

Abstract
Shigella spp. are among the main causative agents of acute diarrheal illness and claim more than 1 million lives per year worldwide. There are multiple bacterial genes that control the pathogenesis of Shigella, but the virF gene may be the most important. This gene, located on the primary pathogenicity island of Shigella, encodes VirF, an AraC-family transcriptional activator that is responsible for initiating the pathogenesis cycle in Shigella. We have previously shown that it is possible to attenuate the virulence of Shigella flexneri via small molecule inhibition of VirF. In this study, we probed the mechanism of action of our small molecule inhibitors of VirF. To enable these studies, we have developed a homologous and efficient expression and purification system for VirF and have optimized two different in vitro VirF-DNA binding assays. We have determined that one of our HTS hit compounds inhibits VirF binding to DNA with a calculated K i similar to the effective doses seen in our transcriptional activation and virulence screens. This is consistent with inhibition of DNA binding as the mechanism of action of this hit compound. We have also screened 15 commercially sourced analogs of this compound and deduced an initial SAR from the approximately 100-fold range in activities. Our four other HTS hit compounds do not inhibit DNA binding and yet they do block VirF activity. This suggests that multiple agents with different molecular mechanisms of inhibition of VirF could be developed. Pursuing hits with different mechanisms of action could be a powerful approach to enhance activity and to circumvent resistance that could develop to any one of these agents.
Introduction
The misuse of prescription antibiotics and the overuse of antibiotics in livestock feed have greatly contributed to the rapid increase in drug-resistant bacteria in the environment. The most recent World Health Organization report on antimicrobial resistance states, “A post-antibiotic era-in which common infections and minor injuries can kill, far from being an apocalyptic fantasy, is instead a very real possibility for the 21st century.” [1]. Current thinking is that one potentially effective approach to overcoming this growing problem is to target bacterial virulence rather than bacterial viability [2–5].
To validate this approach, we have begun identifying/developing an anti-virulence therapy to combat Shigella spp. infections. VirF is an AraC-family transcriptional activator that regulates (directly and indirectly) the transcription of all downstream virulence factors in Shigella spp. [6–9]. Both VirF expression and activity are tightly regulated by environmental signals (pH, temperature, osmolarity), specifically signals commonly encountered in the host cell environment [10–13]. Only under these favorable conditions can VirF directly activate the transcription of two downstream virulence genes, virB and icsA [14, 15]. VirB is a secondary transcriptional activator that is responsible for activating the transcription of other virulence genes, such as ipaB, ipaC, and, ipaD [12], whose gene products are involved in the construction of the Type III Secretion System and the escape from host-cell defense systems [16–18]. IcsA assembles actin polymerase on one pole of the bacterium and propels the bacterium through the infected host cells via the polymerization of host cell actin; allowing the bacterium to spread to adjacent cells [19–21]. Gene silencing studies have shown that the lack of VirB expression leads to a loss of virulence [6, 22], and that the lack of IcsA expression blocks the intra- and inter-cellular movement of Shigella [23, 24]. Additionally, in the infected host, Shigella utilizes VirF-induced IpaB to escape from macrophages [17]. These results suggest that inhibition of VirF with a small molecule should block not only the initial cellular invasion, but also prevent an active Shigella infection from continuing to spread from cell-to-cell and increase the efficiency of macrophage killing Shigella.
The exact mechanism by which VirF activates transcription is not presently understood. Like AraC and most AraC family members, VirF has two domains, an N-terminal dimerization domain and C-terminal DNA binding domain. Both of these domains are necessary for in vivo transcriptional activation [25]. As shown in Fig 1, in order for VirF to activate transcription it must bind to the correct promoter region (either the virB promoter (pvirB) or the icsA promoter (picsA)), dimerize, and recruit RNA polymerase. The order of these events, indeed if they are ordered at all, is presently unknown. Our small molecule inhibitors could be disrupting any of these steps of the VirF gene activation process. In fact, there have been reports indicating that VirF, and/or homologous AraC-family members, can be inhibited through the blockade of DNA binding [4, 26] or self-dimerization [5]. A clearer understanding of the mechanism of action of AraC-family inhibitors would provide critical insight for furthering their development.
Fig 1. Schematic Depicting Presumptive Steps in the Activation of Transcription by VirF.
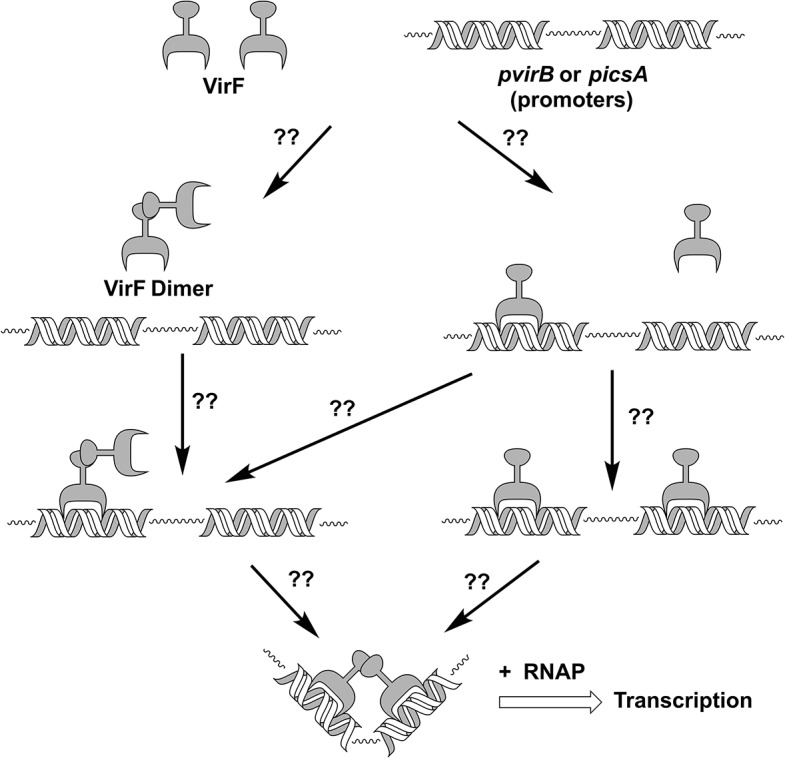
By analogy to AraC and family members, it is presumed that two VirF proteins bind to two proximal DNA binding sites in a dimeric form to activate transcription. The question marks indicate that the exact steps in this process and their order (if ordered) are not yet understood.
It has recently been shown that S. flexneri virulence can be attenuated via treatment with small molecules that inhibit VirF [26, 27]. VirF appears to be an ideal target for an anti-virulence therapy because a number of factors suggest that the likelihood of resistance to VirF inhibitors developing environmentally (outside of an infected host) should be quite low. For example, absent conditions that mimic those of an infected host, there should be little or no expression of VirF [10–13], therefore; the VirF-selective inhibitors should have no effect on Shigella spp. in the environment. Additionally, targeting virulence gene expression does not impair bacterial viability [28, 29], and hence, there should be little selective pressure in the environment for resistance development. Finally, in the infected host, Shigella utilize VirF-induced IpaB to escape from macrophages [17]. Inhibition of VirF should block this and increase the efficiency of macrophage killing Shigella and thereby reduce the development of resistance. Of course, these are postulates and require experimental testing to determine their validity.
We have previously reported the identification of five promising small molecule inhibitors of VirF (see Fig 2) from a high-throughput screening campaign (over 140,000 compounds screened) and a series of follow-up assays, including tissue-culture based invasion and cell-to-cell spread assays that model aspects of the infection process [27, 30]. All five compounds inhibited VirF-driven transcriptional activation in a Shigella-based, β-galactosidase reporter assay with IC50 values in the low micromolar range (14–66 μM). Furthermore, at concentrations at or below their IC50s in the reporter assay, three of the compounds (19615, 144092, 153578) inhibited the spread of an active S. flexneri infection by approximately 75% in a tissue-culture based plaque efficiency assay, and one of the compounds (144092) also inhibited initial S. flexneri invasion by approximately 50% in a gentamicin protection assay. These results, supported by similar results recently published by other groups [4, 5, 26], validate our approach by providing proof of principle that small molecules can attenuate virulence; however, the mechanism by which our compounds inhibit the VirF transcriptional activation process remained unclear (see Fig 1).
Fig 2. Structures of Previously Identified Small Molecule HTS Hit Compounds.
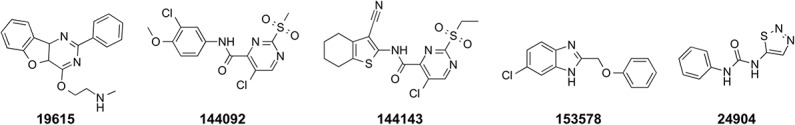
The compounds are referenced by their numbers in the University of Michigan, Center for Chemical Genomics (CCG) compound libraries.
In the studies herein reported, we have probed the mechanism of action of the small molecule inhibitors of VirF that we have discovered [27, 30]. To enable these studies, we have developed a homologous and efficient expression and purification system for VirF and have optimized two different in vitro assays which monitor VirF binding to the DNA promoter region for VirB (pvirB). We make the first report (to our knowledge) of a dissociation constant for VirF binding to DNA (i.e., pvirB) and provide strong evidence that inhibition of VirF binding to DNA is the mechanism of action of one of our hit compounds (19615, Fig 2). Finally, we screened a series of analogs of 19615 and have deduced an initial structure-activity relationship that we are using as a basis to further optimize our inhibitor and work towards achieving our goal of developing a novel therapy for treating shigellosis.
Results and Discussion
To determine the mechanism of action of the compounds and further validate this approach, an efficient preparation of VirF was developed and in vitro assays that directly monitor VirF binding to DNA were optimized.
MalE-VirF Purification
VirF is an AraC-family transcriptional activator. AraC transcriptional activators can function monomerically or as homodimers [31]. VirF is a member of the homodimer class. Unfortunately, AraC-family members, especially those of the homodimer class, are difficult to isolate due to low solubility and poor heterologous expression [32]. Recently, Egan and co-workers reported a VirF purification protocol that utilized a previously developed maltose binding protein (MalE) fusion [14] which improves VirF solubility and allows for purification via amylose resin chromatography [26]. We had previously attempted many different protocols to purify this MalE-VirF fusion protein, but had very little success due to poor heterologous expression in various E. coli strains. The recently published protocol expressed MalE-VirF in E. coli KS1000 from an IPTG-inducible pMALvirF vector [26]. Our initial attempts to purify VirF which relied on this expression/purification system produced results typically seen in other E. coli heterologous expression systems: low overall yield and large amounts of MalE impurity (as shown via SDS-PAGE analysis in Fig 3A).
Fig 3. SDS-PAGE of MalE-VirF Purifications (Heterologous vs. Homologous Expression).

A) SDS-PAGE analysis of MalE-VirF expression in E. coli KS1000 cells via pMALvirF expression vector. MalE-VirF is 73 kDa in weight and the main impurity, MalE is 43 kDa in weight. Numbers at the top of the gel represent the fraction (1 mL volume) number eluting off the amylose resin column. LMW stands for low molecule weight ladder; the numbers on the right-hand side of the gel correspond to the sizes of the LMW bands. B) SDS-PAGE analysis of MalE-VirF expression in S. flexneri BS103 cells via pBAD202-MALvirF expression vector. MalE-VirF is 73 kDa in weight and the main impurity, MalE, is 43 kDa in weight. Numbers at the top of the gel represent the fraction (1 mL volume) number eluting off the amylose resin column. LMW stands for low molecule weight ladder; the numbers on the right-hand side of the gel correspond to the sizes of the LMW bands.
To improve the expression and purification, an arabinose-inducible vector, pBAD202-MALvirF was constructed that allowed for homologous expression of MalE-VirF in S. flexneri BS103 cells. As shown in Fig 3B, this expression system, when used with the same amylose resin purification procedure, greatly improved overall yield (approximately 1.5–2.5 mg per liter of culture) and purity of the MalE-VirF preparation (over 80% pure). Analytical gel filtration chromatography indicates that the purified MalE-VirF exists in solution as a monomer at concentrations used in subsequent experiments (see Fig A in S1 File).
Inhibition of DNA Binding via Electrophoretic Mobility Shift Assay (EMSA)
Researchers at Paratek Pharmaceuticals recently identified a series of benzimidazole compounds [3], similar to 153578, that inhibited the ability of LcrF, an AraC-family transcriptional activator in Yersinia (and subsequently a number of other AraC-family proteins), to bind to its DNA promoter region [4]. Egan and co-workers also recently found a small molecule that inhibited the ability of multiple AraC-family members, including VirF, to bind DNA [26]. These studies prompted us to focus our initial efforts on developing in vitro assays to monitor VirF binding to its DNA promoter site to determine if our compounds inhibit DNA binding.
To monitor MalE-VirF binding to the virB promoter, an EMSA was optimized that utilized a fluorescently-labeled pvirB DNA fragment. The pvirB fragment was 74 bp long, with 60 bp corresponding to the previously determined [14] pvirB region, and 14 bp corresponding to a 5’-Cy5 labeled LUEGO (labeled universal electrophoretic gel shift oligonucleotide) site. Control experiments were conducted to verify that MalE-VirF was specifically binding to the 5’Cy5-labeled pvirB DNA probe (see Fig B in S1 File) and that this binding was dose-dependent. It was observed that binding of MalE-VirF to the virB promoter could not be monitored unless the pH of the gel/running buffer was 9.5. If the pH was lower than 9.5, the protein/DNA complex would not enter the gel matrix (data not shown). Also, the MalE tag was left on the N-terminal of VirF to help prevent VirF precipitation and aggregation during the course of the assay. The MalE-tag has been previously shown to have little effect on VirF activity [14, 27, 30] and did not prevent visualization of binding in the EMSA (see Fig 4).
Fig 4. Electrophoretic Mobility Shift Assay (EMSA) PAGE of MalE-VirF Binding to pvirB DNA Probe in Presence of Small Molecule Hit Compounds.

EMSA image shows the retardation of the 5’Cy5-pvirB DNA probe (0.25 μM) when incubated in the presence of MalE-VirF (1 μM). Image also shows the effect each of the five hit compounds have on MalE-VirF binding. All compounds were dosed at 100 μM, and only one compound, 19615, appeared to inhibit MalE-VirF binding to the pvirB DNA probe.
After the EMSA conditions were optimized, it was used to determine if our five previously identified hit compounds (see Fig 2) could inhibit binding of MalE-VirF to the pvirB at 100 μM. As shown in Fig 4, one compound, 19615, dramatically reduced binding, while the other four compounds were indistinguishable from the DMSO negative control. Surprisingly, 153578, the benzimidazole-derivative hit compound from our HTS, had little to no effect on DNA binding under these conditions. To determine if 19615 is a totally non-specific inhibitor of protein-DNA binding, we performed a control EMSA that revealed no effect on E. coli RNA polymerase binding to the lac promoter in the presence and absence of 19615 (see Fig C in S1 File).
DNA Binding via Fluorescence Polarization (FP)
To confirm and quantitate the results of the EMSA, a Fluorescence Polarization (FP) assay was developed. The FP assay utilized reaction conditions similar to the EMSA, with the only major change being to the pvirB DNA fragment. For the FP assay, the pvirB fragment was shortened to 60 bp with the LUEGO site being removed and was labeled with a 5’Fluoroscein instead of Cy5. These changes increased the FP signal generated in the assay by decreasing the molecular weight of the fragment and increasing the fluorescent lifetime of the probe. Control experiments were conducted to verify that: equilibrium was reached (anisotropy signal did not increase after 20 minutes), binding was specific (MalE-VirF binding to labeled pvirB DNA probe could be competed by unlabeled pvirB DNA probe and MalE-VirF would not bind to sequence scrambled DNA probe, see Fig B in S1 File), and maximum anisotropy signal was achieved. Once optimized, the FP assay was utilized to determine the K D of MalE-VirF binding to the pvirB DNA probe. The data shown in Fig 5 was fitted with a specific binding equation (see Methods) and the experimental K D was determined to be 2.8 ± 1.0 μM. To our knowledge this is the first reported K D for VirF binding either of its promoter regions. It has been shown that VirF only activates the transcription of the virB gene when supercoiled DNA is used as a template [33]. Therefore, we should note that it is possible that our experimental K D, determined with a linear DNA fragment may be different than the in vivo K D, under physiological conditions where VirF is activating supercoiled DNA.
Fig 5. Plot of MalE-VirF Binding to the pvirB DNA Probe.

For the assay, 5’Fluoroscein-pvirB DNA probe concentration was held constant at 50 nM, while MalE- VirF concentration was varied from 45 μM to 0.12 μM. The observed binding max for MalE-VirF binding was approximately r = 75, while the observed baseline (no MalE-VirF, only free pvirB DNA probe) was r = 42. The assay was conducted in duplicate. The inset depicts the reaction being monitored, VirF (not known if monomer or dimer) binding to the virB promoter.
Inhibition of DNA Binding via Fluorescence Polarization (FP)
The FP assay protocol was used to test the ability of our HTS hit compounds to inhibit MalE-VirF binding to the pvirB DNA probe. MalE-VirF was held constant at 20 μM, a concentration higher than its K D value while still sub-saturating, to balance the magnitude of the anisotropy signal and the sensitivity to inhibition. Consistent with the EMSA study, the five compounds were tested at 100 μM, and only 19615 inhibited DNA binding (63% inhibition, see Fig 6). These results confirmed our findings in the EMSA assay, and suggest that the other four compounds are inhibiting VirF activity at different steps of the gene activation process subsequent to DNA binding. (Another, albeit unlikely possibility is that the four other compounds may be metabolized in vivo to species that do inhibit VirF-DNA binding. This will be determined as we continue to investigate their mechanisms of action.) If different molecular mechanisms of inhibition for the different compounds are confirmed, then the development of multiple compounds may circumvent any resistance and/or toxicity issues that could arise during further optimization of any one of the compounds.
Fig 6. Results of 19615 Analog Testing in FP Assay.

To further evaluate the potency of 19615 in the FP assay, a dose-response study was performed (see Fig 7A); from which an IC50 of 46 ± 2.2 μM was determined. Applying this IC50 to the Cheng-Prusoff equation produces a K i for 19615 of 5.6 μM. In our previous study [27], we determined an IC50 for 19615 in a Shigella-based, VirF-driven, β-galactosidase reporter assay to be 14 μM and showed that 19615 inhibited the spread of an active S. flexneri infection by 75% at 6.25 μM (see Fig 7B). The correlation between these results and the K i for inhibition of VirF binding to DNA strongly suggest that 19615 attenuates the virulence of S. flexneri by decreasing VirF-driven transcriptional activation via inhibition of VirF-DNA binding. This, combined with the fact that 19615 was not toxic to mammalian cells or S. flexneri at the tested concentrations [27], makes 19615 an attractive candidate for further exploration.
Fig 7. Characterization of Hit Compound 19615.
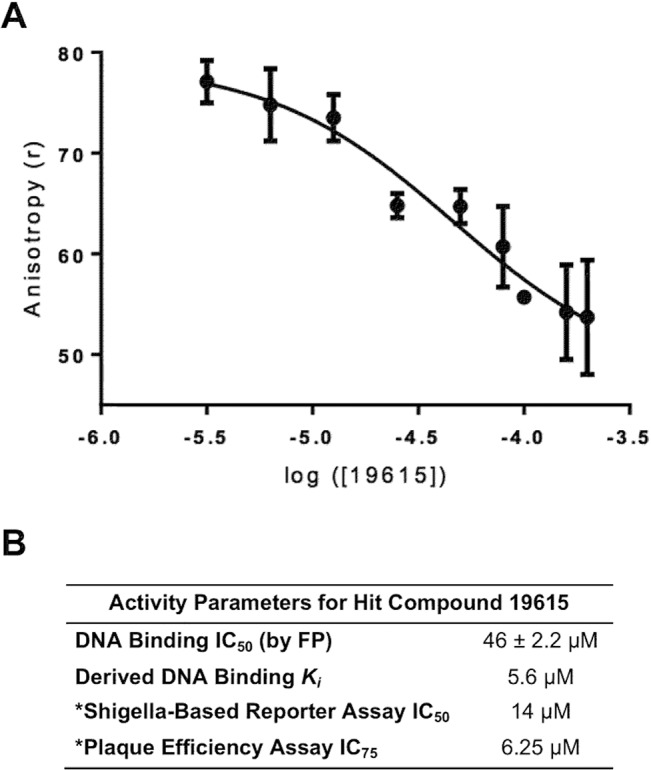
A) Plot of inhibition of MalE-VirF binding to the pvirB DNA probe by 19615. Assayed as in Fig 5 with the MalE-VirF concentration constant at 20 μM. B) Parameters for 19615. An IC50 of 46 ± 2.2 μM for DNA binding was determined, which, by the Cheng-Prusoff equation, yields a derived K i = 5.6 μM. The assay was conducted in duplicate. * Data from Emanuele et al. J. Antibiotics 67:379–386 (2014).
DNA Affinity Determination via a Fluorescence Intercalator Displacement (FID) Assay
To determine if 19615 was inhibiting VirF from binding to the pvirB by directly binding to the DNA a FID assay was conducted. FID assays are commonly used to establish the DNA binding affinity and selectivity of small molecules [34, 35]. In this case, the assay tested the ability of 19615 and Berenil, a known minor groove binder with a preference for AT-rich sequences [34, 36], to displace ethidium bromide from pvirB DNA probes. Two different pvirB probes were used in the study: a full length 60 bp probe, pvirB FID, and a 10 bp probe, pvirB 51–60 FID. A preliminary FID screen was conducted to determine which segment of the pvirB to use for the 10 bp probe and the results are shown in Table A in S1 File. As shown in Table 1, Berenil was able to displace ethidium bromide from the pvirB DNA probes and lower the fluorescence signal generated in the assay to produce low %fluorescence values for all experimental conditions; on the other hand, 19615 was not able to displace ethidium bromide from the pvirB DNA probes resulting in high %fluorescence values for all experimental conditions. These results indicate that 19615 has low affinity for the pvirB at concentrations used throughout the course of this study and suggest that 19615 inhibits VirF from binding to the pvirB via direct interaction with VirF.
Table 1. Results of the Fluorescence Intercalator Displacement Assay.
| Compound | 60 bp pvirB FID probe %fluorescence | 10 bp pvirB 51–60 FID probe %fluorescence |
|---|---|---|
| 12.5 μM 19615 | 93% ± 3% | 97% ± 2% |
| 25 μM 19615 | 97% ± 2% | 97% ± 3% |
| 50 μM 19615 | 95% ± 2% | 97% ± 2% |
| 100 μM 19615 | 93% ± 3% | 95% ± 1% |
| 12.5 μM Berenil | 76% ± 2% | 52% ± 1% |
| 25 μM Berenil | 67% ± 2% | 51% ± 2% |
| 50 μM Berenil | 58% ± 1% | 51% ± 1% |
| 100 μM Berenil | 48% ± 2% | 46% ± 1% |
Initial SAR of 19615 Inhibition of VirF-DNA Binding
The next logical step towards achieving our goal of developing an anti-virulence therapy for treating shigellosis is to structurally optimize the hit compounds to improve their potency. To construct an initial SAR, a small library of analogs of compound 19615 were purchased and screened in the FP assay. The FP assay was chosen over other assays previously developed (e.g., Shigella-based reporter assay, tissue-culture based virulence assays) because it eliminates a number of complicating factors associated with measuring efficacy in the other assays, such as permeability and cytotoxicity. As shown in Fig 6, one of the 19615 analogs (598089) was essentially equal in potency to 19615 (68% versus 63% inhibition, respectively) and the rest of the compounds displayed lower %inhibition values (ranging from 43% to -5%).
It is worth noting that the goal of testing this panel of analogs was primarily to provide supporting evidence for the mechanism of action of 19615 and secondarily to find a compound that was more potent than compound 19615. Some notable trends identified from the data include (a) preference of a phenyl ring substituent over smaller alkyl chains attached to the pyrimidine (e.g., 19615 vs. 582610 or 587469), (b) preference of side chain ether rather than amine linkage to pyrimidine (e.g., 587109 vs. 048373), and (c) preference for the dimethyl amine over the diethyl amine on the ether side chain (19615 vs. 587109 and 582610 vs. 050908). The data also suggest that there is considerable space for favorable SAR development through the installation of additional basic side chain ethers and aromatic moieties off the pyrimidine ring. Fig 8 depicts the most promising trends that will serve as the starting point for later generations of 19615 analogs with the goal of developing a more potent inhibitor.
Fig 8. Figure Depicting Deduced SAR.

Structure shown is a hybrid compound containing most promising substituents and will serve as a starting point for future synthetic SAR studies that will probe the core heterocycle. Region A: SAR data suggest the preferred substituent at this position is an ether chain with a 2–3 carbon spacer and small aminoalkyl headpiece. Region B: SAR data suggest the preferred substituent at this position is an aromatic ring, although further probing with substituted phenyl and heteroaromatic moieties is needed.
Conclusion
In this report, we have determined the mechanism of VirF inhibition by compound 19615 and have constructed an initial SAR to be used for further development. In the course of the study, we made the first report of a dissociation constant for VirF binding to the virB promoter, and refined binding assays and a homologous expression system that will be useful in our further studies of VirF. However, at the present time, we do not yet have direct evidence to indicate that 19615's effects are specific to VirF versus other AraC-family transcription factors, although we have determined that 19615 does not interfere with RNAP binding to DNA (see Fig C in S1 File). There is precedent for small molecules to exhibit cross-reactivity against multiple AraC-family AraC-family members [2, 3, 37]. To examine the specificity of 19615, we plan to test it (or a more potent analogue) in EMSAs against a variety of other AraC-family regulators that are known to activate virulence cascades in other organisms, such as ToxT from Vibrio cholera and LcrF from Yersinia pestis. Once specificity is determined, we will conduct mutagenesis studies to further probe the interactions between the compound and its target(s). We also plan on determining the mechanism of action of the other hit compounds from our high-throughput screen, conducting a synthetic SAR study of 19615 to probe the core heterocyclic structure, and continuing our work towards the development of an anti-virulence therapy to treat shigellosis.
Material and Methods
Reagents
All reagents were purchased from Fisher Scientific (Hampton, NH), unless otherwise specified. Compound 19615 was purchased from Vitas-M Laboratories (Moscow, Russia), while compounds 144092, 144143, 153578, and 24904 were purchased from ChemDiv (San Diego, CA). Analogs of 19615 were purchased through Aldrich Market Select (St. Louis, MO). Berenil was purchased from Sigma Aldrich (St. Louis, MO). Vendors for other reagents are specified throughout the text.
Strains and Plasmids
Escherichia coli KS1000 was purchased from New England Biolabs (Ipswich, MA). A virulence plasmid-cured derivative of wild-type S. flexneri, BS103 [38], was also used in this study. Two different expression vectors were used in this study: pMALvirF and pBAD202-MALvirF. Both vectors encode for a maltose binding protein–VirF fusion protein, MalE-VirF. pMALvirF was constructed by cloning the virF gene into the vector, pMAL-c2x (New England Biolabs), as previously described [14, 30]. pBAD202-MALvirF was constructed using the pBAD directional TOPO expression kit (Invitrogen, Carlsbad, CA). Briefly, the malE-virF fusion gene was amplified from pMALvirF via polymerase chain reaction to include a 5’-NcoI restriction site before the start codon of malE-virF. The amplified gene was then subcloned into pBAD202 via a directional TOPO® cloning reaction. The resulting vector was then subjected to NcoI restriction digestion (5 units, 1 hour, 37°C) to remove the N-terminal His-Patch thioredoxin leader sequence from pBAD202, and form pBAD202-MALvirF. The sequences of both expression vectors were confirmed by DNA sequencing (DNA Sequencing Core Facility, University of Michigan).
Expression and Purification of MalE-VirF
Initial expression and purification experiments were conducted using pMALvirF and E. coli KS1000 as previously described [26]. Subsequent experiments utilized pBAD202-MALvirF to express MalE-VirF in S. flexneri BS103 as follows. Using a MicroPulser Electroporator (BioRad, Hercules, CA), pBAD202-MALvirF was transformed into electrocompetent S. flexneri BS103 cells. Starter cultures (10 mL) of pBAD202-MalVirF BS103 were grown overnight in 2xTY broth (16 g bactotryptone, 10 g yeast extract, 5 g NaCl per liter of water) supplemented with kanamycin (50 μg/mL) at 37°C with shaking. The following day the starter culture was used to inoculate 1 L of 2xTY broth supplemented with kanamycin. The cells were grown to an OD600 = 0.5. Expression of MalE-VirF was induced with the addition of arabinose (0.2% final concentration) and the culture continued to shake at 37°C for an additional 5 hours. Cells were then harvested via centrifugation (6000 X g, 4°C, 15 minutes) and were stored overnight at -20°C. The next day the cells were resuspended in 20 mL of amylose resin binding buffer (20 mM Tris-HCl, 500 mM NaCl, 1 mM EDTA, 1 mM, pH = 7.4) supplemented with phenylmethylsulfonyl fluoride (0.1 mM) and 20 μL of lysonase bioprocessing reagent (EMD Millipore, Billerica, MA). Cells were slowly stirred for 10 minutes at room temperature and were then immediately placed on ice and kept on ice or at 4°C for the remainder of the procedure. Cells were lysed via sonication (8 cycles, 10 second pulse time, 2 minute intervals, max pulse setting) utilizing a ultrasonic XL2020 sonicator (Misonix, Farmingdale, NY). Following sonication, cellular debris were removed via centrifugation (25,000 X g, 4°C, 40 minutes). The resultant supernatant was then applied to a 10 mL column of amylose resin (New England Biolabs) by gravity flow. Before addition of the supernatant the column was washed with 8 column volumes of amylose resin binding buffer. Following addition of the supernatant, the column was washed with 12 column volumes of amylose resin binding buffer. MalE-VirF was eluted from the column in 1 mL fractions of amylose resin elution buffer (amylose resin bind buffer plus 15% glycerol (wt/vol) and 10 mM maltose). Fractions were analyzed by SDS-PAGE. Fractions containing MalE-VirF were pooled, concentrated to approximately 6.5 mg/mL using Amicon Ultra-15 centrifugal units (EMD Millipore), and stored in liquid nitrogen for future use.
Analytical Gel Filtration
Analytical gel filtration chromatography was used to determine the oligomeric state of purified MalE-VirF. Briefly, MalE-VirF (0.75 mg/mL) was applied to a Superose 12 column, which was equilibrated with amylose resin binding buffer using an AKTA FPLC system (both from GE Healthcare Life Sciences, Piscataway, NJ). The sample was run through the column at a flow rate of 0.75 mL/min using amylose resin binding buffer. Eluted proteins were detected spectrophotometrically at 280 nm. The oligomeric state of MalE-VirF was determined by comparison to a previously generated 4-point molecular weight calibration curve specific to the Superose 12 column.
DNA Probe Hybridization
DNA probes were utilized in both EMSA, FP, and FID assays. The sequences of the oligonucleotides were based on previous studies [26, 39] and are listed in Table 2. All oligonucleotides were purchased from Invitrogen, except LUEGO which was purchased from Integrated DNA Technologies (Coralville, IA). Each oligonucleotide was brought up to a final concentration of 10 μM in TE/NaCl buffer (10 mM Tris-HCl, 1 mM EDTA, 50 mM NaCl, pH = 8.0). For EMSA experiments, oligonucleotides were mixed at the following ratio: 10 volumes LUEGO, 5 volumes Top, 1 volume Bottom. For FP and FID experiments, oligonucleotides were mixed 1:1 (Top:Bottom). Annealing was performed using a Mastercycler Nexus thermocycler (Eppendorf, Hauppauge, NY) with the following program: 94°C for 2 minutes, cool down at 2°C/sec to 70°C and hold for 2 minutes, cool down at 0.1°C/sec to 20°C and hold for 2 minutes.
Table 2. Oligonucleotides used in EMSA, FP, and FID experiments.
| Name | Sequence (5’-3’) | Modification | Length (bases) |
|---|---|---|---|
| pvirB Top EMSA | AGAATATTATTCTTTTATCCAATAAAGATAAATTGCATCAATCCAGCTATTAAAATAGTA | None | 60 |
| pvirB Bottom EMSA | TACTATTTTAATAGCTGGATTGATGCAATTTATCTTTATTGGATAAAAGAATAATATTCTCCAGACCAGGGCAC | None | 74 |
| pScram Top EMSA | TAAGTCCTAAATGGAAATTAAATTACGTAATTCACAAATATAGTATGATCATTTATATCA | None | 60 |
| pScram Bottom EMSA | TGATATAAATGATCATACTATATTTGTGAATTACGTAATTTAATTTCCATTTAGGACTTACCAGACCAGGGCAC | None | 74 |
| LUEGO | GTGCCCTGGTCTGG | 5’-Cy5 | 14 |
| pvirB Top FP | AGAATATTATTCTTTTATCCAATAAAGATAAATTGCATCAATCCAGCTATTAAAATAGTA | 5’-Fluoroscein | 60 |
| pvirB Bottom FP | TACTATTTTAATAGCTGGATTGATGCAATTTATCTTTATTGGATAAAAGAATAATATTCT | None | 60 |
| pScram Top FP | TAAGTCCTAAATGGAAATTAAATTACGTAATTCACAAATATAGTATGATCATTTATATCA | 5’-Fluoroscein | 60 |
| pScram Bottom FP | TGATATAAATGATCATACTATATTTGTGAATTACGTAATTTAATTTCCATTTAGGACTTA | None | 60 |
| pvirB Top FID | AGAATATTATTCTTTTATCCAATAAAGATAAATTGCATCAATCCAGCTATTAAAATAGTA | None | 60 |
| pvirB Bottom FID | TACTATTTTAATAGCTGGATTGATGCAATTTATCTTTATTGGATAAAAGAATAATATTCT | None | 60 |
| pvirB (51–60) Top FID | TAAAATAGTA | None | 10 |
| pvirB (51–60) Bottom FID | TACTATTTTA | None | 10 |
DNA Binding via Electrophoretic Mobility Shift Assay (EMSA)
Reactions for the EMSAs were incubated in a 37°C water bath for 15 minutes. Reactions (15 μL total volume) were composed of 6 μL pvirB EMSA DNA probe (0.25 μM), 6 μL of either MalE-VirF (1.0 μM) or native gel loading buffer (Tris-HCl 0.3 M, 50% glycerol, 0.05% bromophenol blue, pH 7.0), 1 μL of salmon sperm DNA (0.7 mg/mL, Invitrogen), 0.5 μL BSA (0.07 mg/mL), and 1.5 μL of either DMSO or compound (100 μM). A 6% native polyacrylamide gel (29:1 acrylamide to bis-acrylamide ratio) was made with 0.25X TBE buffer (22 mM Tris Base, 22 mM boric acid, 0.5 mM EDTA, pH 9.5) for the EMSA. The gel was electrophoresed for 1 hour at 150 V in 0.25X TBE buffer at 4°C before samples were loaded. After the reaction solutions (12 μL) were loaded onto the gel, the gel was electrophoresed for an additional hour at 150 V and 4°C. The gel was then visualized using FluorChem M gel imager (Protein Simple, Santa Clara, CA) with a 607 nm excitation wavelength and a 710 nm emission filter.
DNA Binding via Fluorescence Polarization (FP)
The FP assays were conducted in duplicate in black, half-area, 96-well plates (Corning, Twerksbury, MA). First, 30 μL of 5’-Fluoroscein-pvirB DNA probe working standard was added to appropriate wells of the microplate. The pvirB DNA working standard was in TE/NaCl buffer (see above) supplemented with BSA (0.07 mg/mL) and salmon sperm DNA (0.7 mg/mL). Next, 30 μL of varying concentrations of MalE-VirF in amylose resin elution buffer (see above) was added to appropriate wells of the microplate. The final concentration of pvirB DNA was either 50 nM (for test wells) or none (for blank wells) and the final concentrations of MalE-VirF ranged from 45 μM to 0.12 μM (for test wells) or none (for control wells). For each concentration of MalE-VirF tested, a blank well was also set up that included all reagents except pvirB DNA. The microplate was then incubated for 20 minutes at 37°C and anisotropy was determined using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Data was acquired using an excitation wavelength equal to 490 nm and an emission wavelength equal to 520 nm. To determine anisotropy the following equation was used:
where: Fǁ = fluorescence intensity parallel to excitation source after blanking, F┴ = fluorescence intensity perpendicular to excitation source after blanking, and G = G-factor (correction for polarization bias of the detection system). After anisotropy was determined, data were plotted using GraphPad Prism (La Jolla, Ca) and fit to the following equation:
where Y = specific binding, KD = dissociation constant, BMAX = maximum binding, X = MalE-VirF concentration, and b = y-intercept.
On average, there was a 65% increase in anisotropy when VirF was added to the DNA. Within a single set of experiments, the DNA (alone) fluorescence anisotropy values varied ~4%., whereas, between experiments these anisotropy values varied ~15%. The latter variation is most likely due to differences in the efficiency of duplex DNA formation.
Inhibition of DNA Binding via Fluorescence Polarization (FP), IC50 Determination
The FP protocol listed above was adapted to determine the IC50 of the small molecule hit compounds. Compounds (ranging from 200 μM to 3.1 μM) or DMSO for controls (1% final concentration) were added to the pvirB DNA probe working standard prior to addition to the microplates. For IC50 determination, the MalE-VirF and pvirB DNA probe concentrations were held constant at 20 μM and 50 nM, respectively. Positive controls were established to determine 100% VirF inhibition (no MalE-VirF present, 50 nM pvirB DNA probe) and negative controls were set up to determine no VirF inhibition (1% DMSO, 20 μM MalE-VirF, and 50 nM pvirB DNA probe). Once again blanks were set up to include all reagents, except pvirB DNA probe. All reactions were run in duplicate. Anisotropy was calculated as listed above and data were plotted using GraphPad Prism then fit to the following equation:
where: Y = anisotropy, X = log of compound concentration (M), Top and Bottom = plateaus in anisotropy units, and LogIC50 = log of the concentration that inhibits 50% of VirF binding. In the presence of the compounds (no VirF), a small increase (~6%) in the DNA anisotropy was observed. This did not change over the concentrations tested and may be due to electrostatic interactions between the compounds and the DNA (assuming that the lowest compound concentration of ~3 μM would saturate the 50nM DNA probe).
Inhibition of DNA Binding via FP, %Inhibition Determination
The FP protocol used for IC50 determination was adapted to test a library of small molecule inhibitors of VirF. All steps were identical to the above protocol except compounds were tested at only 100 μM final concentration and positive control samples included 100 μM of compound. Once again blanks were set up to include all reagents, except pvirB DNA probe, and all reactions were run in duplicate. Anisotropy was calculated as listed above, and %inhibition was calculated as follows:
where: rneg = average anisotropy for negative controls and rtest = average anisotropy in presence of compound. Note: All rneg and rtest values were normalized to positive control values.
Fluorescence Intercalator Displacement (FID) Assay
The FID protocol was based on previously published studies [34, 40] and was conducted in triplicate. For the assay, black, half-area, 96-well plates (Corning, Twerksbury, MA) were used. First, 70 μL of ethidium bromide in Tris Buffer (0.1 M Tris HCl, 0.1 M NaCl, pH 8.0) was added to all wells. The concentration of ethidium bromide was contingent upon the length of the DNA probe that was used. For the 60 bp pvirB FID DNA probe, the final ethidium bromide concentration used was 45 μM; whereas, for the 10 bp pvirB (51–60) FID DNA probe, the final ethidium bromide concentration used was 7.5 μM. Next, 10 μL of either pvirB FID DNA probes (10 or 60 bp) in TE/NaCl buffer (see above) or 10 μL of TE/NaCl buffer (background fluorescence control) were added to the appropriate wells of the plate. The final concentrations of the pvirB FID DNA probes were 1.5 μM. Lastly, 20 μL of test compound (either 19615 or Berenil) in 10% DMSO Tris Buffer or 20 μL of 10% DMSO Tris Buffer (100% fluorescence control) were added to the appropriate wells of the plate. The final concentrations of each test compound ranged from 12.5 μM to 100 μM. After the addition of all reagents, the plates were protected from light and allowed to equilibrate for 30 min on an orbital shaker at room temperature. Following equilibration, fluorescence was measured (545 nm emission, 595 nm excitation) using a Biotek Synergy H1 plate reader (Winooski, VT). The average background fluorescence was subtracted from all data generated and %fluorescence was calculated for all wells containing compound via the following formula:
where: Ftest = the average fluorescence value obtained for each test compound and F100% = the average fluorescence value obtained for each 100% fluorescence control.
Supporting Information
I) Chromatogram depicting elution of MalE-VirF (11.04 mL) from Superose 12 column. II) Chromatogram and four-point calibration curve for Superose 12 column used to determine molecular weight of MalE-VirF in S1-A Fig. Fig B: Negative Controls for EMSA and FP assay. I) EMSA image shows the retardation of the 5’Cy5-pvirB DNA probe (0.25 μM) when incubated in the presence of MalE-VirF (1 μM) and shows no retardation of the 5’Cy5-pScram DNA probe (0.25 μM) when incubated in the presence of MalE-VirF (1 μM). II) Graph depicting the anisotropy values generated in the FP assay for the 5’Fluorescein pScram probe alone (r = 39) and in the presence of MalE-VirF (r = 36). Experiments were conducted in duplicate with 50 nM pScram and 20 μM MalE-VirF. Table A: Fluorescence Intercalator Displacement Assay with 10 bp pvirB Probes. *pvirB 51–60 was selected for use in the dose-response FID assay since it was sensitive to Berenil (67%) and was the most sensitive to 19615 (92%) in this study. The differential affinity of Berenil for the various 10 BP fragments reflects its preference for specific AT-rich sequences. Fig C: EMSA depicting E. coli RNA Polymerase (RNAP) Binding to the lac Promoter (plac) in the Presence of 19615. EMSA image shows the retardation of a 5’Cy5-plac DNA probe (0.25 μM) when incubated in the presence of E. coli RNAP (2.7 μM) and also shows that compound 19615 has no effect on RNAP binding when tested at 100 μM. For the EMSA a hybrid 2% acrylamide, 1% agarose gel was used which was made with and ran in a 1X TGE buffer (25 mM Tris base, 190 mM glycine, 1 mM EDTA, pH 8.3). The sequence of the 5’Cy5-plac DNA probe is as follows: 5’-gtgccctggtctggTTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGAATTGTGAG-3’ (lowercase text represents LUEGO sequence, uppercase text represents lac promoter sequence).
(DOCX)
Acknowledgments
The authors gratefully acknowledge Eric Lachacz (Department of Medicinal Chemistry, University of Michigan) for contributions made to the construction of the homologous expression vector for VirF. We also acknowledge Dr. Amanda Garner (Department of Medicinal Chemistry, University of Michigan) for the use the SpectraMax M5 plate reader in her laboratory and Dr. Hollis Showalter (Department of Medicinal Chemistry, University of Michigan) for valuable input and suggestions regarding the SAR. Finally, we would like to acknowledge Dr. Jennifer Meagher of the Center of Structural Biology, Life Sciences Institute, University of Michigan, for use of the analytical gel filtration chromatography system.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors thank the National Institutes of Health (AI085179 to GAG and GM007767, AAE trainee) for funding.
References
- 1.WHO. Antimicrobial Resistance Global Report On Surveillance. 2014.
- 2. Bowser TE, Bartlett VJ, Grier MC, Verma AK, Warchol T, Levy SB, et al. Novel anti-infection agents: small-molecule inhibitors of bacterial transcription factors. Bioorg Med Chem Lett. 2007; 17(20): 5652–5. 10.1016/j.bmcl.2007.07.072 [DOI] [PubMed] [Google Scholar]
- 3. Kim OK, Garrity-Ryan LK, Bartlett VJ, Grier MC, Verma AK, Medjanis G, et al. N-hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J Med Chem. 2009; 52(18): 5626–34. 10.1021/jm9006577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garrity-Ryan LK, Kim OK, Balada-Llasat JM, Bartlett VJ, Verma AK, Fisher ML, et al. Small Molecule Inhibitors of LcrF, a Yersinia pseudotuberculosis Transcription Factor, Attenuate Virulence and Limit Infection in a Murine Pneumonia Model. Infect Immun. 2010; 78(11): 4683–90. 10.1128/IAI.01305-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Small-Molecule Inhibitor of Vibrio cholerae Virulence and Intestinal Colonization. Science (New York, NY). 2005; 310(5748): 670–4. 10.1126/science.1116739 [DOI] [PubMed] [Google Scholar]
- 6. Adler B, Sasakawa C, Tobe T, Makino S, Komatsu K, Yoshikawa M. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol Microbiol. 1989; 3(5): 627–35. 10.1111/j.1365-2958.1989.tb00210.x [DOI] [PubMed] [Google Scholar]
- 7. Schroeder GN, Hilbi H. Molecular Pathogenesis of Shigella spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clin Microbiol Rev. 2008; 21(1): 134–56. 10.1128/CMR.00032-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter ME, Dorman CJ. Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Mol Gen Genet. 1997; 256(2): 93–103. 10.1007/s004380050550 [DOI] [PubMed] [Google Scholar]
- 9. Sakai T, Sasakawa C, Yoshikawa M. Expression of four virulence antigens of Shigella flexneri is positively regulated at the transcriptional level by the 30 kiloDalton virF protein. Mol Microbiol. 1988; 2(5): 589–97. 10.1111/j.1365-2958.1988.tb00067.x [DOI] [PubMed] [Google Scholar]
- 10. Nakayama S, Watanabe H. Involvement of CpxA, a sensor of a 2-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J Bacteriol. 1995; 177(17): 5062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter ME, Dorman CJ. A role for H-NS in the thermo-osmotic regulation of virulence gene expression in Shigella flexneri . J Bacteriol. 1994; 176(13): 4187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tobe T, Nagai S, Okada N, Adler B, Yoshikawa M, Sasakawa C. Temperature-regulated expression of invasion genes in Shigella flexneri is controlled through the transcriptional activation of the virB gene on the large plasmid. Mol Microbiol. 1991; 5(4): 887–93. 10.1111/j.1365-2958.1991.tb00762.x [DOI] [PubMed] [Google Scholar]
- 13. Falconi M, Colonna B, Prosseda G, Micheli G, Gualerzi CO. Thermoregulation of Shigella and Escherichia coli EIEC pathogenicity. A temperature-dependent structural transition of DNA modulates accessibility of virF promoter to transcriptional repressor H-NS. EMBO J. 1998; 17(23): 7033–43. 10.1093/emboj/17.23.7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tobe T, Yoshikawa M, Mizuno T, Sasakawa C. Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by virF and repression by H-NS. J Bacteriol. 1993; 175(19): 6142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tran CN, Giangrossi M, Prosseda G, Brandi A, Di Martino ML, Colonna B, et al. A multifactor regulatory circuit involving H-NS, VirF and an antisense RNA modulates transcription of the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 2011; 39(18): 8122–34. 10.1093/nar/gkr521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, et al. The Tripartite type III secretion of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol. 1999; 147(3): 683–93. 10.1083/jcb.147.3.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. 1998; 273(49): 32895–900. 10.1074/jbc.273.49.32895 [DOI] [PubMed] [Google Scholar]
- 18. Espina M, Olive AJ, Kenjale R, Moore DS, Ausar FS, Kaminski RW, et al. IpaD Localizes to the tip of the Type III Secretion System Needle of Shigella flexneri . Infect Immun. 2006; 74(8): 4391–400. 10.1128/IAI.00440-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989; 86(10): 3867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. May KL, Morona R. Mutagenesis of the Shigella flexneri Autotransporter IcsA Reveals Novel Functional Regions Involved in IcsA Biogenesis and Recruitment of Host Neural Wiscott-Aldrich Syndrome Protein. J Bacteriol. 2008; 190(13): 4666–76. 10.1128/JB.00093-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zumsteg AB, Goosmann C, Brinkmann V, Morona R, Zychlinsky A. IcsA is a Shigella flexneri Adhesion Regulated by the Type III Secretion System and Required for Pathogenesis. Cell Host Microbe. 2014; 15(4): 435–45. 10.1016/j.chom.2014.03.001 [DOI] [PubMed] [Google Scholar]
- 22. Colonna B, Casalino M, Fradiani PA, Zagaglia C, Naitza S, Leoni L, et al. H-NS regulation of virulence gene expression in enteroinvasive Escherichia coli harboring the virulence plasmid integrated into the host chromosome. J Bacteriol. 1995; 177(16): 4703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sansonetti PJ, Arondel J, Fontaine A, d'Hauteville H, Bernardini ML. OmpB (osmo-regulation) and icsA (cell-to-cell spread) mutants of Shigella flexneri: vaccine candidates and probes to study the pathogenesis of shigellosis. Vaccine. 1991; 9(6): 416–22. 10.1016/0264-410X(91)90128-S [DOI] [PubMed] [Google Scholar]
- 24. Makino S, Sasakawa C, Kamata K, Kurata T, Yoshikawa M. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell. 1986; 46(4): 551–5. 10.1016/0092-8674(86)90880-9 [DOI] [PubMed] [Google Scholar]
- 25. Porter ME, Dorman CJ. In vivo DNA-binding and oligomerization properties of the Shigella flexneri AraC-like transcriptional regulator VirF as identified by random and site-specific mutagenesis. J Bacteriol. 2002; 184(2): 531–9. 10.1128/JB.184.2.531-539.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koppolu V, Osaka I, Skredenske JM, Kettle B, Hefty PS, Li J, et al. Small-molecule inhibitor of the Shigella flexneri master virulence regulator VirF. Infect Immun. 2013; 81(11): 4220–31. 10.1128/IAI.00919-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emanuele AA, Adams NE, Chen Y-C, Maurelli AT, Garcia GA. Potential novel antibiotics from HTS targeting the virulence-regulating transcription factor, VirF, from Shigella flexneri. J Antibiot. 2014; 67(5): 379–86. 10.1038/ja.2014.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sasakawa C, Kamata K, Sakai T, Makino S, Yamada M, Okada N, et al. Virulence-Associated Genetic Regions Comprising 31 Kilbases of the 230-Kilobase Plasmid in Shigella flexneri 2a. J Bacteriol. 1988; 170(6): 2480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mills JA, Venkatesan MM, Baron LS, Buysse JM. Spontaneous Insertion of an IS1-Like Element into the virF Gene Is Responsible for Avirulence in Opaque Colonial Variants of Shigella flexneri 2a. Infect Immun. 1992; 60(1): 175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hurt JK, McQuade TJ, Emanuele A, Larsen MJ, Garcia GA. High-Throughput Screening of the Virulence Regulator VirF: A Novel Antibacterial Target for Shigellosis. J Biomol Screen. 2010; 15(4): 379–87. 10.1177/1087057110362101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin RG, Rosner JL. The AraC transcriptional activators. Curr Opin Microbiol. 2001; 4(2): 132–7. 10.1016/S1369-5274(00)00178-8 [DOI] [PubMed] [Google Scholar]
- 32. Schleif R. AraC protein: a love-hate relationship. Bioessays. 2003; 25(3): 274–82. 10.1002/bies.10237 [DOI] [PubMed] [Google Scholar]
- 33. Tobe T, Yoshikawa M, Sasakawa C. Thermoregulation of virB transcription in Shigella flexneri by sensing of changes in local DNA superhelicity. J Bacteriol. 1995; 177(4): 1094–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tse WC, Boger DL. A Fluorescent Intercalator Displacement Assay for Establishing DNA Binding Selectivity and Affinity. Acc Chem Res. 2004; 37(1): 61–9. 10.1021/ar030113y [DOI] [PubMed] [Google Scholar]
- 35. Glass LS, Bapat A, Kelley MR, Georgiadis MM, Long EC. Semi-automated high-throughput fluorescent intercalator displacement-based discovery of cytotoxic DNA binding agents from a large compound library. Bioorganic & medicinal chemistry letters. 2010; 20: 1685–8. 10.1016/j.bmcl.2010.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen B, Hamelberg D, Bailly C, Colson P, Stanek J, Brun R, et al. Characterization of a Novel DNA Minor-Groove Complex. Biophys J. 2004; 86: 1028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skredenske JM, Koppolu V, Kolin A, Deng J, Kettle B, Taylor B, et al. Identification of a Small-Molecule Inhibitor of Bacterial AraC Family Activators. Journal of biomolecular screening. 2010; 18(588–598). 10.1177/1087057112474690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maurelli AT, Blackmon B, Curtiss R III. Loss of pigmentation in Shigella flexneri 2a is correlated with loss of virulence and virulence associated plasmid. Infect Immun. 1984; 43: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jullien N, Herman J-P. LUEGO: a cost and time saving gel shift procedure. BioTechniques. 2011; 51: 267–9. 10.2144/000113751 [DOI] [PubMed] [Google Scholar]
- 40. Tse WC, Boger DL. A Fluorescent Intercalator Displacement Assay for Establishing DNA Binding Selectivity and Affinity. Curr Protoc Nucleic Acid Chem. 2005: 8.5.1–8.5.11. 10.1002/0471142700.nc0805s20 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
I) Chromatogram depicting elution of MalE-VirF (11.04 mL) from Superose 12 column. II) Chromatogram and four-point calibration curve for Superose 12 column used to determine molecular weight of MalE-VirF in S1-A Fig. Fig B: Negative Controls for EMSA and FP assay. I) EMSA image shows the retardation of the 5’Cy5-pvirB DNA probe (0.25 μM) when incubated in the presence of MalE-VirF (1 μM) and shows no retardation of the 5’Cy5-pScram DNA probe (0.25 μM) when incubated in the presence of MalE-VirF (1 μM). II) Graph depicting the anisotropy values generated in the FP assay for the 5’Fluorescein pScram probe alone (r = 39) and in the presence of MalE-VirF (r = 36). Experiments were conducted in duplicate with 50 nM pScram and 20 μM MalE-VirF. Table A: Fluorescence Intercalator Displacement Assay with 10 bp pvirB Probes. *pvirB 51–60 was selected for use in the dose-response FID assay since it was sensitive to Berenil (67%) and was the most sensitive to 19615 (92%) in this study. The differential affinity of Berenil for the various 10 BP fragments reflects its preference for specific AT-rich sequences. Fig C: EMSA depicting E. coli RNA Polymerase (RNAP) Binding to the lac Promoter (plac) in the Presence of 19615. EMSA image shows the retardation of a 5’Cy5-plac DNA probe (0.25 μM) when incubated in the presence of E. coli RNAP (2.7 μM) and also shows that compound 19615 has no effect on RNAP binding when tested at 100 μM. For the EMSA a hybrid 2% acrylamide, 1% agarose gel was used which was made with and ran in a 1X TGE buffer (25 mM Tris base, 190 mM glycine, 1 mM EDTA, pH 8.3). The sequence of the 5’Cy5-plac DNA probe is as follows: 5’-gtgccctggtctggTTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGAATTGTGAG-3’ (lowercase text represents LUEGO sequence, uppercase text represents lac promoter sequence).
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.