by statins, an important class of cardiovascular drugs, we investigated a potential link between these biomechanical and pharmacologic stimuli. This investigation led our group, and others, to discover that statins, the most commonly prescribed class of lipid-lowering agents, originally designed as selective inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, act to up-regulate KLF2 expression in cultured human endothelial cells at pharmacologically relevant doses in vitro [59,60] (Fig. 3). Statins block the production of mevalonate, which forms two major downstream products known as isoprenoids: farnesyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP). These isoprenoids can both prenylate distinct sets of proteins in the cells to enable their proper subcellular targeting and signaling. The statin-mediated up-regulation of KLF2 in human endothelial cells is dependent on the depletion of GGPP, which is well known to prenylate several members of the Rho superfamily. Importantly, up-regulation of KLF2 is critical for the many statin-dependent transcriptional changes in endothelial cells, thus implicating KLF2 in the so-called “pleiotropic” beneficial cardiovascular effects of this class of cardiovascular drugs [61]. The endothelial atheroprotective effects of statins in our in vitro model system are dose dependent and appear to be class dependent, rather than agent specific [59]. Importantly, direct comparisons between laminar shear stress and statins as inducers of KLF2 in the context of pro-inflammatory stimuli indicated that shear stress is a stronger stimulus. This seems to be due to differences in the post-transcriptional stability of KLF2 mRNA, since the induction of the gene by the two stimuli is very similar [62]. This observation might be explained by the recently identified microRNA-92a-dependent regulation of KLF2 mRNA in the context of atheroprotective flow [63,64].
8. Future directions
Collectively, the findings of the past decade linking endothelial atheroprotection, biomechanics, and the pathobiology of atherosclerosis have provided a new conceptual framework to better understand the pathophysiology of endothelial cell dysfunction and atherogenesis. This conceptual framework has defined specific hemodynamic environments as “local risk factors” for atherogenesis and has led to the identification of mechano-activated endothelial signaling pathways critical for vascular homeostasis. In particular, the identification of “critical regulatory ” such as KLF2 and Nrf2, has nodes, provided new perspectives on the pathogenesis of cardiovascular disease and potential novel therapeutic strategies (Fig. 3). For example, it will be interesting to define whether mutations in coding sequences or regulatory regions of KLF2 and Nrf2 in human populations confer an increased risk for cardiovascular events, and, in the case of KLF2, whether such mutations modify the beneficial effects of statins. Importantly, the endothelial expression of KLF2 could be used in high-throughput screens designed to identify novel inducers with the ultimate goal of developing new classes of pharmacologic agents designed to mimic flow-mediated atheroprotection. Finally, it is intriguing to consider whether the hemodynamic regulation of endothelial phenotypes may have implications beyond the pathobiology of atherosclerosis, including various congenital and acquired vasculopathies, as well as fundamental aspects of embryonic development of the cardiovascular system.
Acknowledgments
The authors’ research summarized here was supported primarily by grants from the National Heart, Lung and Blood Institute of the National Institutes of Health.
We would like to acknowledge the past and present members of our laboratories for their contributions to the original studies described here and also the long-standing collaboration of Professor C.F. Dewey and his colleagues in the Fluid Mechanics Laboratory at the Massachusetts Institute of Technology.
References
- 1.Virchow R. Der ateromatose Prozess der Arterien. Wien Med Wochenshr. 1856;6:825–41. [Google Scholar]
- 2.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 3.Ross R. Atherosclerosis–an inflammatory disease. N Engl Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 5.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 6.Gimbrone MA., Jr . Endothelial dysfunction and the pathogenesis of atherosclerosis. In: Gotto VAM, editor. Atherosclerosis. New York: Spring; 1980. pp. 415–55. [Google Scholar]
- 7.Gimbrone MA., Jr . Vascular endothelium in health and disease. In: Harber E, editor. Molecular Cardiovascular Medicine. New York: Scientific American Medicine; 1995. pp. 49–61. [Google Scholar]
- 8.Gimbrone MA., Jr . Foreword. In: Lelkes PI, editor. Mechanical forces and the endothelium. London: Hardwood Academic Publishers; 1999. pp. VII–X. [Google Scholar]
- 9.Cornhill JF, Roach MR. A quantitative study of the localization of atherosclerotic lesions in the rabbit aorta. Atherosclerosis. 1976;23:489–501. doi: 10.1016/0021-9150(76)90009-5. [DOI] [PubMed] [Google Scholar]
- 10.Glagov S, Zarins C, Giddens DP, Ku DN. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch Pathol Lab Med. 1988;112:1018–31. [PubMed] [Google Scholar]
- 11.Topper JN, Gimbrone MA., Jr Blood flow and vascular gene expression: fluid shear stress as a modulator of endothelial phenotype. Mol Med Today. 1999;5:40–6. doi: 10.1016/s1357-4310(98)01372-0. [DOI] [PubMed] [Google Scholar]
- 12.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb Vasc Biol. 1993;13:1291–8. doi: 10.1161/01.atv.13.9.1291. [DOI] [PubMed] [Google Scholar]
- 16.Nerem RM, Levesque MJ, Cornhill JF. Vascular endothelial morphology as an indicator of the pattern of blood flow. J Biomech Eng. 1981;103:172–6. doi: 10.1115/1.3138275. [DOI] [PubMed] [Google Scholar]
- 17.Karino T. Microscopic structure of disturbed flows in the arterial and venous systems, and its implication in the localization of vascular diseases. International Angiol: A Jof the Int Angiol. 1986;5:297–313. [PubMed] [Google Scholar]
- 18.Joris I, Zand T, Majno G. Hydrodynamic injury of the endothelium in acute aortic stenosis. Am J Pathol. 1982;106:394–408. [PMC free article] [PubMed] [Google Scholar]
- 19.Bussolari SR, Dewey CF, Gimbrone MA., Jr Apparatus for subjecting living cells to fluid shear stress. Rev Sci Instrum. 1982;53:1851–4. doi: 10.1063/1.1136909. [DOI] [PubMed] [Google Scholar]
- 20.Dewey CF, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–85. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 21.Remuzzi A, Dewey CF, Jr, Davies PF, Gimbrone MA., Jr Orientation of endothelial cells in shear fields in vitro. Biorheology. 1984;21:617–30. doi: 10.3233/bir-1984-21419. [DOI] [PubMed] [Google Scholar]
- 22.Gimbrone MA, Jr, Topper JN. Biology of the vessel wall: endothelium. In: Chien KR, Troy MO, editors. Molecular Basis of Heart Diseases. Harcourt Brace & Co; 1998. pp. 331–48. [Google Scholar]
- 23.Gimbrone MA, Jr, Nagel T, Topper JN. Biomechanical activation: an emerging paradigm in endothelial adhesion biology. J Clin Invest. 1997;99:1809–13. doi: 10.1172/JCI119346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Resnick N, Collins T, Atkinson W, Bonthron DT, Dewey CF, Gimbrone MA., Jr Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive element. Proc Natl Acad Sci U S A. 1993;90:7908. doi: 10.1073/pnas.90.16.7908-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korenaga R, Ando J, Kosaki K, Isshiki M, Takada Y, Kamiya A. Negative transcriptional regulation of the VCAM-1 gene by fluid shear stress in murine endothelial cells. Am J Physiol. 1997;273:C1506–15. doi: 10.1152/ajpcell.1997.273.5.c1506. [DOI] [PubMed] [Google Scholar]
- 26.Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15:1405–14. doi: 10.1089/ars.2010.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topper JN, Cai J, Falb D, Gimbrone MA., Jr Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci U S A. 1996;93:10417–22. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Cardena G, Comander J, Anderson KR, Blackman BR, Gimbrone MA., Jr Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci U S A. 2001;98:4478–85. doi: 10.1073/pnas.071052598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comander J, Weber GM, Gimbrone MA, Jr, Garcia-Cardena G. Argus–a new database system for Web-based analysis of multiple microarray data sets. Genome Res. 2001;11:1603–10. doi: 10.1101/gr.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comander J, Natarajan S, Gimbrone MA, Jr, Garcia-Cardena G. Improving the statistical detection of regulated genes from microarray data using intensity-based variance estimation. BMC Genomics. 2004;5:17. doi: 10.1186/1471-2164-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blackman BR, Garcia-Cardena G, Gimbrone MA., Jr A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J Biomech Eng. 2002;124:397–407. doi: 10.1115/1.1486468. [DOI] [PubMed] [Google Scholar]
- 33.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, García-Cardeña G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci U S A. 2004;101:14871–6. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–7. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A. 2004;101:2482–7. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T, Aizawa K, Matsumura T, Nagai R. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler Thromb Vasc Biol. 2005;25:1135–41. doi: 10.1161/01.ATV.0000165656.65359.23. [DOI] [PubMed] [Google Scholar]
- 37.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Gen Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wani MA, Wert SE, Lingrel JB. Lung Kruppel-like factor, a zinc finger transcription factor, is essential for normal lung development. J Biol Chem. 1999;274:21180–5. doi: 10.1074/jbc.274.30.21180. [DOI] [PubMed] [Google Scholar]
- 39.Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, Kawahara R, Hauner H, Jain MK. The Kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. 2003;278:2581–4. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Srinivasan SV, Neumann JC, Lingrel JB. The KLF2 transcription factor does not affect the formation of preadipocytes but inhibits their differentiation into adipocytes. Biochemistry. 2005;44:11098–105. doi: 10.1021/bi050166i. [DOI] [PubMed] [Google Scholar]
- 41.Wani MA, Means RT, Jr, Lingrel JB. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res. 1998;7:229–38. doi: 10.1023/a:1008809809843. [DOI] [PubMed] [Google Scholar]
- 42.Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, Graf T, MacRae CA, Lepore JJ, Lo CW, Kahn ML. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev Cell. 2006;11:845–57. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJG. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2) Blood. 2002;100:1689–98. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 44.Huddleson JP, Srinivasan S, Ahmad N, Lingrel JB. Fluid shear stress induces endothelial KLF2 gene expression through a defined promoter region. Biol Chem. 2004;385:723–9. doi: 10.1515/BC.2004.088. [DOI] [PubMed] [Google Scholar]
- 45.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, García-Cardeña G, Jain MK. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–15. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJM, Biessen EAL, van Berkel TJC, Pannekoek H, Horrevoets AJG. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609–18. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, Young A, Yuan S, Nguyen P, Wu CC, Chien S. Shear stress regulation of Kruppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun. 2006;341:1244–51. doi: 10.1016/j.bbrc.2006.01.089. [DOI] [PubMed] [Google Scholar]
- 48.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, García-Cardeña G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–63. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 50.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, García-Cardeña G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 51.Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–3. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lingrel JB, Pilcher-Roberts R, Basford JE, Manoharan P, Neumann J, Konaniah ES, Srinivasan R, Bogdanov VY, Hui DY. Myeloid-specific kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ Res. 2012;110:1294–302. doi: 10.1161/CIRCRESAHA.112.267310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bu D-x, Tarrio M, Grabie N, Zhang Y, Yamazaki H, Stavrakis G, Maganto-Garcia E, Pepper-Cunningham Z, Jarolim P, Aikawa M, García-Cardeña G, Lichtman AH. Statin-induced Krüppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J Clin Invest. 2010;120:1961–70. doi: 10.1172/JCI41384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A. 2006;103:6653–8. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA., Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–33. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 56.Fledderus JO, Boon RA, Volger OL, Hurttila H, Yla-Herttuala S, Pannekoek H, Levonen AL, Horrevoets AJ. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1339–46. doi: 10.1161/ATVBAHA.108.165811. [DOI] [PubMed] [Google Scholar]
- 57.Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–13. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- 58.Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJG, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–56. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 59.Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, García-Cardeña G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–9. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
- 60.Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–6. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- 61.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 62.van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, Kootstra NA, Pannekoek H, Horrevoets AJ. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc Res. 2006;72:231–40. doi: 10.1016/j.cardiores.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 63.Wu W, Xiao H, Laguna-Fernandez A, Villarreal G, Wang K-C, Geary GG, Zhang Y, Wang W-C, Huang H-D, Zhou J, Li Y-S, Chien S, García-Cardeña G, Shyy JY-J. Flow-dependent regulation of kruppel-like factor 2 is mediated by MicroRNA-92a. Circulation. 2011;124:633–41. doi: 10.1161/CIRCULATIONAHA.110.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol. 2012;32:979–87. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Won D, Zhu SN, Chen M, Teichert AM, Fish JE, Matouk CC, Bonert M, Ojha M, Marsden PA, Cybulsky MI. Relative reduction of endothelial nitric-oxide synthase expression and transcription in atherosclerosis-prone regions of the mouse aorta and in an in vitro model of disturbed flow. Am J Pathol. 2007;171:1691–704. doi: 10.2353/ajpath.2007.060860. [DOI] [PMC free article] [PubMed] [Google Scholar]