

Abstract
Human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs) offer unprecedented opportunities to study inherited heart conditions in vitro, but are phenotypically immature, limiting their ability to effectively model adult-onset diseases. Cardiomyopathy is becoming the leading cause of death in patients with Duchenne muscular dystrophy (DMD), but the pathogenesis of this disease phenotype is not fully understood. Therefore, we aimed to test whether biomimetic nanotopography could further stratify the disease phenotype of DMD hiPSC-CMs to create more translationally relevant cardiomyocytes for disease modeling applications. We found that anisotropic nanotopography was necessary to distinguish structural differences between normal and DMD hiPSC-CMs, as these differences were masked on conventional flat substrates. DMD hiPSC-CMs exhibited a diminished structural and functional response to the underlying nanotopography compared to normal cardiomyocytes at both the macroscopic and subcellular levels. This blunted response may be due to a lower level of actin cytoskeleton turnover as measured by fluorescence recovery after photobleaching. Taken together these data suggest that DMD hiPSC-CMs are less adaptable to changes in their extracellular environment, and highlight the utility of nanotopographic substrates for effectively stratifying normal and structural cardiac disease phenotypes in vitro.
Keywords: Stem Cell Derived Cardiomyocytes, Duchenne muscular dystrophy, Cytoskeletal Alignment, FRAP
INTRODUCTION
Human induced pluripotent stem cells (hiPSCs) offer the means to generate a potentially limitless supply of a diverse array of human cell types for both basic science and clinical applications 33,35,36. Moreover, isolation of cells from patients with rare genetic conditions now holds the potential to generate disease specific human cells with which to further our understanding of underlying pathologies 33. Integration of these cells with bioengineering technologies has great promise for enabling the development of culture systems that can accurately recapitulate specific inherited disease phenotypes and assess symptomatic progression 39.
Given the prevalence of heart disease in society 22, models of cardiomyopathic conditions using human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have become one of the most intensely investigated tissues by bioengineers seeking to synthesize biomimetic tissues for drug development and cell replacement applications 11,19,21,24,27,28,37. One of the major goals of cardiac tissue engineering using human pluripotent stem cell technology is to create a functional piece of human myocardial tissue to serve as a high fidelity in vitro model of the heart for drug screening applications and the study of specific disease mechanisms 12,38. Unlike animal models, human pluripotent stem cell derived cardiomyocytes (hPSC-CMs) theoretically express the full array of ion channels, sarcomeric proteins, and metabolic machinery found in patient’s cells. To date, there have been a number of different iPSC models of inherited cardiac diseases that have been published, including ion channelopathies 5,13,18, dilated cardiomyopathy 34, and Barth syndrome 39. In addition, our group has previously developed a urine cell-derived model of Duchenne muscular dystrophy (DMD). These dystrophin-null hiPSC-CMs exhibit aberrant metabolic activity, altered calcium (Ca2+) handling, and are more prone to hypotonic mechanical stress 10.
DMD is an X-linked genetic disorder affecting ~1:3500 live male births. Frameshift mutations in the dystrophin gene result in the translation of a truncated dystrophin protein, which is non-functional and essentially unexpressed in cells 3. Under normal conditions, dystrophin is an integral part of the dystrophin-glycoprotein complex (DGC), a major structural and signaling hub that helps link the actin cytoskeleton to the extracellular matrix (ECM) and provide cellular stability 6. With the loss of dystrophin, the DGC is disrupted, leaving myocytes particularly prone to mechanical stress and rupture. Over time this results in muscle degeneration and the deposition of fibrotic tissue, eventually leading to premature death. However, with improved palliative treatment leading to prolonged lifespans, cardiomyopathy is becoming the leading cause of death in patients with DMD. While it is known that patients with DMD exhibit diastolic dysfunction and arrhythmias, the pathogenesis of this disease phenotype in cardiac tissue is not fully understood. The mdx mouse model of DMD has provided valuable insights into the disease pathology in skeletal muscle, but, paradoxically, mdx mice exhibit minimal cardiac dysfunction, limiting their utility for screening translational cardiac therapies 26. In fact, a recent clinical trial with sildenafil, which showed promise in alleviating cardiac dysfunction in the mdx mouse 1, was actually found to exacerbate cardiac symptoms in DMD patients and had to be terminated early 20. Therefore, the need for an accurate human cardiac model of dystrophin cardiomyopathies has never been greater.
In order to be of practical relevance, in vitro cardiac tissue screening assays utilizing hiPSC-CMs must be able to comprehensively stratify healthy and disease states in order to quantify the ability for novel treatments to rescue the disease phenotype. Here we present a simple biomimetic engineered platform for culturing hiPSC-CMs that enhances the structure and morphology of cardiomyocytes so that aberrant disease-specific characteristics relating to cell structure and contractile function in DMD can be detected. Our novel anisotropically nanofabricated substrata (ANFS) consist of 800 nm parallel arrays of grooves and ridges, mimicking the ordered structure of the myocardial extracellular matrix. We have shown previously that these patterned substrates promote the alignment of cultured cardiomyocytes, leading to the formation of anisotropic monolayers closely resembling the in vivo structure of myocardial tissue 14. In this system, both cellular action potential propagation and contractility are highly anisotropic and consistent with the underlying nanotopographic cues, suggesting that ECM-like nanotopographic substrates provide a powerful guidance cue regulating cardiomyocyte alignment and function in vitro 15,17.
In this study, cardiomyocyte structural development on ANFS was compared between DMD hiPSC-CMs and normal controls. In this manner, the capacity for dystrophin null cardiomyocytes to respond to exogenous mechanical cues was evaluated. These presented data demonstrate that ANFS mimicking the native ECM microenvironment help to stratify the DMD disease phenotype in vitro when compared to traditional flat culturing substrates. As such, the described system provides a more accurate and comprehensive model of DMD cardiomyopathy-in-a-dish for use in subsequent mechanistic studies and drug screening applications.
MATERIALS AND METHODS
Capillary Force Lithography
ANFS with 800 nm topographic features were fabricated via UV-assisted capillary force lithography as previously described 23. Briefly, first liquid polyurethane acrylate (PUA) prepolymer was drop dispensed onto a silicon master mold of the desired nanotopographic dimensions. A transparent polyester film (PET) was then placed on top of the dispensed PUA. After exposure to UV radiation (λ = 250–400 nm), the film was peeled away from the silicon master, creating a PUA mold. A polyurethane-based prepolymer (NOA76, Norland Products, Inc.) was then drop dispensed onto standard glass coverslips and the PUA mold was placed on top. The mold was then exposed to UV radiation for curing. After curing, the PUA mold was peeled off, leaving behind an ANFS for cell culture. Figure 1 depicts a schematic representation of this process. Flat polymer control surfaces were fabricated in the same manner, only with a bare PET film instead of a PUA mold. Prior to cell seeding, the ANFS are UV sterilized and coated with human fibronectin at 5μg/mL (Life Technologies).
Figure 1.

Schematic representation of anisotropically nanofabricated substrata (ANFS) fabrication process. (A) A polyurethane-acrylate (PUA) replica mold is created from a silicon (Si) master with the desired 800 nm topographic dimensions. (B) This PUA mold is then used to mass-produce ANFS by placing the PUA mold on top of a liquid polyurethane (PU) photo-curable pre-polymer. (C) The system is then exposed to UV light to polymerize the PU. (D) The PUA mold can then be peeled away, leaving behind the ANFS. The photograph depicts the iridescent surface of the ANFS caused by light diffraction at the nanotopographic surface.
Induced PSC Production and Cardiac Differentiation
Informed consent was obtained from all patients as directed by the Institutional Review Board (IRB) policies. Urine cells were isolated and expanded from a single healthy male participant (normal) and a DMD patient harboring a large dystrophin mutation as previously described 10,43. A polycistronic lentiviral vector encoding human Oct3/4, Sox2, Klf4, and c-Myc 40 was used to reprogram the urine cells into iPSCs 10. The derivative normal and DMD hiPSC lines were karyotyped and shown to be normal 46, XY karyotypes, and were subsequently used for differentiation. A monolayer-based directed differentiation was used as previously published 10. Figure 2A details the specific timing and reagents used in our cardiac differentiation protocol. Briefly, hiPSCs are plated into a monolayer and fed serum-free media supplemented with specific morphogen proteins and small molecule Wnt modulators to guide the cells down the cardiac lineage. At day 20 post-induction, hiPSC-CMs were dissociated with 0.05% Trypsin-EDTA, plated onto flat or ANFS at 175,000 cells/cm2, and cultured for an additional 14 days post-replating before further analysis, unless otherwise stated. The purity of cardiomyocyte populations plated onto experimental surfaces was confirmed by flow cytometry (Fig 2B). In all cell populations used, cardiomyocyte purity was found to lie in the range of 70 to 90% (data not shown).
Figure 2.
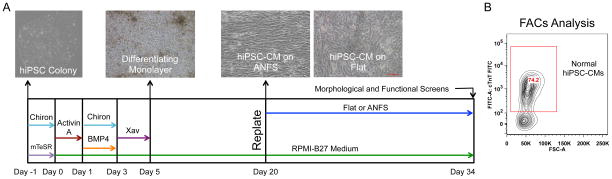
Protocol of hiPSC cardiac differentiation. (A) Timeline and representative images of the monolayer-based cardiac differentiation protocol and plating regimen for these studies. The timing and duration of specific morphogens (Activin-A and Bone Morphogenic Protein-4 [BMP4]) and small molecule Wnt modulators (Chiron and Xav939) specify cardiomyocyte development. (B) Representative flow cytometry result of cardiac purity for a cardiac differentiation prep. Differentiation runs with cardiac purity >70% were used for subsequent analysis.
The stated cell density was established based on preliminary investigation as optimal for producing confluent cardiomyocyte monolayers. Preliminary analysis with hiPSC-CMs revealed not significant structural differences between day 14 and day 100 cultures on ANFS (data not shown). As such, 14 days was selected as a suitable endpoint for these analyses as a sufficient length of time for cells to develop stable structural phenotypes. Cultures were fed every other day with serum-free RPMI-B27 plus L-glutamine.
CRISPR Knockout of Dystrophin
Guide sequences targeting the 3′-end of the first muscle exon of human dystrophin locus were designed using the online CRISPR design tool 2. Forward and reverse single strand oligodeoxynucleotides (ssODN) were synthesized with added BbsI restriction enzyme site at 5′-end (Eurofins Genomics, Huntsville AL). Validated guide sequence was then cloned into the plasmid hSpCas9 (BB)-2A-Puro (px459, addgene # 48139) to construct the targeting plasmid, according to the method described previously 30. Normal male iPSC UC3-4 was electroporated with the target plasmid using the Neon transfection system (Life technologies) according to the manufacturer’s protocol. After electroporation, cells were gently plated in 24-well-plate with 500ul of mTeSR1 medium (Stemcell technologies) and 10 μg/mL of ROCK inhibitor Y-26732 (Selleckchem). Medium was refreshed every day and 0.8 μg/mL puromycin selection was initiated 48 hours post transfection for 2 days. Individual colonies survived puromycin selection were manually picked and passaged into 96-well-plate for further expansion. Genomic mutation of the dystrophin locus was confirmed by Sanger’s sequencing (Genewiz, Seattle). Dystrophin protein knockout in the iPSCs was confirmed via Western blot (data not shown).
Cell Morphological Analysis
Normal and DMD hiPSC-CMs were cultured on flat or ANFS for two weeks and then observed using a Nikon Eclipse TS100 microscope. Phase contrast images were taken from three random fields of view per substrate (N = 5 biological replicates per condition). Morphological parameters such as cell length, width, and area were measured using ImageJ (National Institutes of Health). Once outlined, ImageJ processing tools were used to fit an ellipse to the outlined region, and the major and minor axes of each cell were determined from that shape. Cell morphology was measured by manually outlining clearly defined cell boarders of cells that showed rhythmic beating in videos of the same field of view 14. For all examined conditions, at least 250 cells were measured.
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde for 15 minutes and blocked with 5% goat serum in PBS for 1 hour at room temperature. Cells were then incubated with mouse anti-α-sarcomeric actinin monoclonal antibody (1:500, Sigma-Aldrich) diluted in 1% goat serum in PBS overnight at 4°C. The next day, cells were washed with PBS followed by staining with Alexa-594 conjugated goat-anti-mouse secondary antibody (1:200, Invitrogen) and Alexafluo 488 phalloidin (1:200, Invitrogen). Counterstaining was performed with Vectashied containing DAPI (Vector Labs). Images were captured with a Nikon A1R confocal microscope using a Nikon CFI Plan Apo VC 60X water immersion objective. For each condition, at least 4 biological specimens were prepared and imaged, with at least 4 images taken per specimen.
Cytoskeletal Alignment Measurements
Fluorescent images of F-actin fibers of cardiomyocytes on unpatterned and patterned substrates were analyzed for alignment using a custom MATLAB script as described previously 42. A 2D convolution was performed on each image and a Sobel edge-emphasizing filter was applied to extract the horizontal and vertical edges within the image. These edges were then combined to calculate the gradient magnitude of each pixel. A magnitude threshold was applied to select the major F-actin fibers. Next, the gradient orientation was calculated by determining the angle of the gradient with the x- and y-axes. Finally, the angle that the orthogonal of the gradient makes with the x- and y-axes was calculated. This orthogonal angle (from −90° to +90° with respect to the x-axis) is the principle orientation of the individual F-actin fibers. A probability density histogram of the fiber angle distribution was calculated for each image and averaged per condition.
Sarcomere Length Analysis
The α-actinin confocal images were analyzed using standard analysis plugins in ImageJ. To calculate sarcomere length, a straight line was drawn perpendicular to at least ten consecutive well-defined α-actinin-positive bands. An intensity profile was then plotted across the entire length of the line and the distance between intensity peaks was calculated. Between ten and fifteen measurements were made per image and averaged per condition.
Correlation-based Contraction Quantification (CCQ)
Our CCQ method utilizes Particle Image Velocimetry (PIV) and Digital Image Correlation (DIC) algorithms 25 to provide relevant contractile endpoints from bright field video recordings. Briefly, a reference video frame is divided into a grid of windows of a set size. Each window is run through a correlation scheme with a second frame, providing the new location for that window in the second frame. This displacement is converted into a vector map, which provides contraction angles and, when spatially averaged, contraction magnitudes and velocities. The correlation equation used provides a Gaussian correlation peak with a probabilistic nature that provides sub-pixel accuracy. The videos used to perform this analysis were taken using a Nikon TS100 microscope at 12 frames per second (fps).
Fluorescent Recovery After Photobleaching (FRAP)
Briefly, cardiomyocytes were differentiated as previously described 10. Cells were re-plated on BD matrigel-coated glass bottom dishes as single cells. Cellular actin was labeled with Life Technologies CellLight® Actin-RFP (BacMam 2.0). Imaging was performed on a Nikon A1R confocal using a Nikon CFI Plan Apo VC 60X water immersion objective, and cells were maintained at 37°C using a Tokai Hit Stagetop Incubator. Cardiomyocytes were identified by rhythmic beating. A region of sarcomere was bleached using a 561nm laser at 100% power, and recovery of fluorescence was measured every second for 60 seconds, followed by every 8 seconds until 10 minutes after bleaching. The size of the bleaching area was identical for all experiments. Fluorescence recovery was calculated using pre-bleach fluorescence intensity in the bleached area. Statistics were performed using Graphpad’s Prism software.
Statistical Analysis
Statistical significance between flat and nanopatterned, as well as between normal vs DMD vs KO DMD cultures, was determined using Two-Way ANOVA with Tukey’s pairwise post hoc analysis using SigmaPlot software unless otherwise stated. For the contraction angle analysis, a chi-square test run at 5% significance was utilized to quantify uniformity in alignment distributions. This test was calculated using MATLAB. For all statistical analyses, a p value less than 0.05 was considered significant. Error bars represent standard error of the mean (SEM).
Ethical approval
Procedures performed in studies involving human subjects were approved by the University of Washington Institutional Review Board, approval number 45581. Human subject activities were performed in accordance with the regulatory requirements laid down in U.S. Code of Federal Regulations, Title 45 Department of Health and Human Services Part 46, Protection of Human Subjects.
RESULTS
DMD hiPSC-CMs Display a Blunted Response to Nanotopography Compared to Normal Control
Our previous data with NRVMs demonstrated that 800 nm ANFS can be used to promote in vivo-like structure and function in cultured cardiomyocytes 14. We therefore set out to answer whether ANFS could be used to enhance the structure of hiPSC-CMs in order to further stratify cardiomyocyte phenotypes in a disease model of DMD cardiomyopathy. Phase contrast images of synchronously beating monolayers of normal hiPSC-CMs (Fig 3A and 3B) demonstrate that under healthy conditions hiPSC-CMs align very well with the underlying anisotropic nanotopographic cues (ANFS are oriented horizontally in all images). Both patient-derived DMD hiPSC-CMs and isogenic dystrophin knockout (KO DMD) hiPSC-CMs, on the other hand, demonstrate a less drastic macroscopic alignment on the ANFS compared to normal cardiomyocytes (Fig 3C–F).
Figure 3.
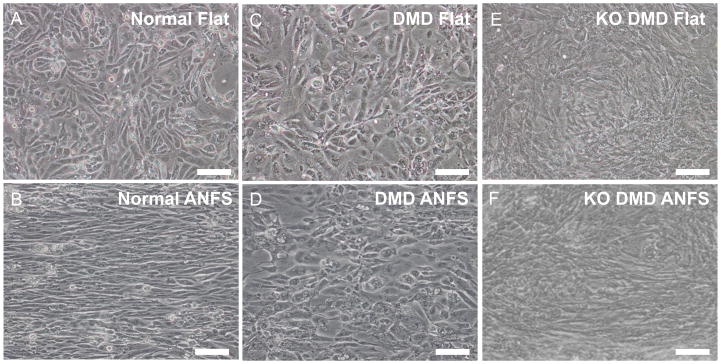
Bright field images of normal (A and B), patient-derived DMD (C and D), and KO DMD hiPSC-CMs (E and F) on flat or ANFS after two weeks of culture. Cells on flat substrata (A, C, and E) are randomly oriented while cells on ANFS (B, D, and F) have a more anisotropic and aligned structure. ANFS are oriented horizontally in the images. Scale bar: 100μm.
Looking at the subcellular structure with immunocytochemistry, again we found that qualitatively, normal hiPSC-CMs showed a higher level of anisotropy and alignment on the ANFS compared to flat control (Fig 4 A and 4B). The actin cytoskeleton in normal cardiomyocytes exhibit local alignment in the flat condition but overall showed no preferential directionality. Conversely, actin cytoskeleton of normal hiPSC-CMs on ANFS were highly aligned in the direction of the ANFS. DMD and KO DMD hiPSC-CMs on flat substrates (Fig 4C and 4E) were randomly organized similar to normal, but showed much less directionality than normal cardiomyocytes on ANFS (Fig 4D and 4F).
Figure 4.

Immunofluorescent confocal images of sarcomere structure in normal (A and B), patient-derived DMD (C and D), and KO DMD hiPSC-CMs (E and F) on flat or ANFS after two weeks of culture. Cells have highly aligned cytoskeletal and sarcomere structures on ANFS compared to flat substrata, but DMD hiPSC-CMs show less alignment than normal hiPSC-CMs. Insets show close-ups of striated sarcomeres. Green = F-actin, Red = α-actinin, Blue = DAPI. Scale bar: 20μm.
In order to quantify the cellular response to the nanotopography, we began by measuring cell morphology and anisotropy from the bright field images. We found that hiPSC-CMs from all the cell lines (normal, DMD, and KO DMD) had a higher anisotropic ratio (cell length:width ratio) on ANFS compared to flat substrates (Fig 5A, p < 0.001). Patient-derived and KO DMD hiPSC-CMs, however, had a lower anisotropic ratio on ANFS (3.73 ± 0.12 and 3.92 ± 0.24, respectively) compared to normal on ANFS (4.83 ± 0.09, p < 0.001 for both comparisons). Next, we measured the distribution of actin fiber angles from the confocal images. hiPSC-CMs from all cell lines exhibited random cytoskeletal alignment as evident by the relatively flat actin fiber angle distribution in Fig 5B (solid lines). Normal hiPSC-CMs on ANFS, on the other hand, were much more aligned based on the peaked distribution of actin fiber angles (indicating most actin fibers were preferentially aligned in a single direction). As the qualitative analysis suggests, however, patient-derived and KO DMD hiPSC-CMs were less aligned and had an actin fiber distribution that fell between the flat and highly aligned distributions of normal control. Taken together these data suggest that DMD hiPSC-CMs have a diminished capacity to respond to topographic cues compared to normal hiPSC-CMs. Cell density was assessed for each examined condition by counting the total cells per field of view during morphological assessment. No significant difference was observed in cell density across all examined conditions (data not shown), indicating that the observed differences in cell structure was not due to variance in population density.
Figure 5.

Morphological analysis of cardiomyocytes show that DMD hiPSC-CMs are less responsive to substrate nanotopographical cues than normal control hiPSC-CMs. (A) Cell elongation was measured by taking the ratio of the major and minor axes of the cell in ImageJ. Patient-derived and KO DMD hiPSC-CMs are less elongated on ANFS compared to normal control. * p < 0.05 via Two-Way ANOVA with Tukey’s Post-Hoc analysis compared to normal within topographic group. # p < 0.05 compared to flat within cell type. (B) Probability density histogram of F-actin fiber orientations highlight that cells on traditional flat substrata have randomly oriented cytoskeletons as evident by the flat distributions (solid lines). Cells on ANFS (dotted lines) are more preferentially aligned in the direction of the nanotopography and have a peaked distribution of F-actin fibers.
DMD hiPSC-CMs Exhibit a Cardiomyopathy-Like Phenotype
The clinical manifestation of the cardiac involvement in DMD is generally described as cardiomyopathy complicated by arrhythmias 8. We measured cell area from bright field images to test whether this phenotype was apparent in our DMD cardiomyopathy models. While normal hiPSC-CMs on ANFS had larger cell areas (591 ± 12.2 μm2) than on flat substrates (465 ± 9.07 μm2), suggesting a greater degree of cellular hypertrophy and/or cell spreading induced by the ANFS (p < 0.001), patient-derived DMD hiPSC-CMs were significantly larger than normal on both flat (796 ± 15.5 μm2, p < 0.001) and ANFS (719 ± 19.0 μm2, p < 0.001) (Fig 6A). In fact, DMD hiPSC-CMs were moderately smaller on ANFS than flat substrates. KO DMD hiPSC-CMs displayed a similar trend on flat (695 ± 19.5 μm2) and ANFS (653 ± 30.6μm2) as the patient-derived DMD hiPSC-CMs and were significantly larger than normal hiPSC-CMs (p < 0.001) (but on average slightly smaller than the patient-derived hiPSC-CMs, p = 0.003).
Figure 6.

DMD hiPSC-CMs have a cardiomyopathy-like phenotype. (A) Cell area measurements show that patient-derived and KO DMD hiPSC-CMs are larger on both flat and ANFS compared to normal control. * p < 0.05 via Two-Way ANOVA with Tukey’s Post-Hoc analysis compared to normal. # p < 0.05 compared to flat within cell type. (B) Sarcomere analysis highlights that patient-derived and KO DMD hiPSC-CMs have atypically long resting sarcomere lengths compared to normal control. * p < 0.05 compared to normal hiPSC-CMs.
Next we measured sarcomere length from the confocal images of the cells. Similar to the results with cell area, patient-derived DMD hiPSC-CMs had significantly longer resting sarcomere lengths (flat = 1.95 ± 0.021 μm, ANFS = 1.93 ± 0.017 μm) than normal on both flat (1.78 ± 0.013 μm) and ANFS (1.78 ± 0.014 μm, p < 0.001) (Fig 6B). Again, this disease trend held true in the KO DMD hiPSC-CMs as cells on both flat and ANFS had significantly longer sarcomere lengths than normal hiPSC-CMs (flat = 1.84 ± 0.018 μm, ANFS = 1.83 ± 0.018 μm, p = 0.011). This abnormally long resting sarcomere length for patient-derived and KO DMD hiPSC-CMs, combined with the increased cell area, indicates that DMD hiPSC-CMs appear to recapitulate some of the structural characteristics of DMD cardiomyopathy.
DMD hiPSC-CMs Demonstrate Alterations in Contractile Properties
Based on our CCQ contraction analysis, patient-derived DMD hiPSC-CMs showed greater variability and lower alignment in contraction angles on nanopatterned substrates compared to normal hiPSC-CMs (Fig 7A–D). Cells on ANFS had slower overall contraction velocities than on flat substrates in all conditions. Interestingly, the patient-derived and KO DMD hiPSC-CMs had faster contraction velocities on both flat and ANFS compared to normal hiPSC-CMs (Fig 7G, *p < 0.05). The chi-square tests showed that each ANFS group showed significantly lower variation in contraction angle than flat with the same cell condition at the 5% significance level. The chi-square test also showed that DMD hiPSC-CMs on ANFS showed higher variation in contraction angle than normal hiPSC-CMs on flat. This can be interpreted as the flat substrates showing less alignment than the ANFS overall, and the DMD hiPSC-CM on ANFS showing less alignment than the normal controls.
Figure 7.

Correlation-based Contraction Quantification (CCQ) analysis of beating monolayers. (A-F) Average contraction angle histograms of normal (A and B), patient-derived DMD (C and D), and KO DMD hiPSC-CMs (E and F) on flat and ANFS. Patient-derived DMD hiPSC-CMs have more random contraction angles on ANFS than normal hiPSC-CMs. (G) Maximum contraction speeds of normal, patient-derived DMD, and KO DMD hiPSC-CMs. In general, DMD hiPSC-CMs have faster contraction speeds than normal hiPSC-CMs (*p < 0.05). (H) Isolated longitudinal contraction velocities in the direction of cell alignment and nanotopography show no significant difference between groups (p = 0.65). (I) Contraction velocity anisotropic ratio (Longitudinal:Transverse Velocity) of normal hiPSC-CMs on flat and ANFS. Normal hiPSC-CMs are highly functionally anisotropic on ANFS. These results, together with the contraction angle histograms, suggest there is greater variability in the DMD hiPSC-CM contractile directions, leading to the larger overall contraction speeds in (G).
When contraction in parallel with the nanotopographic cues (longitudinal velocity) was isolated and analyzed independently, it was found that no significant difference between normal and DMD cells was observed (Fig 7H, p = 0.65). Normal cells on ANFS exhibited highly anisotropic contraction parameters (Fig 7I). As such, the isolated transverse contraction velocities were significantly smaller than those in parallel with the topography (Fig 7I, p = 0.03). A greater level of variability in contraction direction was observed in DMD cells (Fig 7D). No significant difference was observed in longitudinal and transverse contraction velocities in KO DMD cells (data not shown, p = 0.48) or patient-derived DMD cells on ANFS, (data not shown, p = 0.08). Taken together, although the contraction velocities are greater in DMD hiPSC-CMs compared to normal, contraction longitudinal to the ANFS is not different. Since contraction is also highly anisotropic on the ANFS for normal hiPSC-CMs and more variable in direction for DMD cells, these results could lead to larger overall contraction velocities.
No significant differences in spontaneous contraction frequencies were observed between any experimental condition, suggesting the differences observed were due to fundamental changes in cell structure, rather than variation in beat rhythmicity.
Slower Cytoskeletal Turnover May Explain Blunted DMD hiPSC-CM Response to ECM Cues
Dystrophin is a vital component connecting the actin cytoskeleton to the extracellular matrix. Therefore, it is reasonable to predict that DMD hiPSC-CMs would have a blunted response to ECM cues such as nanotopography. However, just as the mechanism behind increased membrane fragility in DMD is debated and not fully understood 3, the reasoning why DMD hiPSC-CMs should be less organized on nanotopography than normal is not straightforward. Thus, to further investigate how the loss of dystrophin leads to this diminished contact guidance, we performed FRAP measurements of sarcomeric actin turnover in normal and DMD hiPSC-CMs (Fig 8). We found that compared to normal, DMD hiPSC-CMs had significantly lower actin turnover in the sarcomere region as measured by lower FRAP levels after 10 minutes. This result is in agreement with our results of cytoskeletal alignment of normal and DMD hiPSC-CMs on ANFS (Fig 5B). With a lower rate cytoskeletal turnover, the cells would have less ability to reorganize their cytoskeleton in response to the contact guidance cues imparted by the nanotopography.
Figure 8.

Fluorescence recovery after photobleaching (FRAP) measurements of sarcomeric actin in normal and DMD hiPSC-CMs demonstrate that DMD hiPSC-CMs have significantly slower actin turnover than normal control. Slower actin turnover may explain why DMD hiPSC-CMs are less responsive to extracellular matrix cues such as nanotopography, as the cells are less able to adapt to changes in their environment.
DISCUSSION
Once cardiomyopathy has developed in patients, there is no known cure outside of heart transplantation, a procedure rarely performed in DMD patients due to weak respiratory muscle function, respiratory insufficiency, and short supplies of donor hearts 31. Therefore, it is imperative to discover new therapies that can slow the progression of diseases (such as DMD) that cause cardiomyopathy, to improve the quality of life for these patients until a more definitive, long-term cure is found 41. Human iPSC disease models have an incredible potential to elucidate important molecular mechanisms driving diseases such as DMD, and their availability opens the door to developing new methods for developing and testing novel therapies. In this study, we aimed to assess the ability of biomimetic nanotopographic cell culture substrates to exacerbate the disease phenotype in an in vitro model of DMD cardiomyopathy so that the diseased state is more distinct from the healthy one. Such a platform could be used to identify simple, high-throughput endpoints with which to screen novel therapeutics for their capacity to ameliorate the cardiac pathology of DMD.
We hypothesized that a biomimetic microenvironment would further stratify the disease phenotype in our dystrophin-null hiPSC-CM model of DMD cardiomyopathy by adding more in vivo-like ECM cues that would necessitate cell-ECM interactions and activation of the DGC. In addition, while single cell analyses often constitute effective in vitro systems for studying cell structure, monolayer cultures were chosen for this study to provide a more accurate representation of myocardial tissue. Eventual application of this technology for drug screening and functional studies will benefit from analysis of whole tissue mechanics and their breakdown in disease.
When normal control, patient-derived DMD, and KO DMD hiPSC-CMs were cultured on traditional flat substrates, there was very little difference between their structures. There was no difference in their actin cytoskeletal alignment and minimal differences in cell shape (Fig 5). When the cells were cultured on the biomimetic ANFS, however, a striking difference in the structure of the DMD cells became apparent compared with normal controls. Both patient-derived and KO DMD hiPSC-CMs failed to align as robustly as normal cells on the ANFS, and there was a larger reduction in elongation between normal and DMD hiPSC-CMs on ANFS than flat substrates. Comparing the isogenic controls (normal and KO DMD), we found that the cell elongation was only significantly different on the ANFS, while the cells on the traditional flat surfaces showed no difference in their elongation. Therefore the ANFS did indeed help to bring out a more severe “disease phenotype” and helped to stratify healthy from diseased hiPSC-CMs.
In this study we measured sarcomere length as a surrogate for sarcomere organization. While we found that patient-derived and KO DMD hiPSC-CMs had longer sarcomere lengths compared to normal control cells, we interpret this data to suggest that this increased sarcomere length did not result from increased maturation, because there did not appear to be a concomitant increase in cellular organization (as evident from Figs 3–5). Unbiased measurements, such as organization order parameter (OOP) 7, may elucidate this increased sarcomere organization in our cultures and are under current investigation by our group. Dilated cardiomyopathy, such as the type observed with DMD patients however, is associated with lower myofilament calcium sensitivity 16,29. In accordance with this calcium insensitivity, the resting sarcomere length of dilated cardiomyopathic cardiomyocytes has been found to be longer than WT control 4. Thus, our findings of increased cell area along with longer resting sarcomere lengths in DMD hiPSC-CMs suggests an accurate model of dilated cardiomyopathy-in-a-dish. Further investigation into the Ca2+ sensitivity of DMD hiPSC-CM myofibrils is necessary.
The most prominent finding of this work was that both patient-derived and KO DMD hiPSC-CMs displayed a blunted cellular response to the topographic cues imparted by the ANFS. The fact that structural phenotypes in patient derived and KO DMD hiPSC-CMs were not significantly different from each other, but were different from normal controls, supports the hypothesis that the abnormal structural phenotype observed in DMD patient cells directly results from dystrophin deficiency, and is not simply due to cell line variability between patient cells and normal controls. Taken together, findings presented here from data gathered in cell lines derived from normal controls, engineered dystrophin-null, and naturally occurring dystrophin-mutants, support the overall hypothesis that dystrophin plays a direct role in regulating cell mechanotransduction and responding to biomimetic nanotopography in vitro.
DMD hiPSC-CMs were less organized both macroscopically and subcellularly on ANFS compared to normal control cardiomyocytes. Although a mechanistic explanation is not known, our findings of lower actin turnover in DMD hiPSC-CMs provide an interesting possibility. The loss of dystrophin is known to disrupt the DGC, but this disruption is not lethal and does not interfere with cardiac differentiation. Therefore, compensatory mechanisms may help to offset the loss of dystrophin. Other groups have postulated that utrophin may play an important roll in compensating for the loss of dystrophin due to its homology with dystrophin 9. Other proteins such as integrins and focal adhesion proteins might also be differentially expressed in DMD cardiomyocytes that help to maintain connection with the ECM 32. These compensatory mechanisms, however, may be less dynamic than the DGC, and thus inhibit actin cytoskeletal remodeling. Thus, when the DMD hiPSC-CMs establish their connections with the ECM, they may be less able to respond to changes to the microenvironment, such as dissociation and replating. The assay system developed in this study represents a useful tool to further test this theory, and our group is working to answer this question in more rigorous detail.
In this study, we focused on the structural changes of DMD patient hiPSC-CMs. However, to utilize hiPSC-CMs for effective and comprehensive disease modeling, we must also be able to measure the functional output of cells directly. To that end, we performed video-based contraction mapping of the synchronously beating monolayers. We found that patient-derived DMD hiPSC-CMs display faster overall contraction and relaxation velocities compared to normal controls (p < 0.001). Additionally, both normal and patient-derived DMD cells on flat substrates manifest faster contraction and relaxation velocities than cells on ANFS (p = 0.01). Although one might assume DMD cells would have slower, weaker contraction kinetics than normal cells, the increase in overall contraction speed may be due to the loss of anisotropic contraction properties. When contraction in parallel with the topographies was isolated, no significant differences between normal and DMD cells was observed (Fig 7H). The variability in transverse contraction levels across the different conditions examined could therefore explain why overall contraction was higher in DMD cultures. The very low transverse contraction speeds in normal cells likely reduced overall measurements in these cultures, whereas the variable level in DMD cultures led to larger overall contraction velocity measurements.
While we are unable to definitively explain this increase in overall contractile performance in DMD hiPSC-CMs, an alternative hypothesis for the observed data is that greater contraction and relaxation kinetics in DMD cells is a result of their less stable attachment to the underlying substrate. Breakdown of the DGC in DMD cells may result in cultures that are less tightly bound to the substrate, and are better able to contract against these rigid surfaces, leading to apparent improvements in function compared with normal cells. Overall, our contractility results indicate that the DMD hiPSC-CMs have less organized beating on ANFS compared to normal controls (F-test showed higher variation in DMD cells than normal at the 5% significance level), but that DMD hiPSC-CMs have inherently faster contractility under the conditions tested (Fig 7).
CONCLUSIONS
In this study, we demonstrate that engineered nanotopographies enable clearer distinction between DMD and normal structural and functional phenotypes in cultured hiPSC-CMs. Specifically, the differences in the observed responses of diseased and normal cardiomyocytes to ANFS are suggestive of a structural cardiomyopathy-like phenotype in DMD cells. Moreover, the collected data accentuate that ANFS can be used to stratify the structural and functional differences between normal and DMD hiPSC-CMs in a way not possible on conventional flat substrates. The developed platform also offers a simple, reliable, and reproducible method for screening phenotypic changes in these cells in response to novel therapeutic treatment. Such technology could be invaluable to the development of a new DMD test bed for drug discovery, enabling the provision of better care to patients suffering from this debilitating condition in the near future.
Acknowledgments
This work was supported by an American Heart Association (AHA) Scientist Development Grant (13SDG14560076 to DHK), a Muscular Dystrophy Association (MDA) research grant (to MKC) and the Senator Paul D Wellstone Muscular Dystrophy Cooperative Research Center, Seattle (NIH U54AR065139). DHK thanks the Department of Bioengineering at the University of Washington for the new faculty startup fund. JM thanks the support from the NIH Bioengineering Cardiovascular Training Grant fellowship.
Biography
SHORT BIOGRAPHY
Dr. Deok-Ho Kim is currently an Assistant Professor in the Department of Bioengineering at the University of Washington. He received his Ph.D. degree in Biomedical Engineering from the Johns Hopkins University in 2010. From March 2000 to June 2005, he worked as a Research Scientist at the Korea Institute of Science and Technology, which included a seven-month position at the Swiss Federal Institute of Technology in Zurich. His research interests include the development and application of advanced biomaterials and micro/nano-technologies for studying cell-matrix mechanobiology, stem cells and tissue engineering. He has received numerous awards including the Samsung Humantech Thesis Award (2009), the Harold M. Weintraub Award in Biological Sciences (2010), the Perkins Coie Award for Discovery (2011), the American Heart Association National Scientist Development Award (2012), the Springer Award for Most Downloaded and Most Cited Review Article from The Annals of Biomedical Engineering (2013), and the BMES-CMBE Rising Star Award (2013). Dr. Kim is currently an Associate Editor for Biomedical Microdevices and IEEE Transactions on NanoBioscience, and serves as a member of the editorial boards of numerous journals, including Scientific Reports, International Journal of Nanomedicine, and Journal of Laboratory Automation.
Footnotes
CONFLICTS OF INTEREST
J.M., X.G., R.L., S.C., M.K.C., and D.L.M. declare they have no conflicts of interest. A.S.T.S. is a paid consultant for NanoSurface Biomedical Inc. D-H.K. is an uncompensated cofounder of NanoSurface Biomedical Inc., and has uncompensated stock options in this company.
ETHICAL STANDARDS
Procedures performed in studies involving human subjects were approved by the University of Washington Institutional Review Board, approval number 45581. Human subject activities were performed in accordance with the regulatory requirements laid down in U.S. Code of Federal Regulations, Title 45 Department of Health and Human Services Part 46, Protection of Human Subjects.
No animal studies were carries out by the authors for this article.
References
- 1.Adamo CM, et al. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proceedings of the National Academy of Sciences. 2010;107:19079–19083. doi: 10.1073/pnas.1013077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatric Neurology Elsevier. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Du CK, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circulation Research Lippincott Williams & Wilkins. 2007;101:185–194. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- 5.Egashira T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovascular Research. 2012;95:419–429. doi: 10.1093/cvr/cvs206. [DOI] [PubMed] [Google Scholar]
- 6.Ervasti J. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2007;1772:108–117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Feinberg AW, et al. Controlling the contractile strength of engineered cardiac muscle by hierarchal tissue architecture. Biomaterials Elsevier Ltd. 2012;33:5732–5741. doi: 10.1016/j.biomaterials.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finsterer J, Cripe L. Treatment of dystrophin cardiomyopathies. 2014;11:168–179. doi: 10.1038/nrcardio.2013.213. Available from: http://dx.doi.org/10.1038/nrcardio.2013.213. [DOI] [PubMed] [Google Scholar]
- 9.Gordon BS, Lowe DA, Kostek MC. Exercise increases utrophin protein expression in the mdxmouse model of Duchenne muscular dystrophy. Muscle Nerve. 2014;49:915–918. doi: 10.1002/mus.24151. [DOI] [PubMed] [Google Scholar]
- 10.Guan X, et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Research. 2014;12:467–480. doi: 10.1016/j.scr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen A, et al. Development of a Drug Screening Platform Based on Engineered Heart Tissue. Circulation Research. 2010;107:35–44. doi: 10.1161/CIRCRESAHA.109.211458. [DOI] [PubMed] [Google Scholar]
- 12.Hirt MN, Hansen A, Eschenhagen T. Cardiac Tissue Engineering: State of the Art. Circulation Research. 2014;114:354–367. doi: 10.1161/CIRCRESAHA.114.300522. [DOI] [PubMed] [Google Scholar]
- 13.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature Nature Publishing Group. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 14.Kim D-H, et al. Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proceedings of the National Academy of Sciences. 2010;107:565–570. doi: 10.1073/pnas.0906504107. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0906504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim DH, et al. Nanopatterned cardiac cell patches promote stem cell niche formation and myocardial regeneration. Integr Biol. 2012;4:1019. doi: 10.1039/c2ib20067h. [DOI] [PubMed] [Google Scholar]
- 16.Kimura A. Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. Journal of Human Genetics Nature Publishing Group. 2010;55:81–90. doi: 10.1038/jhg.2009.138. [DOI] [PubMed] [Google Scholar]
- 17.Kshitiz, et al. Control of Stem Cell Fate and Fucntion by Engineering Physical Microenvironments. Integr Biol. 2012:1–20. doi: 10.1039/c2ib20080e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Disease Models & Mechanisms. 2012;5:220–230. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Razu ME, Wang X, Lacerda C, Kim J. Biomimetic cardiac microsystems for pathophysiological studies and drug screens. Journal of Laboratory Automation. 2014:1–24. doi: 10.1177/2211068214560903. [DOI] [PubMed] [Google Scholar]
- 20.Leung DG, et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann Neurol. 2014;76:541–549. doi: 10.1002/ana.24214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang P, et al. Drug Screening Using a Library of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals Disease-Specific Patterns of Cardiotoxicity. Circulation. 2013;127:1677–1691. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lozano R, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet Elsevier Ltd. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macadangdang J, et al. Capillary Force Lithography for Cardiac Tissue Engineering. JoVE. 2014:e50039. doi: 10.3791/50039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsa E, et al. Drug evaluation in cardiomyocytes derived fromhuman induced pluripotent stem cells carrying along QT syndrome type 2 mutation. European Heart Journal. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milde F, Franco D, Ferrari A, Kurtcuoglu V, Poulikakos D, Koumoutsakos P. Cell Image Velocimetry (CIV): boosting the automated quantification of cell migration in wound healing assays. Integr Biol. 2012;4:1437. doi: 10.1039/c2ib20113e. [DOI] [PubMed] [Google Scholar]
- 26.Mourkioti F, et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. 2013;15:895–904. doi: 10.1038/ncb2790. Available from: http://dx.doi.org/10.1038/ncb2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarrete EG, et al. Screening Drug-Induced Arrhythmia Events Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Low-Impedance Microelectrode Arrays. Circulation. 2013;128:S3–S13. doi: 10.1161/CIRCULATIONAHA.112.000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada JI, et al. Screening system for drug-induced arrhythmogenic risk combining a patch clamp and heart simulator. Science Advances. 2015;1:e1400142–e1400142. doi: 10.1126/sciadv.1400142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajan S, et al. Dilated Cardiomyopathy Mutant Tropomyosin Mice Develop Cardiac Dysfunction With Significantly Decreased Fractional Shortening and Myofilament Calcium Sensitivity. Circulation Research. 2007;101:205–214. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]
- 30.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roger VL, et al. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sen S, Tewari M, Zajac A, Barton E, Sweeney HL, Discher DE. Upregulation of paxillin and focal adhesion signaling follows Dystroglycan Complex deletions and promotes a hypertensive state of differentiation. European Journal of Cell Biology Elsevier GmbH. 2010;90:249–260. doi: 10.1016/j.ejcb.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sipp D. Challenges in the clinical application of induced pluripotent stem cells. Stem Cell Research Therapy. 2010:1–3. doi: 10.1186/scrt9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun N, et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Science Translational Medicine. 2012;4:1–14. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Svendsen CN. Back to the future: how human induced pluripotent stem cells will transform regenerative medicine. Human Molecular Genetics. 2013;22:R32–R38. doi: 10.1093/hmg/ddt379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi K, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 37.Turnbull IC, Lieu DK, Li RA, Costa KD. Cardiac tissue engineering using human stem cell-derived cardiomyocytes for disease modeling and drug discovery. Drug Discovery Today: Disease Models. 2012 doi: 10.1016/j.ddmod.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vunjak-Novakovic G, et al. Challenges in Cardiac Tissue Engineering. Tissue Engineering. 2010;16:169–187. doi: 10.1089/ten.teb.2009.0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med Nature Publishing Group. 2014;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warlich E, et al. Lentiviral Vector Design and Imaging Approaches to Visualize the Early Stages of Cellular Reprogramming. Mol Ther Nature Publishing Group. 2009;19:782–789. doi: 10.1038/mt.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang HS, et al. Nanopatterned muscle cell patches for enhanced myogenesis and dystrophin expression in a mouse model of muscular dystrophy. Biomaterials Elsevier Ltd. 2014;35:1478–1486. doi: 10.1016/j.biomaterials.2013.10.067. [DOI] [PubMed] [Google Scholar]
- 42.Yang HS, et al. Electroconductive Nanopatterned Substrates for Enhanced Myogenic Differentiation and Maturation. Adv Healthcare Mater. 2015 doi: 10.1002/adhm.201500003. n/a n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, et al. Urine Derived Cells are a Potential Source for Urological Tissue Reconstruction. The Journal of Urology Elsevier. 2008;180:2226–2233. doi: 10.1016/j.juro.2008.07.023. [DOI] [PubMed] [Google Scholar]