

Abstract
Apoptosis is a cellular death process involving the sequential activation of a series of caspases, endonucleases, and other enzymes. The initiation of apoptosis can be inhibited by overexpression of bcl-2 and certain other members of a related family of proteins. We examined the effects of bcl-2 overexpression on the apoptotic response to photodynamic therapy (PDT), using aluminum phthalocyanine as the photosensitizing agent. In this study, we compared the immortalized human breast epithelial cell line MCF10A with a subline (MCF10A/bcl-2) transfected with the human bcl-2 gene. The latter was ~2-fold more sensitive to the phototoxic effects of PDT. At a 50 mJ/cm2 light dose, photodamage to MCF-10A/bcl-2 resulted in a greater loss of the mitochondrial membrane potential (ΔΨm), enhanced release of mitochondrial cytochrome c, a more rapid and greater activation of caspase-3, and a greater apoptotic response. Western blot analysis revealed that the transfected cell line showed overexpression of both bcl-2 and bax, and that PDT caused selective destruction of bcl-2, leaving bax unaffected. The greater apoptotic response by the transfected line is, therefore, attributed to the higher bax:bcl-2 ratio after photodamage.
INTRODUCTION
PDT3 involves the irradiation of photosensitized cells, leading to photodamage at subcellular sites where the photosensitizing agent has accumulated (1). The first report of apoptotic cell death after PDT appeared in 1991 (2). At the time, little was known regarding mechanisms involved in programmed cell death, but we now know that initiation of the apoptotic program can be triggered by the translocation of cytochrome c from mitochondria to the cytosol (3). PDT using a photosensitizing agent that mediates selective mitochondrial photo-damage can initiate a rapid apoptotic response if other proteins required for expression of apoptosis are undamaged (4, 5); otherwise, a necrotic outcome can result (6). Overexpression of bcl-2 inhibits apoptosis by antagonizing release of mitochondrial cytochrome c (7, 8). In a finding consistent with these observations, He et al. (9) reported that bcl-2 transfection of a Chinese hamster ovary cell line led to partial resistance to apoptotic cell death after PDT. In this report, we describe the effect of bcl-2 transfection on the immortalized human breast epithelial cell line MCF10A (10), a near-diploid cell line that appeared during long-term culture of breast tissue in low-calcium medium. We had anticipated that this procedure would also antagonize PDT-induced apoptosis in this system, but a contrary result provided the rationale for this investigation.
MATERIALS AND METHODS
Cell Culture Conditions
The development and characterization of the MCF10A cell line have been described elsewhere (10). We reported previously a procedure for preparation of bcl-2-overexpressing MCF10A clones (11). Briefly, the bcl-2 gene, under the control of cytomegalovirus promoter (provided by Dr. S. Korsmeyer), was introduced into MCF10A cells using Lipofectin (Sigma). Stable transfectants were selected in the presence of 400 μg/ml G418, and individual clones were isolated and characterized.
The human bax cDNA in a pSFFV-neo expression vector (provided by Dr. S. Korsmeyer, Washington University School of Medicine, St. Louis, MO) was introduced into MCF10A cells using FuGENE 6 reagent (Boehringer Mannheim). Fifty % confluent cells in a 60-mm dish were transfected in 3 ml of culture medium containing 100 μl of FuGENE 6 reaction mixture. The FuGENE 6 mixture was prepared as follows; 2 μg of expression vector or control vector was mixed with 5 μl of FuGENE6 in 100 μl of serum free-medium. The mixture was incubated at room temperature for 30 min before transfection. Cells were harvested 36–48 h later.
Cells were cultured in 5% CO2 in DMEM/F12 supplemented with 5% horse serum, 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 20 ng/ml epidermal growth factor, 0.1 μg/ml cholera enterotoxin, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, and 0.5 μg/ml fungizone.
Photosensitizer
AlPc was obtained from Kodak and recrystallized from α-chloronaphthalene. Stock solutions of AlPc in ethanol were diluted 1:1000 into cell suspensions for these studies to yield a final concentration of 0.3 μM. Accumulation of AlPc by the two cell lines was measured after incubation for 15 min at 37°C. The ratio of intracellular versus extracellular AlPc was determined by comparing the fluorescence in media versus extracts of washed cells.
Fluorescent Probes
Fluorescent probes for the mitochondrial membrane potential (MTO) and for nuclear morphology (HO342) were purchased from Molecular Probes (Eugene, OR). Fluorescence studies were carried out with a Nikon Labophot microscope fitted with a digital video camera (Photometrics, Tucson, AZ). A 600-nm low-pass filter was inserted into both the excitation path and the camera entry port to reduce transmission of infra-red light from the mercury source, which will otherwise fog the CCD detector. Images were processed with MetaMorph software (Universal Imaging Corp., West Chester, PA).
Photodynamic Therapy and Its Consequences
Photosensitized cell cultures were irradiated at 10°C, using specified light doses. Light was provided by a 600 W quartz-halogen lamp with IR radiation removed by 10 cm of water and an 850-nm cutoff filter. The bandwidth was further confined to 660 ± 5 nm by an interference filter (Oriel). The effect of photodamage on the mitochondrial membrane potential (ΔΨm) was assessed directly after irradiation or 4 h later, using the fluorescent probe MTO. PDT-induced changes in nuclear morphology were examined 4 and 24 h after irradiation by labeling cells with HO342. For the latter determination, three fields of 100 cells were surveyed, and the percentage of apoptotic nuclei was reported. These procedures have been described previously (4, 12). Viability studies were carried out using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 96 h after PDT (13).
Immunoblot Analysis
Extracts were prepared from 106 cells in 125 mM Tris-HCl (pH 6.8) buffer containing 2% SDS and 10% glycerol. The protein concentration was measured using BCA protein assay reagents (Pierce, Inc., Rockford, IL). Protein samples were heated to 100°C for 10 min in the presence of 5% β-mercaptoethanol, chilled on ice, and subjected to SDS-PAGE analysis, followed by electrophoretic transfer to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in 100 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.02% NaN3, and 0.2% Tween-20 (T-TBS) for 1 h at room temperature and incubated with anti-bcl-2, anti-bax, or anti-β-actin antibodies in T-TBS. After several washes with T-TBS, the blot was incubated with a horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibody. The antigen was detected using the ECL detection system (Pierce) according to the manufacturer’s instruction. Release of cytochrome c from mitochondria into the cytosol was assessed by Western blot analysis (5) using control cells and in cells photosensitized and treated with a light dose of 50 mJ/cm2.
Caspase-3 Activity
Cells were collected after incubation at 37°C after irradiation (50 or 100 mJ/cm2) and then lysed in 50 mM Tris buffer (pH 7.5) containing 0.03% Nonidet and 1 mM DTT. Nuclei were removed by low-speed centrifugation (800 × g, 5 min), and the cytosol fraction was incubated with 40 μM DEVD-afc, 10 mM HEPES (pH 7.5), 50 mM NaCl, and 2.5 mM DTT in a total volume of 200 μl for 120 min at 37°C. Fluoromethylcoumarin fluorescence, released by caspase activity, was measured using 360-nm excitation. A CCD device (Instaspec IV; Oriel, Stratford, CT) fitted with a monochromator was used to measure the fluorescence emission spectrum. The intensity at the optimum (~450 nm) was measured. Units of enzyme activity are measured as pmols of substrate hydrolyzed/mg cells/mg protein/60 min. In all cases, control determinations were made on extracts of untreated cells. The system was calibrated with known levels of trifluoromethylcoumarin.
RESULTS
MCF10A is an adhering human breast epithelial cell line derived from human fibrocystic tissue and immortalized by long-term passage in calcium-deficient medium (10). We used this line, and a stable subline transfected with bcl-2, to examine the effect of bcl-2 overexpression on the apoptotic response to PDT. The photosensitizing agent was AlPc, an agent that initiated apoptosis in either of two murine leukemia cell lines, with mitochondria being the predominant target (6). After a 15-min incubation, AlPc was concentrated ~110-fold by MCF10A or MCF10A/bcl-2, i.e., there was no difference in concentrative ability after transfection. There was, however, a marked difference in PDT responsiveness; bcl-2 transfection resulted in a decrease in the light dose required for 90% loss of viability (Fig. 1).
Fig. 1.

Loss of viability of MCF10A (○) and MCF10A/bcl-2 (●) after photodynamic therapy. Cells were incubated with 0.3 μM AlPc and then irradiated with the specified light dose. Survival is expressed as a percentage of control (cells exposed to drug in the dark) using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 96 h after irradiation. Data are reported as means of three experiments; bars, SD.
Four h after irradiation, using a light dose of 50 mJ/cm2, we observed <5% apoptotic cells in MCF10A and small but detectable loss in ΔΨm, as distinguished by the MTO labeling pattern (Fig. 2). In contrast, a substantial number of apoptotic cells were detected in the transfected subline, along with a markedly diminished intensity of MTO fluorescence (Fig. 2). When the incubation time was prolonged to 24 h after irradiation, greater numbers of apoptotic nuclei were detected in both cell lines (Table 1). Increasing the light dose promoted the apoptotic response in both cell lines, but MCF10A/bcl always showed the greater response.
Fig. 2.
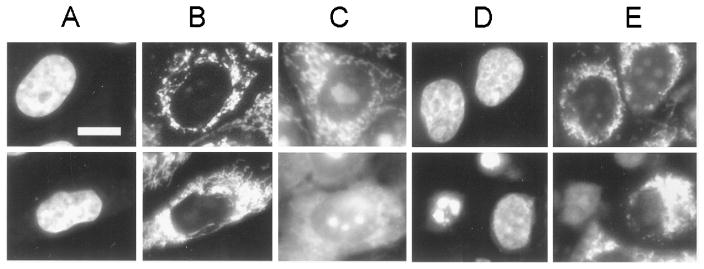
Effects of photodynamic therapy with AlPc (light dose, 50 mJ/cm2) on MCF10A (top) and MCF10A/bcl2 (bottom) labeled with HO342 or MTO. A, HO342 control; B, MTO control; C, MTO label directly after irradiation; D, HO342 label 4 h after irradiation; E, MTO label 4 h after irradiation. Bar, 10 μm.
Table 1. DEVDase activity and apoptotic morphology in control and photodamaged cells.
Cells were treated with 0.3 μM AlPc for 15 min and then irradiated with 50 or 100 mJ/cm2 of red light. DEVDase activity (units = pmols/mg protein/60 min) was measured after an additional incubation for 4 or 24 h in growth medium at 37°C. Results are mean ± SD of three determinations. Dark controls were assayed 24 h after transfection. The percentage of apoptotic cells was estimated from a survey of three fields of 100 cells labeled with HO342.
| MCF10Aa
|
||||||
|---|---|---|---|---|---|---|
| 0 mJ/cm2b
|
50 mJ/cm2b
|
100 mJ/cm2b
|
||||
| DEVDase | % apoptotic | DEVDase | % apoptotic | DEVDase | % apoptotic | |
| Dark controls | 0.5 ± 0.1 | <2 | ||||
| PDT + 4 h | 1.4 ± 0.3 | <5 | 7 ± 3 | 31 ± 6 | ||
| PDT + 24 h | 26.3 ± 2.7 | 25 | 65 ± 6 | 60 ± 5 | ||
|
| ||||||
| MCF10A/bcl-2a | ||||||
|
| ||||||
| Dark controls | 0.6 ± 0.3 | <2 | ||||
| PDT + 4 h | 13 ± 3.2 | 41 ± 4 | 24 ± 5 | 75 ± 4 | ||
| PDT + 24 h | 102 ± 16 | 65 ± 7 | 110 ± 9 | 83 ± 3 | ||
|
| ||||||
| MCF10A/baxa | ||||||
|
| ||||||
| Dark controls | 25 ± 4.1 | 21 ± 4 | ||||
| PDT + 4 h | 36 ± 5.9 | 52 ± 5 | ||||
Cell line.
Light dose.
Western blot analysis for bcl-2 and bax was carried out before and immediately after irradiation. Compared with the parent MCF10A, the transfected line exhibited enhanced levels of both bcl-2 and bax (Fig. 3). After irradiation at 10°C, there was a complete loss of the bcl-2 signal in MCF10A, and the appearance of a band of lower molecular weight (Fig. 3, Lane 2), representing a photodegradation product. In the transfected line, the bcl-2 signal was markedly diminished, and the degradation product was also detected. At a 150 mJ/cm2 light dose, signals from both bcl-2 and the degradation product were abolished in both cell lines (not shown). In no case was there any effect on bax expression. Western blot analysis for cytosolic cytochrome c indicated that the level of translocation from the mitochondria was promoted by PDT and further promoted when MCF10A/bcl-2 cells were used (Fig. 3).
Fig. 3.
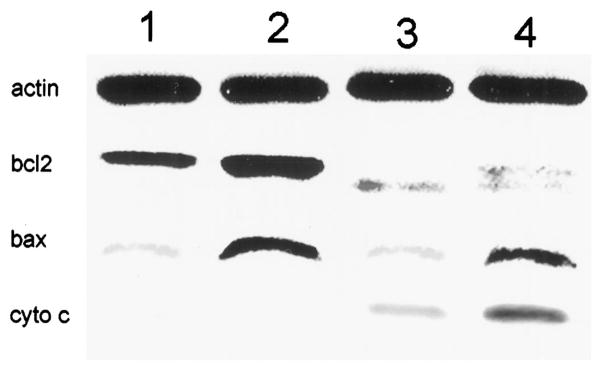
Western blot analysis of actin, bcl-2 (bcl2), bax, and cytochrome c (cyto c) in control cells and in cells directly after PDT was carried out at 10°C. Cells were photosensitized with AlPc and then irradiated with a light dose of 100 mJ/cm2.
1 = control, 2 = transfected, 3 = control + PDT, 4 = transfected + PDT
Using the fluorogenic substrate DEVD-afc, we could not detect caspase-3-like activity in either line before irradiation (Table 1). Four h after a 50 mJ/cm2 light dose to AlPc-sensitized cells, DEVDase activity was detected only in the transfected cell line. After 24 h, both lines exhibited enhanced levels of DEVDase activity; this was ~8-fold higher in the transfected cell line. Use of a 100 mJ/cm2 light dose increased the level of DEVDase activity in both cell lines.
Transfection of MCF10A with bax led to production of an unstable cell line, with DEVDase activation and apoptotic nuclear morphology detectable within 48 h (Table 1). The transfection efficiency was ~20%. These cells showed an enhanced apoptotic response to mitochondrial photodamage. In contrast, levels of DEVDase activity and numbers of apoptotic cells in control cultures and in MCF10A/bcl-2 cells were not elevated significantly.
DISCUSSION
The first report on PDT-initiated apoptosis (2) contained a proposal for a possible mechanism; mitochondrial photodamage could result in degradation of bcl-2 and related proteins that normally function as apoptotic suppressors (14). A later study demonstrated that transfection of a Chinese hamster ovary cell line with bcl-2 yielded a cell line 1.5-fold more resistant to PDT phototoxicity than the parent cell line (9).
In contrast to the latter report, we observed a different pattern. A subline of MCF10A transfected with bcl-2 was more sensitive to the lethal effects of PDT (Fig. 1), showed a greater apoptotic response (Fig. 2), and a more rapid and extensive activation of caspase-3-like activity (Table 1), compared with the parent cell line. We had reported previously that PDT, resulting in mitochondrial photodamage, caused the release of cytochrome c into the cytosol (5), a translocation that can initiate the apoptotic program (3).
Because bcl-2 inhibits the loss of cytochrome c from mitochondria (7, 8), it might be anticipated that bcl-2 overexpression would protect cells from PDT-induced apoptosis. An explanation for the enhanced PDT responsiveness of MCF10A/bcl-2 was suggested by the finding that the transfection led to overexpression of both bcl-2 and bax (Fig. 3). The stabilization of bax by bcl-2 overexpression has been reported before (15). Stabilization of bax in the MCF10A/bcl-2 cell line did not affect the inhibition of apoptosis initiated by a variety of stimuli including ionizing radiation, hydrogen peroxide, and calcium ionophores (11, 16). Because mitochondrial photodamage catalyzed by AlPc resulted in the selective degradation of bcl-2 without affecting bax, the enhanced apoptotic response to PDT in the MCF10A/bcl-2 cell line can therefore be attributed to the elevated bax:bcl-2 ratio after PDT. A high ratio is known to promote the initiation of apoptosis (17).
The critical function of bcl-2 gene product is the prevention of cytochrome c release from mitochondria, inhibiting activation of caspases at the outer mitochondrial membrane (8). However, increasing evidence suggests a pleiotropic role for bcl-2 in apoptosis regulation (16, 18, 19). In addition to the mitochondrial membrane, bcl-2 is also localized to the outer nuclear and endoplasmic reticulum. We showed previously that bcl-2 increased calcium uptake through the up-regulation of the endoplasmic reticulum calcium pump expression and enhanced lumenal Ca2+ concentration of endoplasmic reticulum in MCF10A cells (16). A recent report showed that bcl-2 activates the transcription factor nuclear factor-κB through degradation of its inhibitor IκBa (19). bcl-2 was also shown to inhibit activation of c-Jun NH2-terminal kinases, which are known to play a role in apoptosis and gene expression (18). These results suggest that inhibition of apoptosis by bcl-2 may involve regulation of gene expression through modulation of central signaling molecules, including Ca2+ flux, transcription factors, and kinase activity. Consistent with this, we showed previously that bcl-2 down-regulates p21WAF1/CIP1 expression, an inhibitor of cyclin-dependent kinases, in MCF10A cells. Although bcl-2 down-regulated p21WAF1/CIP1 expression at confluent cells or after irradiation, serum-induced p21WAF1/CIP1 was not inhibited by bcl-2 (11). Modulation of p21WAF1/CIP1 and calcium pump expression by bcl-2 suggests that the role of bcl-2 in the inhibition of apoptosis may be related to its ability to regulate cell cycle progression. However, it is unlikely that PDT-induced apoptosis depends on cell cycle distribution, because apoptosis is rapidly induced after mitochondrial photodamage.
In the present study, we propose that the enhanced PDT responsiveness of MCF10A/bcl-2 results from bcl-2-mediated increase in the stability of bax, a proapoptotic gene product. A transient bax transfection was found to lead to both spontaneous apoptosis and an enhanced apoptotic response to PDT (Table 1). Bax may promote apoptosis by triggering pore-forming activity in the mitochondrial membrane (20) and can promote release of cytochrome c from isolated mitochondria (21). Moreover, bcl-2 can antagonize bax-induced initiation of apoptosis after release of cytochrome c (22). It was reported previously that the caspase-mediated conversion of bcl-2 to a bax-like molecule can also increase the apoptotic response (23). Caspase-induced bcl-2 degradation after PDT is not likely to be significant under conditions reported here, because the degradation was observed immediately after irradiation at 10°C (Fig. 3) and is considered to be the direct result of photodamage.
The suggestion made by Agarwal et al. (2), that photodamage to bcl-2 could promote an apoptotic response to PDT, can now be put into the proper context. PDT-induced mitochondrial photodamage is now known to cause release of cytochrome c into the cytosol and the triggering of apoptosis (4–6). Moreover, bcl-2 overexpression can lead to impaired apoptosis after PDT (9). If, however, bcl-2 overexpression also leads to stabilization of bax, selective bcl-2 photodamage can result in a high bax:bcl-2 ratio and an enhanced apoptotic response to mitochondrial photodamage.
Acknowledgments
Excellent technical assistance was provided by Ann Marie Santiago and Kathleen Gallagher.
Footnotes
This investigation was supported by Grants CA 641349, CA 23378, and CA 65561 from the National Cancer Institute, NIH.
The abbreviations used are: PDT, photodynamic therapy; AlPc, aluminum phthalocyanine; MTO, MitoTracker Orange (Molecular Probes); DEVD-afc, asp-glu-val-asp-aminofluoro coumarin; HO342, Höchst dye 33342 (bis-benzamide).
References
- 1.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal ME, Clay ME, Harvey EJ, Evans HH, Antunez AR, Oleinick N. Photodynamic therapy induces rapid cell death by apoptosis in L5178Y mouse lymphoma cells. Cancer Res. 1991;51:5993–5996. [PubMed] [Google Scholar]
- 3.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 4.Kessel D, Luo Y. Mitochondrial photodamage and PDT-induced apoptosis. J Photochem Photobiol B Biol. 1998;42:89–95. doi: 10.1016/s1011-1344(97)00127-9. [DOI] [PubMed] [Google Scholar]
- 5.Kessel D, Luo Y. Photodynamic therapy: a mitochondrial inducer of apoptosis. Cell Death Differ. 1999;6:28–35. doi: 10.1038/sj.cdd.4400446. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Kessel D. Initiation of apoptosis vs. necrosis by photodynamic therapy with chloroaluminum phthalocyanine. Photochem Photobiol. 1997;66:479–483. doi: 10.1111/j.1751-1097.1997.tb03176.x. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Liu X, Bhalla K, Kim CN, Ibrado A, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science (Washington DC) 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 8.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science (Washington DC) 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 9.He J, Larkin HE, Friedman LR, Xue LY, Oleinick NL. The induction of partial resistance to photodynamic therapy by the protooncogene BCL-2. Photochem Photobiol. 1996;64:845–852. doi: 10.1111/j.1751-1097.1996.tb01845.x. [DOI] [PubMed] [Google Scholar]
- 10.Soule H, Maloney TM, Wolman SR, Peterson WD, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 11.Upadhyay S, Li G, Liu H, Chen YQ, Sarkar FH, Kim H-RC. bcl-2 suppresses expression of p21WAF1/CIP1 in breast epithelial cells. Cancer Res. 1995;55:4520–4524. [PubMed] [Google Scholar]
- 12.Kessel D, Luo Y, Deng Y, Chang CK. The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem Photobiol. 1997;65:422–426. doi: 10.1111/j.1751-1097.1997.tb08581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHale AP, McHale L. Use of a tetrazolium based colorimetric assay in assessing photoradiation therapy in vitro. Cancer Lett. 1988;30:315–321. doi: 10.1016/0304-3835(88)90293-5. [DOI] [PubMed] [Google Scholar]
- 14.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyachita T, Kitadam A, Krajewski S, Horne WA, Delia D, Reed JC. Overexpression of the bcl-2 protein increases the half life of p21 bax. J Biol Chem. 1995;270:26049–26052. doi: 10.1074/jbc.270.44.26049. [DOI] [PubMed] [Google Scholar]
- 16.Kuo TH, Kim H-RC, Zhu L, Yu Y, Lin H-M, Tsang W. Modulation of endoplasmic reticulum calcium pump by bcl-2. Oncogene. 1998;17:1903–1910. doi: 10.1038/sj.onc.1202110. [DOI] [PubMed] [Google Scholar]
- 17.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 18.Frisch SM, Vuori K, Kelaita D, Sicks S. A role for Jun-N-terminal kinase in anoikis: suppression by bcl-2 and crmA. J Cell Biol. 1996;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Moissac D, Mustapha S, Greenberg AH, Kirshenbaum LA. Bcl-2 activates the transcription factor NFκB through the degradation of the cytoplasmic inhibitor IκBα. J Biol Chem. 1998;273:23946–23951. doi: 10.1074/jbc.273.37.23946. [DOI] [PubMed] [Google Scholar]
- 20.Antonsson B, Conti F, Ciavatta A, Montessiut S, Lewis S, Martinou I, Bernasconi I, Bernard A, Mermod J-J, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou J-C. Inhibition of bax channel-forming activity by Bcl-2. Science (Washington DC) 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 21.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature (Lond) 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 23.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science (Washington DC) 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]