

Abstract
Nicotine addiction drives tobacco use by one billion people worldwide, causing nearly six million deaths a year. Nicotine binds to nicotinic acetylcholine receptors that are normally activated by the endogenous neurotransmitter acetylcholine. The widespread expression of nicotinic receptors throughout the nervous system accounts for the diverse physiological effects triggered by nicotine. A crucial influence of nicotine is on the synaptic mechanisms underlying learning that contribute to the addiction process. Here, we focus on the acquisition phase of smoking addiction and review animal model studies on how nicotine modifies dopaminergic and cholinergic signaling in key nodes of the reinforcement circuitry: ventral tegmental area, nucleus accumbens (NAc), amygdala, and hippocampus. Capitalizing on mechanisms that subserve natural rewards, nicotine activates midbrain dopamine neurons directly and indirectly, and nicotine causes dopamine release in very broad target areas throughout the brain, including the NAc, amygdala, and hippocampus. In addition, nicotine orchestrates local changes within those target structures, alters the release of virtually all major neurotransmitters, and primes the nervous system to the influence of other addictive drugs. Hence, understanding how nicotine affects the circuitry for synaptic plasticity and learning may aid in developing reasoned therapies to treat nicotine addiction.
Keywords: dopamine, acetylcholine, VTA, hippocampus, nucleus accumbens, memory
Introduction
The nervous system has evolved reward‐ and punishment‐based learning to guide individuals to select actions that sustain life and species. Addictive drugs activate and modify these innate learning circuits. Nearly a billion people around the globe smoke tobacco, driven mainly by the addictive plant alkaloid nicotine.1, 2 Tobacco use, most commonly by smoking, initiates a powerful addictive process, ensuring its persistent use despite willful attempts humans make to break out of the addiction. Consequently, tobacco use is the leading cause of preventable death, causing nearly six million deaths per year worldwide.3 Why is the human nervous system, in particular, susceptible to such a phenomenon? What are the key neural systems that nicotine influences to initiate the acquisition of this addictive drug use?
Nicotine is the primary addictive substance in tobacco.2, 4 In addition, environmental stimuli and sensory experiences associated with smoking play an important role in the addiction process.5, 6, 7 When smoked, nicotine enters the blood stream mainly through the lungs and reaches the brain in tens of seconds.8 In the bloodstream at pH 7.4, nicotine exists in charged (69%) and uncharged forms (31%).8 The uncharged form can pass through lipid membranes and directly alter intracellular signaling.9 It is the charged form that binds to the nicotinic acetylcholine receptors (nAChRs), which are ligand‐gated ion channels formed by homo‐ or heteropentameric subunit combinations. The subunits are grouped into two classes, α and β. In the mammalian brain there are eight α subunits (α2–α7, α9, and α10) and three β subunits (β2–β4). The nAChRs are distributed widely in the nervous system, including cortical and limbic regions that contribute to the addiction process, such as the cerebral cortex, hippocampus, amygdala, ventral tegmental area (VTA), and nucleus accumbens (NAc).10, 11, 12, 13, 14 In individual neurons, nAChRs are located in preterminal, presynaptic, axonal, dendritic, and soma compartments.12, 15 Hence, the influence of nicotine on brain function is determined, at least in part, by the receptors’ subtype‐specific localization, density, and functional properties.
Associative learning and addiction
Because mammals function within broad and highly variable environments in the world, they must perform behaviors and voluntary movements in order to achieve success and to avoid danger. It is also advantageous for them to learn the causal relationship between specific actions and outcomes. Behavioral psychologists have made significant progress in understanding the structure of the associations among stimuli (S), actions or responses (R), and outcomes (O). Learning these associations is one of the most fundamental capabilities of the nervous system of animals, ranging from worms to humans. When animals perform rewarded instrumental actions in the presence of a stimulus, they learn all combinations of binary associations among S, R, and O: namely, R–O, S–R, and S–O (Fig. 1). In addition, they learn the higher order association S–(R–O), where the stimulus becomes associated with the R–O relation.16 This higher order association of instrumental learning can be viewed as a Pavlovian association of S with R–O.
Figure 1.
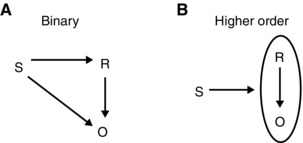
Animals learn multiple associations among stimuli (S), response (R), and outcome (O). These associations can be binary (A) such as S–R, R–O, and S–O, or higher order (B) such as S–(R–O). Adapted with permission from Ref. 16.
In general, these associations play a significant role in addiction. In particular, the nicotine addiction process resembles instrumental learning in some respects. For example, the smoker performs the instrumental action of lighting the cigarette and smoking it, with the outcome being (in some cases) positive effects such as a sense of well‐being, feeling more relaxed, reduced anxiety, enhanced working memory, increased alertness, better attention, increased cognition, relief of withdrawal symptoms, and so on, depending on the smoker and circumstances. In such a case, the smoker's nervous system likely learns several associations. First, it learns the R–O association between lighting the cigarette and the positive outcomes, motivating the smoker to repeat the action. Second, it learns the Pavlovian S–O and S–(R–O) associations, whereby the location and other environmental input from the smoking episodes gain salience and eventually lead to the initiation of smoking. Although these associations are rather simple, a diverse set of neuronal systems, including reward‐related structures, are involved in this process. Because an explicit comparison between natural learning and addiction at the mechanistic level is beyond the scope of this review, we will only briefly touch upon it. Our main focus instead will be on the mechanistic learning processes in the acquisition phase of the addictive process. Topics related to the maintenance and withdrawal phases of nicotine addiction are not considered here, but they have been reviewed elsewhere.13, 17, 18, 19, 20, 21
Learning circuitry modified by nicotine
In modern neurobiological studies, the two classically evaluated learning paradigms—Pavlovian conditioning and instrumental conditioning—have been brought under a common conceptual framework called reinforcement learning, where rewards and punishments act as positive and negative reinforcers, respectively. When a goal directed action is pursued, such as foraging for food, successful outcomes are marked by reward‐related signals. The reward signals are thought to play a crucial role in linking the precedent action sequence and cues with the positive experience of the outcome. The main sources of these reinforcement signals are the midbrain dopaminergic neurons located in the VTA and substantia nigra pars compacta (SNc).22, 23, 24, 25 Several crucial brain structures in the addiction process that receive dopamine (DA) include the prefrontal cortex, hippocampus, amygdala, and dorsal and ventral striatum (which includes the NAc). The response of dopaminergic neurons has been modeled as a reward prediction error that is thought to send a teaching signal that initiates plasticity in key learning and memory circuits.26, 27, 28 When appropriately cued, the learned sequences are repeated in order to receive the reward. This learning is important for survival, as it provides an efficient means of obtaining essential resources based on previous successful outcomes. Unfortunately, it is these learned associations among stimuli, action sequences, and rewards that all drugs of abuse (including nicotine) usurp to achieve their strong undesired effects on behavior. In the following, we will review how nicotine engages some of the key reward‐related structures to achieve its addictive influence.
Influences of nicotine over DA signaling by the VTA
The VTA consists of heterogeneous group of cells: the paranigral nucleus, the parainterfascicular nucleus, the parabrachial pigmented nucleus, and the rostral VTA.29 It is located bilaterally near the midline at the floor of the midbrain (Fig. 2).30 It is an important node in the reward‐based learning circuit (Fig. 2, top) and projects to many brain structures including the NAc, PFC, and hippocampus.31 The VTA dopaminergic neurons spontaneously fire at a tonic average rate of around 4 Hz (2–10 Hz range) with randomly interspersed bursts.32 During the acquisition phase of reward‐based learning, they fire briefly in phasic bursts, signaling and broadcasting the arrival of reward.33 Once the association between reward predicting cues and rewards is learned, the arrival of reward no longer elicits burst firing. Instead, the conditioned incentive stimuli that predict reward elicit burst firing,26, 28, 33, 34 and many conditioned behaviors depend on this burst firing.35 It should be noted that there are similarities and differences between natural rewards and drug reward.36 For example, in rats that have learned to self‐administer cocaine, similar to associative learning in food rewards,37 the presentation of a cue paired with cocaine elicits phasic DA release in the NAc.38 However, unlike natural rewards such as food, cocaine acting as a primary reward continues to elicit prolonged DA release even after learning the association between cocaine reward and the cues that predict it.39 Although such studies do not exist for nicotine addiction, nicotine likely acts in a similar fashion because nicotine also elicits prolonged burst firing in VTA DA neurons and causes prolonged DA release in the NAc.40, 41 This unnatural non‐habituating DA release has been hypothesized to be an important contributor to the excessive motivational power attributed to the cues and contexts associated with drug use.36, 42, 43
Figure 2.
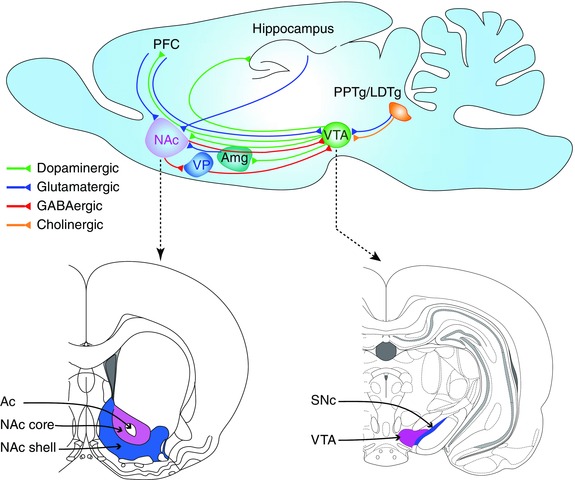
A simplified and incomplete schematic diagram (top panel) that highlights components of the circuitry contributing to reinforcement learning in rodents. Top panel: Sagittal section of a rodent brain didactically depicting strategic connections. Bottom panels: Coronal sections approximately at the anterior–posterior axis levels indicated by the dashed arrows, adapted from Figs. 20 and 74 of Ref. 29. Ac, anterior commissure; Amg, amygdala; LDTg, lateral dorsal tegmentum; NAc, nucleus accumbens; PFC, prefrontal cortex; PPTg, pedunculopontine tegmentum; SNc, substantia nigra pars compacta; VP, ventral pallidum; VTA, ventral tegmental area.
Although the VTA receives convergent inputs from many areas, prominent input sources, when considering reward‐based learning, include the pedunculopontine/lateral dorsal tegmentum (PPTg/LDTg)44, 45, 46, 47, 48, 49 and the PFC.50, 51, 52, 53 The PPTg sends glutamatergic/cholinergic projections to the VTA, and activation of PPTg elicits burst firing in VTA DA neurons.54 The PPTg is, in turn, activated by cortical and limbic inputs, including PFC and extended amygdala,55 and by sensory inputs from visual,56 auditory,56, 57 and somatosensory modalities.58 This convergence of inputs marks the PPTg as a crucial structure through which various behaviorally relevant inputs control the burst firing of VTA DA neurons.59 For instance, the PPTg/LDTg has been shown to control the conditioned responses of VTA DA neurons in behaving rats.56 The LDTg sends glutamatergic/cholinergic/ GABAergic projections to the VTA44, 60, 61 and plays a permissive role in the burst firing. This conclusion is supported by the observation that inactivation of the LDTg leads to severe reductions in burst firing62 and leads to tonic pacemaker‐like activity in DA neurons, similar to that found in brain slices.63 In addition, NMDA receptors play an essential role in burst firing, as suggested by the finding that the NMDA receptor antagonist AP‐5, infused into the VTA, reduces the burst firing markedly, whereas the non‐NMDA receptor antagonist CNQX has less influence over bursting.64 Although the PPTg and LDTg inputs significantly regulate burst firing in VTA neurons, the number of DA neurons that undergo burst firing is likely controlled by the ventral pallidum (VP). The VTA receives tonic inhibition from the VP,54, 65 leading to hyperpolarization of the DA neurons. This hyperpolarization likely results in Mg2+ block of the NMDA receptors, which in turn diminishes the burst firing. Removing the inhibition from the VP not only leads to an increase in the number of spontaneously active DA neurons54 but also likely leads to removal of Mg2+ block, thereby, permitting afferent inputs to elicit burst firing.66
When nicotine arrives in the brain, among its many actions is to excite the midbrain dopaminergic centers of the VTA and SNc.41 Nicotinic receptors are found on the GABAergic interneurons and DA neurons within the VTA and on the excitatory and inhibitory input axons and/or terminals.67, 68, 69, 70, 71 Although there are many types of nAChRs located on VTA DA neurons,72 the β2 subunit in combination with α4 and/or α6 subunits have been shown to be essential for nicotine addiction in animal models.73, 74, 75, 76, 77 In vivo recording from VTA DA neurons in anesthetized mice lacking the β2 subunit (Chrnb2 –/–) showed that spontaneous burst firing was absent in all of the recorded neurons.78 However, re‐expression of the β2 subunit selectively in the VTA restored the spontaneous firing patterns. In addition, the re‐expression also restored the nicotine‐induced increase in burst firing and intracranial self‐administration (ICSA) of nicotine into the VTA. Various subtypes of β2 containing nAChRs are located on both dopaminergic and GABAergic interneurons of the VTA. Interestingly, using in vivo recording in anesthetized Chrnb2 –/– mice, a recent study79 showed that re‐expression of the β2 subunit on DA neurons or GABA interneurons alone did not restore nicotine‐induced burst firing in DA neurons, whereas re‐expression in both cell types restored nicotine‐induced burst firing. In addition, re‐expression of the β2 subunit in either cell population alone did not sustain the ICSA behavior whereas re‐expression on both cell populations restored ICSA to the wild‐type level. These results show that the concerted activation of β2* nAChRs located on both dopaminergic and GABAergic interneurons is involved in nicotine self‐administration.59, 79
Under natural circumstances, usually processed information such as reward, novelty, and behaviorally salient stimuli gain access to the VTA to enable phasic burst firing.80 However, nicotine bypasses such preprocessing and directly activates VTA DA neurons to cause burst firing. In vivo tetrode recording from VTA DA neurons in behaving rats showed that an acute nicotine dose induces prolonged burst firing (Fig. 3) that can continue intermittently for more than 30 min,41 and causes DA release in the NAc (Fig. 4) that lasts for more than an hour.40, 81 When nicotine binds to nAChRs located on the soma of DA neurons, it causes direct depolarization, resulting in an increased firing rate. However, some nAChRs located on the soma desensitize quickly,71, 82 raising the question of how nicotine causes prolonged DA release.
Figure 3.
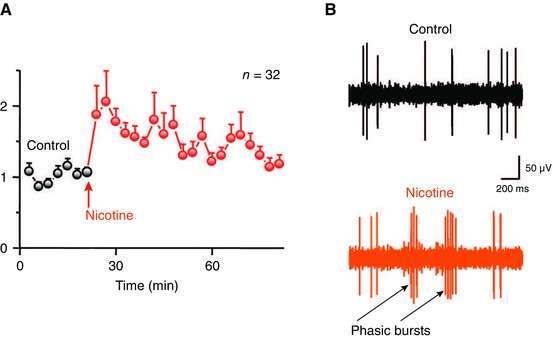
Nicotine increases burst firing in putative DA neurons within the VTA of freely moving rats. (A) Normalized average firing rate before (control, black) and after (nicotine, red) intraperitoneal (i.p.) injection of 0.4–0.5 mg/kg nicotine. Error bars represent SEM. (B) Representative traces from a single putative DA neuron before (black) and after (red) nicotine injection. Reproduced from Ref. 41.
Figure 4.
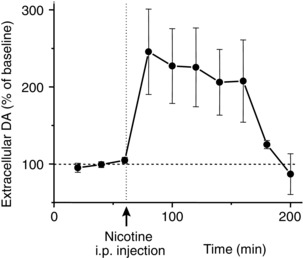
Nicotine (0.6 mg/kg, i.p.) increases DA release in the nucleus accumbens shell of freely moving rats as measured by microdialysis with HPLC. Error bars represent SEM. Modified with permission from Ref. 40.
Early in vitro studies indicated that both pre‐ and postsynaptic mechanisms contribute to nicotine‐induced prolonged DA release.40, 83, 84 The α7‐containing nAChRs (α7*) located at presynaptic glutamatergic terminals are highly permeable to calcium85 and are less prone to desensitization at the nicotine concentrations experienced by smokers.71 The increased Ca2+ entry acting through the vesicle release machinery enhances glutamate release onto DA neurons. Conversely, the high‐affinity α4β2 receptors located on the GABAergic neuron somas and terminals desensitize quicker, thereby, reducing the inhibitory drive onto the DA neurons.79 The net effect was thought to provide strong presynaptic drive to the DA neurons.83 Accordingly, when brief nicotine application was paired with postsynaptic depolarization, long‐term synaptic potentiation (LTP) was induced that lasted for many minutes and hours. Both the α7 antagonist methyllycaconitine (MLA) and the NMDA antagonist AP‐5 prevented the nicotine‐induced LTP in vitro,84 but in vivo, the role of α7 is more subtle.74 These experiments inspired studies in which nicotine was first given to the animals and VTA slices were then cut after various time periods. This approach revealed that nicotine causes its effect on VTA neurons in the intact animal, while still allowing in vitro techniques to measure the outcome. Consistent with the in vitro studies, a single dose of systemic nicotine increased the ratio of synaptic currents mediated by AMPA relative to NMDA receptors (AMPA/NMDA ratio) in VTA DA neurons, indicating increased numbers of AMPA receptors induced by LTP.86, 87, 88 This increase lasted up to 3 days (Fig. 5A) with a single nicotine dose and up to 8 days (Fig. 5B) after the cessation of 1 week of a daily nicotine dose.86 Although the early studies pointed out the crucial role of α7* presynaptic receptors in LTP, their roles are more subtle, as α7* gene knockout mice self‐administered nicotine to wild‐type levels.75
Figure 5.
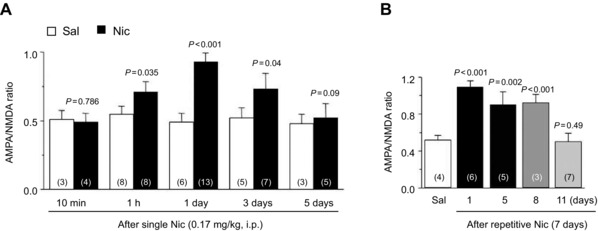
AMPA/NMDA ratio measured in slices prepared from mice that were given a single (A) or seven daily doses (B) of nicotine (0.17 mg/kg, i.p, filled bars) or saline (open bars). Error bars represent SEM. Modified with permission from Ref. 86.
What is the role of this increased AMPA current measured by the AMPA/NMDA ratio in nicotine addiction? An increased AMPA/NMDA ratio does not appear to be sufficient for nicotine addiction, as Chrnb2 –/– mice, which do not self‐administer nicotine, still show an increased AMPA/NMDA ratio.86 This indicates that the nicotine‐induced increased AMPA current on DA neurons by itself cannot establish addiction and additional factors are needed. It must also be recalled that β2‐containing nAChRs not only play a role in DA neuron activation in the midbrain, they also are vitally important in controlling DA release arising from action potentials reaching the DA neuron target areas.89, 90 Moreover, future studies need to establish the causal role of the potentiated AMPA currents (i.e., DA neuron LTP) in nicotine addiction. It is also currently unknown which synapses onto VTA DA neurons contribute to the increased AMPA current.89, 90 Although PFC glutamatergic inputs have been the focus of many studies, it is surprising that those inputs actually synapse onto VTA neurons that project mainly back to PFC, not to the NAc.50 On the contrary, DA neurons that receive inputs from PPTg project to the NAc,49 and it has been shown that those glutamatergic projections are modulated by nicotinic mechanisms.40 Hence, it is important for future studies to elucidate which synapses are actually strengthened by nicotine‐induced LTP. The role of metabotropic glutamate receptors (mGluRs) in the regulation nicotine‐mediated DA release also is important and has been recently reviewed.91
In addition, to understand the long‐lasting DA signal induced by nicotine, we must consider that nAChR desensitization to low nicotine concentrations is incomplete. The DA neurons from the posterior VTA that projection to the NAc commonly express the α6 subunit with the α4 and β2 subunits.14 The presence of the α6 subunit, particularly in α6α4β2* nAChRs, slows the rate and degree of desensitization seen with the higher affinity α4β2 nAChRs.92 Thus, the subtype of nAChR commonly expressed in DA neurons (i.e., α6α4β2*) is not as susceptible to desensitization as the higher affinity α4β2‐nAChR subtype, which is more common on GABA neurons.
Learning the smoking context through hippocampal processing
The hippocampus is involved in many diverse functions, including spatial navigation, working memory, transitive inference, arbitrary association, and temporal ordering of events.93, 94, 95 The hippocampus is anatomically in a strategic position to perform these functions because it receives highly processed information from diverse cortical and subcortical areas. While spatial information is conveyed mainly from the medial entorhinal cortex through the medial perforant path (Fig. 6), object information mainly from the lateral entorhinal cortex reaches the hippocampus through the lateral perforant path.96, 97 In addition, the hippocampus receives several neuromodulatory inputs, which include noradrenergic input from the locus coeruleus,98 serotonergic input from the raphe,99, 100 cholinergic input from the medial septum,101, 102, 103 and dopaminergic input mainly from the VTA.104 In addition, the dorsal (septal) and ventral (temporal) halves of the hippocampus have different input‐output configurations.105 Not surprisingly, the hippocampus has been implicated in a multitude of functions ranging from spatial navigation to anxiety. How does nicotine engage the hippocampus with such diverse functions in achieving its addictive influence?
Figure 6.
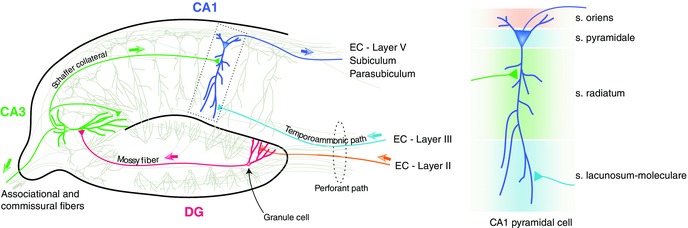
Greatly simplified and incomplete schematic diagram of hippocampal connections. EC, entorhinal cortex; CA, cornu ammonis; DG, dentate gyrus. The pyramidal neuron in the dotted rectangle is shown on the right with the different strata labeled (s = stratum in the labels).
Given that a wide variety of nAChR subtypes have been reported throughout the hippocampus,12 one important likely role played by the hippocampus in addiction is to process the contextual information associated with nicotine use. Nicotine has been shown to induce LTP in the medial perforant path‐DG synapses, the mossy fiber CA3 synapses, and the Schaffer collateral CA1 synapses (reviewed in Ref. 106). A large number of behavioral studies also support the idea that nicotine improves learning and memory performance in various tasks (reviewed in Ref. 107). It is interesting to note that in the hippocampus the GABAergic interneurons express much higher densities of nAChRs than the pyramidal cells,108, 109, 110, 111 and recent studies112, 113 have started examining the crucial role of a special class of inhibitory neurons in hippocampal plasticity. These neurons are called oriens lacunosum moleculare, or OLM interneurons, as their cell bodies reside in the stratum oriens and their axonal projections go up to the stratum lacunosum moleculare (Fig. 6, right). Activation of these GABAergic neurons by nicotine results in facilitating the Schaffer collateral pathway by inhibiting the inhibitory interneurons that synapse onto the pyramidal cell dendrites at the stratum radiatum (Fig. 7). On the other hand, OLM interneurons inhibit the temporoammonic (TA) pathway by directly synapsing at principal neuron dendrites in the stratum lacunosum moleculare. The Schaffer collateral pathway is hypothesized to bring information retrieved from CA3 to CA1, while the temporoammonic pathway brings sensory information directly from the entorhinal cortex.114, 115 Accordingly, CA1 is thought to compare these two streams of inputs in order to determine the novelty of the information.116 Controlling the information flow in this way by OLM interneurons using endogenous acetylcholine may serve specific functions. For example, selectively blocking the TA pathway may facilitate retrieval of stored representations from CA3. However, future studies need to address how such control of information flow leads to nicotine addiction.
Figure 7.
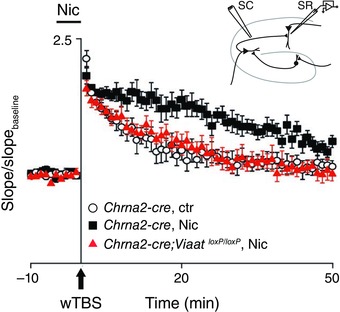
Nicotine's facilitation of LTP in the Schaffer collateral pathway depends on OLM interneurons. The inset shows the stimulating electrode placed in the Schaffer collateral (SC) path and the recording electrode in the stratum radiatum (SR) in a hippocampal slice. LTP was induced by weak theta burst stimulation (wTBS) and was quantified by comparing the slopes of field EPSPs in the presence and absence (baseline) of nicotine in the perfusate. Compared to the control condition (open circles), nicotine (filled squares) enhanced LTP in the Chrna2‐cre mice. Removing inhibition from OLM cells (filled triangles) abolished the LTP‐enhancing effect of nicotine. Error bars represent SEM. Modified with permission from Ref. 112.
Being mobile organisms, humans experience various events throughout the day under different contexts. Yet, not all these events and contexts are worth committing to memory. How does the hippocampus “choose” what to store in memory? One idea is that the VTA sends DA signals to the hippocampus to “stamp” behaviorally relevant information.116 This idea would predict that the hippocampus might play a similar role in committing drug‐associated contexts to memory if the hippocampus received DA signals when the context was experienced. Accordingly, when nicotine activates VTA DA neurons, DA will be released in the hippocampus and the contextual information will be marked for memory storage.117, 118 First, in freely moving mice, systemic nicotine injection potentiates (LTP) the medial perforant path input to the dentate gyrus (Fig. 8A), likely allowing more effective transmission of contextual information into the hippocampus. Second, this effect was abolished when DA function was disrupted in the hippocampus indirectly by inactivation of VTA (Fig. 8B), or more directly by infusion of SCH23390 (a D1‐type DA receptor antagonist) locally into the hippocampus (Fig. 8C). In addition, systemic injection of the same D1 antagonist abolished nicotine‐induced conditioned place preference (CPP).117 A subsequent study showed that this plasticity was mediated in part by nicotine's ability to diminish the feedforward and feedback inhibition onto DG granule cells.119 These results underscore the importance of DA signals in enabling the association of contextual information with nicotine use.
Figure 8.
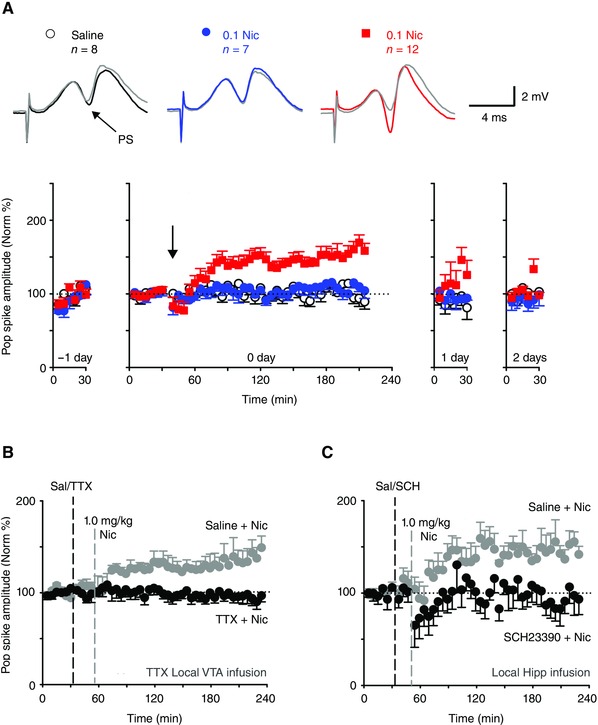
Nicotine's ability to enhance LTP in the medial perforant path‐DG synapse depends on DA in freely moving mice. (A) Nicotine enhances LTP in vivo. Top row: field potential traces showing population spike (PS). Black, blue, and red traces indicate evoked responses after saline, 0.1 mg/kg nicotine, and 1 mg/kg nicotine injection (i.p.), respectively. Gray traces show the baseline evoked response. Bottom row: Normalized PS amplitude measured a day prior to nicotine injection (–1 day), on the day of nicotine injection (arrow, 0 day), and 1 (1 day), or 2 days (2 days) after the treatment. (B) Blocking VTA neuronal action potential with tetrodotoxin (TTX) abolishes nicotine‐induced LTP (n = 5). (C) Blockade of DA D1 receptors by local hippocampal infusion of SCH23390 also abolishes nicotine‐induced LTP (n = 6). Error bars represent SEM. Modified with permission from Ref. 117.
Nucleus accumbens
One of the key targets that receives potent nicotine‐induced DA release from the VTA is the NAc. The NAc has been hypothesized to serve an important role in translating motivation to action.120 It has earned a distinction in addiction because all major addictive drugs, including nicotine, cause DA release into the NAc.91, 121, 122, 123, 124, 125 The NAc is implicated in a diverse set of functions that include stress response, motor function, sensory‐motor gating, and instrumental learning.126
The NAc is divided into core and shell sub‐regions based on anatomy and neurochemical makeup.127, 128 The core region is surrounded on the medial, lateral, and ventral sides by the shell region (Fig. 2). On the dorsal side, the core is directly continuous with the dorsal part of the striatum. In many respects, the core boundary cannot be easily distinguished from the dorsal part of the striatum. The core and shell regions receive distinct but overlapping inputs.129, 130 Anatomical connections also exist between the core and shell, allowing interactions between them.131 Given the distinction between the core and the shell, many studies have explored the relative role of these two regions in instrumental learning in general and nicotine addiction in particular. In acquiring simple instrumental learning tasks such as lever pressing for food reward, the core region has been shown to be involved.132 Furthermore, this learning depended on coincident activation of NMDA and DA D1 receptors in the core region.133 The consolidation of this learning also depends on protein synthesis in the NAc core, not shell.134 If the acquisition of smoking behavior engaged instrumental learning, those results would predict a greater involvement of the core region. Paradoxically, initial intravenously injected nicotine was shown to increase DA release in the shell region, not the core.41, 123 These microdialysis studies showed that the initial large nicotine‐induced DA increase was in the shell. The DA signal did initially change in the core, but microdialysis does not see changes in DA afferent activity well owing to its slow sampling over a large (averaged) area of the background DA concentration. Experimentation showed that the NAc shell was best able to capitalize on afferent burst firing by DA neurons to increase DA release.41 Repeated noncontingent injections of nicotine, however, lead to a sensitized DA release in the core, whereas the shell, although it showed a significantly enhanced DA release, did not show such sensitization.124, 135 This sensitized response in the core was thought to underlie the enhanced locomotor response observed in rodents following repeated nicotine administration.136 However, subsequent studies showed a dissociation between the sensitized DA release in the core and the locomotor sensitization.137, 138 It should be noted that in the above nicotine studies the rodents did not perform any instrumental action to receive nicotine. In a nicotine self‐administration (SA) study,139 rats were trained to nose poke to receive 0.03 mg/kg intravenous nicotine. It was found that starting from the first week, when the rats were learning to self‐administer nicotine, the DA level increased in the NAc shell. Consistently, significant enhancement of DA release in the core region was only observed from the fourth week, well after the animals learned to nose‐poke the nicotine‐delivering port. The authors also showed that over the entire 6‐week period of SA, there was no significant change in the dialysate concentration of DA measured during SA in the core or the shell when the intake of nicotine was taken as a covariate, suggesting very little sensitization in both sub‐regions of the NAc. These observations suggest that response‐contingent and non‐contingent nicotine administrations may lead to different DA release sensitization outcomes. Another study140 that examined nicotine‐induced CPP found that infusion of D1 antagonist in the shell region impaired acquisition of CPP; infusion into the core region did not have an effect. The basis for the difference in DA signaling to the NAc shell by nicotine was found to arise from the differences in DA release induced by phasic burst firing when decoded in the NAc shell versus the dorsal striatum (at least during the initial exposures to nicotine).41 These studies indicate that, initially, nicotine preferentially engages the shell region of the NAc to bring about its reinforcing properties. The core may become important for the later stages in the addiction process, such as cue‐induced reinstatement of nicotine seeking.141 However, the role of nicotine‐induced DA release in the core region in nicotine addiction is largely unexplored. The studies summarized above and other studies suggest that acquisition of instrumental learning reinforced by food and nicotine may engage different neural circuits.142, 143, 144
Nicotine modulates DA levels in the NAc in two ways. First, nicotine activates VTA neurons, which leads to DA release in the NAc. Second, nicotine modulates nicotinic activity in the target areas of VTA, including the NAc.89, 90, 145, 146, 147, 148 Ongoing nAChR activity located at the dopaminergic terminals in the NAc augments DA release.89, 90, 149, 150 While nAChRs containing the β2 subunit are highly important for DA release in the striatum,89, 90, 145, 148 the α7 subunit–containing nAChRs also may have some role since NAc infusions of either α‐bungarotoxin or the α7* nAChR antagonist methyllycaconitine reduces the DA level in the NAc.151 In addition, nicotine enhances the contrast between tonic and phasic DA signals in the NAc by suppressing DA release resulting from tonic presynaptic inputs, while enhancing DA release from phasic burst‐like presynaptic activity.90 Potential functional roles of nicotine‐induced DA release in the NAc have been addressed in a recent review.91
The main projection neurons out of the NAc are the medium spiny neurons (MSNs), which receive convergent inputs from the medial prefrontal cortex (mPFC), ventral subiculum of the hippocampus, and basolateral amygdala (BLA).152, 153, 154, 155 The mPFC inputs carry information about executive and motor plans. The hippocampal inputs provide contextual information, while the BLA provides discrete cue information and emotional salience information. These areas express two general types of DA receptors (D1 and D2), and their activation is hypothesized to lead to facilitation of one input over another in the NAc. Both kinds of DA receptors are found pre‐ and post‐synaptically in the NAc.156, 157, 158 The DA affinity of D1 receptors is lower than that of D2 receptors.159, 160 As a consequence, while activation of D1 receptors requires higher DA levels afforded by phasic release, tonic levels of DA more continuously activate D2 receptors. D1 activation in the NAc has been shown to favor hippocampal inputs. On the other hand, activation of D2 receptors leads to suppression of PFC inputs.161 Hence, to overcome the suppression by D2 activation during phasic DA release, stronger PFC inputs are required for them to activate the NAc. Interestingly, there is another condition, which may potentially allow PFC inputs to activate the NAc. When the tonic DA level goes below the baseline level, for example when reward is withheld,26 the D2 activation level will decrease, allowing PFC inputs to activate NAc neurons. In summary, these mechanisms allow the NAc to engage in different information processing: (1) phasic DA signaling during reward to integrate hippocampal contextual inputs and behaviorally relevant PFC motor plans, and (2) engage PFC inputs to enable behavioral flexibility mediated through the NAc. Since nicotine increases burst firing in VTA neurons,41 we hypothesize it may then favor hippocampal inputs while suppressing PFC inputs. Since the PFC is implicated in executive function and behavioral flexibility,162 the shift from the PFC may partly explain the pursuit of tobacco despite its harmful effects. This shift from PFC processing to more limbic, hippocampal processing may also lead to the inflexibility in changing the smoking behavior to better alternatives.
Many observations indicate that the hippocampus and NAc work closely in learning and memory of place‐reward associations. First, under anesthetized conditions, the MSNs of the NAc exhibit a relatively depolarized “up” state and a hyperpolarized “down” state. The MSNs fire action potentials more readily in their “up” state. Inactivating the hippocampus completely abolishes the state transition, indicating that the hippocampal inputs are necessary for the bistable MSNs to enter the depolarized “up” state.163 Second, the NAc exhibits hippocampus‐entrained theta rhythm.164 Place cells, which are most commonly identified within the hippocampus,165 are also observed in the NAc166 and show phase precession similar to hippocampal place cells.167 Finally, reward‐related information is reactivated in the NAc during quiet wakefulness and slow wave sleep,168 similar to the findings in the hippocampus.169 Although the hippocampus and NAc have been studied separately in nicotine addiction, the interaction between the two has not been well explored. Since both areas express nAChRs, it would be valuable to explore the above‐mentioned interactions to understand how drug associated memories gain abnormal strength and persistence.
The amygdala
The amygdala is traditionally associated with fear and emotion processing and memory. Although studied mainly for fear processing, the amygdala also mediates positive affect;170 hence, its relevance to addiction is a fertile area for exploration. In addition to the context in which smoking is carried out, discrete cues also play a role in reinstating smoking behavior.2 For example, the click of a cigarette lighter or the smell of smoke can trigger a smoker's desire for tobacco. The effect of nicotine on the plasticity in the lateral amygdala was recently explored in mice.171 A 7‐day oral intake of nicotine led to enhanced LTP in the entorhinal cortex–lateral amygdala synapses that lasted up to 72 hours. This enhancement was pathway specific since the effect was not present in thalamus–lateral amygdala or lateral BLA synapses. The effect was mediated by α7‐ and β2‐containing nicotinic receptors. Reduction of inhibition was found to contribute partially to the LTP enhancement. It remains to be seen if long term changes in nicotinic receptors also played a role in the enhanced LTP. Since the entorhinal cortex provides sensory information to the lateral amygdala, enhanced neurotransmission in this pathway may enhance processing of discrete cues associated with smoking.
The function of the amygdala is not simply to associate stimulus with reward (or negative reward). Rather, it is involved in associating a stimulus with the value of the reward. This expectation arises from the observations that lesions of the amygdala lead to insensitivity to reward devaluation.170, 172 While the hippocampus processes contextual information associated with drug use, the amygdala has been implicated in adding and updating the value of cues associated with reward. The amygdala is reciprocally connected with the orbitofrontal cortex (OFC) and works closely with it to assign value to S–O associations. Working together these regions update S–O values in the face of changes in the value of the outcomes.170 Once the updating is complete, the value of the expected reward outcome is stored in the OFC.173 Hence the amygdala–OFC axis may be pivotal in the addiction process. In chronic nicotine use, the positive effects of nicotine substantially decrease, changing the value of the outcome of smoking. A question of future exploration is whether nicotine addiction or addiction in general involves impairment in the amygdala–OFC‐dependent value updating process.
Nicotine‐induced long‐term changes in synaptic potentiation
Smoking continues for years in addicted smokers. This results in long lasting modifications in the nervous system, ranging from changes in receptor subtype composition and expression levels to systems level adaptations.2, 13 These changes contribute to the continued smoking. From the perspective of learning, another important issue is which areas of the brain undergo elaborate changes orchestrated by chronic nicotine that lead to smoking addiction. Although many areas of the brain are affected, there is accumulating evidence that chronic use of cocaine leads to habit learning in the dorsal striatum.174 Future studies are needed to address the extent of habit learning driven in the dorsal striatum by nicotine. Finally, many studies have started exploring the role of the insular cortex in nicotine dependence.175, 176, 177, 178
Apart from being an addictive substance on its own, nicotine upon continued use primes the nervous system for addiction to other substances of abuse such as cocaine and alcohol.179, 180, 181 In support for this gateway role of nicotine, a recent mouse study182 showed that a single low dose of cocaine or a 7‐day chronic oral nicotine administration did not enhance LTP. However, the same single dose of cocaine when given after the 7‐day chronic nicotine exposure increased LTP substantially as measured using in vitro brain slices. Interestingly, this effect was also dependent on D1/D5 DA receptor activation as seen for nicotine‐induced LTP in the hippocampus.117, 183 Similar priming effects of nicotine in LTP enhancement by cocaine have been shown for the striatum184 and amygdala.185 Also, pre‐exposure to nicotine increased alcohol SA, while alcohol‐induced DA responses measured in the NAc shell decreased.186 The blunted DA response was due to increased inhibitory synaptic transmission onto DA neurons. Inhibiting stress hormone receptors prior to nicotine exposure prevented the decreased alcohol‐induced DA responses and the increased alcohol SA. Those results indicated that pre‐exposure to nicotine recruited neuroendocrine systems to influence neurotransmission and behavior associated with alcohol reinforcement.186 It remains to be seen whether these gateway effects of nicotine are widely generalizable to other drugs of addiction.
Summary
Nicotine addiction, still a growing health problem, is the leading cause of preventable death in developed countries.10, 14, 91, 187, 188 Many of its addictive influences can be understood within the framework of reinforcement learning. Nicotine orchestrates changes in key learning‐related structures such as the VTA, NAc, hippocampus, and PFC. However, there are many psychopharmacological factors that are involved in nicotine addiction. For example, nicotine is known to alter the release of virtually all major neurotransmitters.12, 189, 190, 191, 192, 193, 194 In addition, due to the widespread expression of nAChRs throughout the nervous system, structures outside the traditional reward learning pathways are likely involved in nicotine addiction. Recent studies point out yet another way nicotine could affect cellular function: in its uncharged form, nicotine can pass through the lipid bilayer without the need to bind to nAChRs, and then alter intracellular signaling of targeted cells.9 The importance of understanding and preventing nicotine addiction is further underscored by nicotine's ability to act as a gateway drug.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Mariella De Biasi, Kechun Yang, Alyse Thomas, and Christopher Hayworth for helpful discussions and comments on the manuscript. The authors are supported in part by grants from the NIH, NIDA R01DA009411, and NINDS R01NS021229.
References
- 1. Ng, M. et al 2014. Smoking prevalence and cigarette consumption in 187 countries, 1980–2012. JAMA 311: 183–192. [DOI] [PubMed] [Google Scholar]
- 2. Dani, J.A. , Kosten T.R. & Benowitz N.L.. 2009. “The pharmacology of nicotine and tobacco.” In Principles of Addiction Medicine, 4th ed. Ries R.K., Fiellin D.A., Miller S.C. & Saitz R., Eds.: 179–192. Wolters Kluwer, Lippincott, Williams & Wilkins. [Google Scholar]
- 3.World Health Organization. 2015. Tobacco fact sheet 339. July 6, 2015. Accessed Aug. 5, 2015., authors http://www.who.int/mediacentre/factsheets/fs339/en/
- 4. Marti, F. et al 2011. Smoke extracts and nicotine, but not tobacco extracts, potentiate firing and burst activity of ventral tegmental area dopaminergic neurons in mice. Neuropsychopharmacology 36: 2244–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crombag, H.S. et al 2008. Review. Context‐induced relapse to drug seeking: a review. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363: 3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bevins, R.A. & Palmatier M.I.. 2004. Extending the role of associative learning processes in nicotine addiction. Behav. Cogn. Neurosci. Rev. 3: 143–158. [DOI] [PubMed] [Google Scholar]
- 7. Naqvi, N.H. & Bechara A.. 2009. The hidden island of addiction: the insula. Trends Neurosci. 32: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benowitz, N.L. , Hukkanen J. & P. Jacob, III . 2009. “Nicotine chemistry, metabolism, kinetics and biomarkers” In Handbook of Experimental Pharmacology: Nicotine Psychopharmacology. J.E. Henningfield, E.D. London & S. Pogun, Eds.: 29–60. Springer‐Verlag. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rezvani, K. et al 2007. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J. Neurosci. 27: 10508–10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dani, J.A. & Harris R.A.. 2005. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat. Neurosci. 8: 1465–1470. [DOI] [PubMed] [Google Scholar]
- 11. Dani, J.A. & Heinemann S.. 1996. Molecular and cellular aspects of nicotine abuse. Neuron 16: 905–908. [DOI] [PubMed] [Google Scholar]
- 12. Dani, J.A. & Bertrand D.. 2007. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Ann. Rev. Pharmacol. Toxicol. 47: 699–729. [DOI] [PubMed] [Google Scholar]
- 13. De Biasi, M. & Dani J.A.. 2011. Reward, addiction, withdrawal to nicotine. Annu. Rev. Neurosci. 34: 105–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leslie, F.M. , Mojica C.Y. & Reynaga D.D.. 2013. Nicotinic receptors in addiction pathways. Mol. Pharmacol. 83: 753–758. [DOI] [PubMed] [Google Scholar]
- 15. Albuquerque, E.X. et al 2009. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rescorla, R.A. 1991. Associative relations in instrumental learning: the eighteenth Bartlett memorial lecture. Quart. J. Exp. Psychol. 43B: 1–23. [Google Scholar]
- 17. McLaughlin, I. , Dani J.A. & De Biasi M.. 2015. Nicotine withdrawal. Curr. Top. Behav. Neurosci. 24: 99–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dani, J.A. & De Biasi M.. 2013. Mesolimbic dopamine and habenulo‐interpeduncular pathways in nicotine withdrawal. Cold Spring Harb. Perspect. Med. 3: a012138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jackson, K.J. et al 2015. New mechanisms and perspectives in nicotine withdrawal. Neuropharmacology 96: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antolin‐Fontes, B. et al 2015. The habenulo‐interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology 96: 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paolini, M. & De Biasi M.. 2011. Mechanistic insights into nicotine withdrawal. Biochem. Pharmacol. 82: 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schultz, W. 1997. Dopamine neurons and their role in reward mechanisms. Curr. Opin. Neurobiol. 7: 191–197. [DOI] [PubMed] [Google Scholar]
- 23. Wise, R.A. & Rompre P.P.. 1989. Brain dopamine and reward. Annu. Rev. Psychol. 40: 191–225. [DOI] [PubMed] [Google Scholar]
- 24. Wise, R.A. 2009. Roles for nigrostriatal–not just mesocorticolimbic–dopamine in reward and addiction. Trends Neurosci. 32: 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schultz, W. 2002. Getting formal with dopamine and reward. Neuron 36: 241–263. [DOI] [PubMed] [Google Scholar]
- 26. Schultz, W. , Dayan P. & Montague P.R.. 1997. A neural substrate of prediction and reward. Science 275: 1593–1599. [DOI] [PubMed] [Google Scholar]
- 27. Schultz, W. 2010. Dopamine signals for reward value and risk: basic and recent data. Behav. Brain Funct. 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montague, P.R. , Dayan P. & Sejnowski T.J.. 1996. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J. Neurosci. 16: 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paxinos, G. & Watson C.. 2007. The Rat Brain in Stereotaxic Coordinates. Hard Cover Edition. Elsevier Science. [Google Scholar]
- 30. Oades, R.D. & Halliday G.M.. 1987. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 434: 117ȁ165. [DOI] [PubMed] [Google Scholar]
- 31. Swanson, L.W. 1982. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res. Bull. 9: 321–353. [DOI] [PubMed] [Google Scholar]
- 32. Grace, A.A. & Bunney B.S.. 1983. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—1. Identification and characterization. Neuroscience 10: 301–315. [DOI] [PubMed] [Google Scholar]
- 33. Ljungberg, T. , Apicella P. & Schultz W.. 1992. Responses of monkey dopamine neurons during learning of behavioral reactions. J. Neurophysiol. 67: 145–163. [DOI] [PubMed] [Google Scholar]
- 34. Mirenowicz, J. & Schultz W.. 1994. Importance of unpredictability for reward responses in primate dopamine neurons. J. Neurophysiol. 72: 1024–1027. [DOI] [PubMed] [Google Scholar]
- 35. Zweifel, L.S. et al 2009. Disruption of NMDAR‐dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine‐dependent behavior. Proc. Natl. Acad. Sci. U. S. A. 106: 7281–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Di Chiara, G. 2000. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur. J. Pharmacol. 393: 295–314. [DOI] [PubMed] [Google Scholar]
- 37. Day, J.J. et al 2007. Associative learning mediates dynamic shifts in dopamine signaling in the nucleus accumbens. Nat. Neurosci. 10: 1020–1028. [DOI] [PubMed] [Google Scholar]
- 38. Phillips, P.E. et al 2003. Subsecond dopamine release promotes cocaine seeking. Nature 422: 614–618. [DOI] [PubMed] [Google Scholar]
- 39. Stuber, G.D. et al 2005. Rapid dopamine signaling in the nucleus accumbens during contingent and noncontingent cocaine administration. Neuropsychopharmacology 30: 853–863. [DOI] [PubMed] [Google Scholar]
- 40. Pidoplichko, V.I. et al 2004. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn. Mem. 11: 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang, T. et al 2009. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J. Neurosci. 29: 4035–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Redish, A.D. 2004. Addiction as a computational process gone awry. Science 306: 1944–1947. [DOI] [PubMed] [Google Scholar]
- 43. Schultz, W. 2011. Potential vulnerabilities of neuronal reward, risk, and decision mechanisms to addictive drugs. Neuron 69: 603–617. [DOI] [PubMed] [Google Scholar]
- 44. Oakman, S.A. et al 1995. Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J. Neurosci. 15: 5859–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sugimoto, T. & Hattori T.. 1984. Organization and efferent projections of nucleus tegmenti pedunculopontinus pars compacta with special reference to its cholinergic aspects. Neuroscience 11: 931–946. [DOI] [PubMed] [Google Scholar]
- 46. Cornwall, J. , Cooper J.D. & Phillipson O.T.. 1990. Afferent and efferent connections of the laterodorsal tegmental nucleus in the rat. Brain Res. Bull. 25: 271–284. [DOI] [PubMed] [Google Scholar]
- 47. Hallanger, A.E. & Wainer B.H.. 1988. Ascending projections from the pedunculopontine tegmental nucleus and the adjacent mesopontine tegmentum in the rat. J. Comp. Neurol. 274: 483–515. [DOI] [PubMed] [Google Scholar]
- 48. Geisler, S. et al 2007. Glutamatergic afferents of the ventral tegmental area in the rat. J. Neurosci. 27: 5730–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Omelchenko, N. & Sesack S.R.. 2005. Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. J. Comp. Neurol. 483: 217–235. [DOI] [PubMed] [Google Scholar]
- 50. Carr, D.B. & Sesack S.R.. 2000. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J. Neurosci. 20: 3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Christie, M.J. et al 1985. Excitotoxin lesions suggest an aspartatergic projection from rat medial prefrontal cortex to ventral tegmental area. Brain Res. 333: 169–172. [DOI] [PubMed] [Google Scholar]
- 52. Sesack, S.R. et al 1989. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract‐tracing study with Phaseolus vulgaris leucoagglutinin. J. Comp. Neurol. 290: 213–242. [DOI] [PubMed] [Google Scholar]
- 53. Sesack, S.R. & Pickel V.M.. 1992. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J. Comp. Neurol. 320: 145–160. [DOI] [PubMed] [Google Scholar]
- 54. Floresco, S.B. et al 2003. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci. 6: 968–973. [DOI] [PubMed] [Google Scholar]
- 55. Semba, K. & Fibiger H.C.. 1992. Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro‐ and antero‐grade transport and immunohistochemical study. J. Comp. Neurol. 323: 387–410. [DOI] [PubMed] [Google Scholar]
- 56. Pan, W.X. & Hyland B.I.. 2005. Pedunculopontine tegmental nucleus controls conditioned responses of midbrain dopamine neurons in behaving rats. J. Neurosci. 25: 4725–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reese, N.B. , Garcia‐Rill E. & Skinner R.D.. 1995. Auditory input to the pedunculopontine nucleus: II. Unit responses. Brain Res. Bull. 37: 265–273. [DOI] [PubMed] [Google Scholar]
- 58. Grunwerg, B.S. , Krein H. & Krauthamer G.M.. 1992. Somatosensory input and thalamic projection of pedunculopontine tegmental neurons. Neuroreport 3: 673–675. [DOI] [PubMed] [Google Scholar]
- 59. Faure, P. et al 2014. Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neuroscience 282C: 86–100. [DOI] [PubMed] [Google Scholar]
- 60. Jia, H.G. et al 2003. Colocalization of gamma‐aminobutyric acid and acetylcholine in neurons in the laterodorsal and pedunculopontine tegmental nuclei in the cat: a light and electron microscopic study. Brain Res. 992: 205–219. [DOI] [PubMed] [Google Scholar]
- 61. Clements, J.R. & Grant S.. 1990. Glutamate‐like immunoreactivity in neurons of the laterodorsal tegmental and pedunculopontine nuclei in the rat. Neurosci. Lett. 120: 70–73. [DOI] [PubMed] [Google Scholar]
- 62. Lodge, D.J. & Grace A.A.. 2006. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 103: 5167–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grace, A.A. & Onn S.P.. 1989. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J. Neurosci. 9: 3463–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chergui, K. et al 1993. Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur. J. Neurosci. 5: 137–144. [DOI] [PubMed] [Google Scholar]
- 65. Wu, M. , Hrycyshyn A.W. & Brudzynski S.M.. 1996. Subpallidal outputs to the nucleus accumbens and the ventral tegmental area: anatomical and electrophysiological studies. Brain Res. 740: 151–161. [DOI] [PubMed] [Google Scholar]
- 66. Grace, A.A. et al 2007. Regulation of firing of dopaminergic neurons and control of goal‐directed behaviors. Trends Neurosci. 30: 220–227. [DOI] [PubMed] [Google Scholar]
- 67. Jones, I.W. & Wonnacott S.. 2004. Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J. Neurosci. 24: 11244–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klink, R. et al 2001. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 21: 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Charpantier, E. et al 1998. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport 9: 3097–3101. [DOI] [PubMed] [Google Scholar]
- 70. Yang, K. et al 2011. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J. Neurosci. 31: 2537–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wooltorton, J.R. et al 2003. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 23: 3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Azam, L. et al 2002. Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J. Comp. Neurol. 444: 260–274. [DOI] [PubMed] [Google Scholar]
- 73. Tapper, A.R. et al 2004. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science 306: 1029–1032. [DOI] [PubMed] [Google Scholar]
- 74. Mameli‐Engvall, M. et al 2006. Hierarchical control of dopamine neuron‐firing patterns by nicotinic receptors. Neuron 50: 911–921. [DOI] [PubMed] [Google Scholar]
- 75. Pons, S. et al 2008. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self‐administration. J. Neurosci. 28: 12318–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Drenan, R.M. et al 2010. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J. Neurosci. 30: 9877–9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Picciotto, M.R. et al 1998. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391: 173–177. [DOI] [PubMed] [Google Scholar]
- 78. Maskos, U. et al 2005. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436: 103–107. [DOI] [PubMed] [Google Scholar]
- 79. Tolu, S. et al 2013. Co‐activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol. Psychiatry 18: 382–393. [DOI] [PubMed] [Google Scholar]
- 80. Schultz, W. 2010. Multiple functions of dopamine neurons. F1000 Biol. Rep. 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Imperato, A. , Mulas A. & Di Chiara G.. 1986. Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur. J. Pharmacol. 132: 337–338. [DOI] [PubMed] [Google Scholar]
- 82. Pidoplichko, V.I. et al 1997. Nicotine activates and desensitizes midbrain dopamine neurons. Nature 390: 401–404. [DOI] [PubMed] [Google Scholar]
- 83. Mansvelder, H.D. , Keath J.R. & McGehee D.S.. 2002. Synaptic mechanisms underlie nicotine‐induced excitability of brain reward areas. Neuron 33: 905–919. [DOI] [PubMed] [Google Scholar]
- 84. Mansvelder, H.D. & McGehee D.S.. 2000. Long‐term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27: 349–357. [DOI] [PubMed] [Google Scholar]
- 85. Seguela, P. et al 1993. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J. Neurosci. 13: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gao, M. et al 2010. Mechanisms involved in systemic nicotine‐induced glutamatergic synaptic plasticity on dopamine neurons in the ventral tegmental area. J. Neurosci. 30: 13814–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Placzek, A.N. , Zhang T.A. & Dani J.A.. 2009. Age dependent nicotinic influences over dopamine neuron synaptic plasticity. Biochem. Pharmacol. 78: 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Saal, D. et al 2003. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582. [DOI] [PubMed] [Google Scholar]
- 89. Zhou, F.M. , Liang Y. & Dani J.A.. 2001. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 4: 1224–1229. [DOI] [PubMed] [Google Scholar]
- 90. Zhang, L. et al 2009. Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol. Pharmacol. 76: 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Balfour, D.J. 2015. The role of mesoaccumbens dopamine in nicotine dependence. Curr. Top. Behav. Neurosci. 24: 55–98. [DOI] [PubMed] [Google Scholar]
- 92. Liu, L. et al 2012. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing alpha4 and alpha6 subunits. Mol. Pharmacol. 81: 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hartley, T. et al 2014. Space in the brain: how the hippocampal formation supports spatial cognition. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369: 20120510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bannerman, D.M. et al 2014. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 15: 181–192. [DOI] [PubMed] [Google Scholar]
- 95. Eichenbaum, H. 2013. Memory on time. Trends Cogn. Sci. 17: 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hargreaves, E.L. et al 2005. Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science 308: 1792–1794. [DOI] [PubMed] [Google Scholar]
- 97. Knierim, J.J. , Neunuebel J.P. & Deshmukh S.S.. 2014. Functional correlates of the lateral and medial entorhinal cortex: objects, path integration and local‐global reference frames. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369: 20130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Loy, R. et al 1980. Noradrenergic innervation of the adult rat hippocampal formation. J. Comp. Neurol. 189: 699–710. [DOI] [PubMed] [Google Scholar]
- 99. Moore, R.Y. & Halaris A.E.. 1975. Hippocampal innervation by serotonin neurons of the midbrain raphe in the rat. J. Comp. Neurol. 164: 171–183. [DOI] [PubMed] [Google Scholar]
- 100. Azmitia, E.C. & Segal M.. 1978. An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J. Comp. Neurol. 179: 641–667. [DOI] [PubMed] [Google Scholar]
- 101. Lewis, P.R. & Shute C.C.. 1967. The cholinergic limbic system: projections to hippocampal formation, medial cortex, nuclei of the ascending cholinergic reticular system, and the subfornical organ and supra‐optic crest. Brain 90: 521–540. [DOI] [PubMed] [Google Scholar]
- 102. Teles‐Grilo Ruivo, L.M. & Mellor J.R.. 2013. Cholinergic modulation of hippocampal network function. Front. Synaptic Neurosci. 5: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Frotscher, M. & Leranth C.. 1985. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J. Comp. Neurol. 239: 237–246. [DOI] [PubMed] [Google Scholar]
- 104. Scatton, B. et al 1980. Origin of dopaminergic innervation of the rat hippocampal formation. Neurosci. Lett. 18: 125–131. [DOI] [PubMed] [Google Scholar]
- 105. Fanselow, M.S. & Dong H.W.. 2010. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gould, T.J. & Leach P.T.. 2014. Cellular, molecular, and genetic substrates underlying the impact of nicotine on learning. Neurobiol. Learn. Mem. 107: 108–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Davis, J.A. & Gould T.J.. 2008. Associative learning, the hippocampus, and nicotine addiction. Curr. Drug Abuse Rev. 1: 9–19. [DOI] [PubMed] [Google Scholar]
- 108. Frazier, C.J. et al 1998. Acetylcholine activates an alpha‐bungarotoxin‐sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J. Neurosci. 18: 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jones, S. & Yakel J.L.. 1997. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J. Physiol. 504(Pt 3): 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McQuiston, A.R. & Madison D.V.. 1999. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J. Neurosci. 19: 2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ji, D. & Dani J.A.. 2000. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J. Neurophysiol. 83: 2682–2690. [DOI] [PubMed] [Google Scholar]
- 112. Leao, R.N. et al 2012. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat. Neurosci. 15: 1524–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nakauchi, S. et al 2007. Nicotine gates long‐term potentiation in the hippocampal CA1 region via the activation of alpha2* nicotinic ACh receptors. Eur. J. Neurosci. 25: 2666–2681. [DOI] [PubMed] [Google Scholar]
- 114. Rolls, E.T. & Kesner R.P.. 2006. A computational theory of hippocampal function, and empirical tests of the theory. Prog. Neurobiol. 79: 1–48. [DOI] [PubMed] [Google Scholar]
- 115. Treves, A. 2004. Computational constraints between retrieving the past and predicting the future, and the CA3‐CA1 differentiation. Hippocampus 14: 539–556. [DOI] [PubMed] [Google Scholar]
- 116. Lisman, J.E. & Grace A.A.. 2005. The hippocampal‐VTA loop: controlling the entry of information into long‐term memory. Neuron 46: 703–713. [DOI] [PubMed] [Google Scholar]
- 117. Tang, J. & Dani J.A.. 2009. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron 63: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang, S. et al 2014. Interlamellar CA1 network in the hippocampus. Proc. Natl. Acad. Sci. U. S. A. 111: 12919–12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhang, T.A. et al 2010. Addictive nicotine alters local circuit inhibition during the induction of in vivo hippocampal synaptic potentiation. J. Neurosci. 30: 6443–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mogenson, G.J. , Jones D.L. & Yim C.Y.. 1980. From motivation to action: functional interface between the limbic system and the motor system. Prog. Neurobiol. 14: 69–97. [DOI] [PubMed] [Google Scholar]
- 121. Di Chiara, G. & Imperato A.. 1988. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U. S. A. 85: 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pontieri, F.E. , Tanda G. & Di Chiara G.. 1995. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc. Natl. Acad. Sci. U. S. A. 92: 12304–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pontieri, F.E. et al 1996. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382: 255–257. [DOI] [PubMed] [Google Scholar]
- 124. Benwell, M.E. & Balfour D.J.. 1992. The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br. J. Pharmacol. 105: 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Balfour, D.J. 1994. Neural mechanisms underlying nicotine dependence. Addiction 89: 1419–1423. [DOI] [PubMed] [Google Scholar]
- 126. Goto, Y. & Grace A.A.. 2008. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 31: 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Voorn, P. et al 2004. Putting a spin on the dorsal‐ventral divide of the striatum. Trends Neurosci. 27: 468–474. [DOI] [PubMed] [Google Scholar]
- 128. Zahm, D.S. 1999. Functional‐anatomical implications of the nucleus accumbens core and shell subterritories. Ann. N. Y. Acad. Sci. 877: 113–128. [DOI] [PubMed] [Google Scholar]
- 129. Heimer, L. et al 1997. The accumbens: beyond the core‐shell dichotomy. J. Neuropsychiatry Clin. Neurosci. 9: 354–381. [DOI] [PubMed] [Google Scholar]
- 130. Groenewegen, H.J. et al 1999. Convergence and segregation of ventral striatal inputs and outputs. Ann. N. Y. Acad. Sci. 877: 49–63. [DOI] [PubMed] [Google Scholar]
- 131. van Dongen, Y.C. et al 2005. Anatomical evidence for direct connections between the shell and core subregions of the rat nucleus accumbens. Neuroscience 136: 1049–1071. [DOI] [PubMed] [Google Scholar]
- 132. Kelley, A.E. , Smith‐Roe S.L. & Holahan M.R.. 1997. Response‐reinforcement learning is dependent on N‐methyl‐d‐aspartate receptor activation in the nucleus accumbens core. Proc. Natl. Acad. Sci. U. S. A. 94: 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Smith‐Roe, S.L. & Kelley A.E.. 2000. Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J. Neurosci. 20: 7737–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hernandez, P.J. , Sadeghian K. & Kelley A.E.. 2002. Early consolidation of instrumental learning requires protein synthesis in the nucleus accumbens. Nat. Neurosci. 5: 1327–1331. [DOI] [PubMed] [Google Scholar]
- 135. Cadoni, C. & Di Chiara G.. 2000. Differential changes in accumbens shell and core dopamine in behavioral sensitization to nicotine. Eur. J. Pharmacol. 387: R23‐25. [DOI] [PubMed] [Google Scholar]
- 136. Boye, S.M. , Grant R.J. & Clarke P.B.. 2001. Disruption of dopaminergic neurotransmission in nucleus accumbens core inhibits the locomotor stimulant effects of nicotine and D‐amphetamine in rats. Neuropharmacology 40: 792–805. [DOI] [PubMed] [Google Scholar]
- 137. Shoaib, M. et al 1994. Behavioural and neurochemical adaptations to nicotine in rats: influence of NMDA antagonists. Br. J. Pharmacol. 111: 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Balfour, D.J. et al 1996. Effects of acute D‐CPPene on mesoaccumbens dopamine responses to nicotine in the rat. Eur. J. Pharmacol. 316: 153–156. [DOI] [PubMed] [Google Scholar]
- 139. Lecca, D. et al 2006. Preferential increase of extracellular dopamine in the rat nucleus accumbens shell as compared to that in the core during acquisition and maintenance of intravenous nicotine self‐administration. Psychopharmacology 184: 435–446. [DOI] [PubMed] [Google Scholar]
- 140. Spina, L. et al 2006. Nicotine‐conditioned single‐trial place preference: selective role of nucleus accumbens shell dopamine D1 receptors in acquisition. Psychopharmacology 184: 447–455. [DOI] [PubMed] [Google Scholar]
- 141. D'Souza, M.S. & Markou A.. 2014. Differential role of N‐methyl‐D‐aspartate receptor‐mediated glutamate transmission in the nucleus accumbens shell and core in nicotine seeking in rats. Eur. J. Neurosci. 39: 1314–1322. [DOI] [PubMed] [Google Scholar]
- 142. Bassareo, V. , Musio P. & Di Chiara G.. 2011. Reciprocal responsiveness of nucleus accumbens shell and core dopamine to food‐ and drug‐conditioned stimuli. Psychopharmacology 214: 687–697. [DOI] [PubMed] [Google Scholar]
- 143. Di Chiara, G. & Bassareo V.. 2007. Reward system and addiction: what dopamine does and doesn't do. Curr. Opin. Pharmacol. 7: 69–76. [DOI] [PubMed] [Google Scholar]
- 144. Di Chiara, G. 2002. Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav. Brain Res. 137: 75–114. [DOI] [PubMed] [Google Scholar]
- 145. Zhang, H. & Sulzer D.. 2004. Frequency‐dependent modulation of dopamine release by nicotine. Nat. Neurosci. 7: 581–582. [DOI] [PubMed] [Google Scholar]
- 146. Nisell, M. , Nomikos G.G. & Svensson T.H.. 1994. Systemic nicotine‐induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse 16: 36–44. [DOI] [PubMed] [Google Scholar]
- 147. Exley, R. et al 2008. Alpha6‐containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology 33: 2158–2166. [DOI] [PubMed] [Google Scholar]
- 148. Rice, M.E. & Cragg S.J.. 2004. Nicotine amplifies reward‐related dopamine signals in striatum. Nat. Neurosci. 7: 583–584. [DOI] [PubMed] [Google Scholar]
- 149. Mifsud, J.C. , Hernandez L. & Hoebel B.G.. 1989. Nicotine infused into the nucleus accumbens increases synaptic dopamine as measured by in vivo microdialysis. Brain Res. 478: 365–367. [DOI] [PubMed] [Google Scholar]
- 150. Kleijn, J. et al 2011. Direct effect of nicotine on mesolimbic dopamine release in rat nucleus accumbens shell. Neurosci. Lett. 493: 55–58. [DOI] [PubMed] [Google Scholar]
- 151. Fu, Y. et al 2000. Local alpha‐bungarotoxin‐sensitive nicotinic receptors in the nucleus accumbens modulate nicotine‐stimulated dopamine secretion in vivo. Neuroscience 101: 369–375. [DOI] [PubMed] [Google Scholar]
- 152. Phillipson, O.T. & Griffiths A.C.. 1985. The topographic order of inputs to nucleus accumbens in the rat. Neuroscience 16: 275–296. [DOI] [PubMed] [Google Scholar]
- 153. Raisman, G. , Cowan W.M. & Powell T.P.. 1966. An experimental analysis of the efferent projection of the hippocampus. Brain 89: 83–108. [DOI] [PubMed] [Google Scholar]
- 154. De Olmos, J.S. & Ingram W.R.. 1972. The projection field of the stria terminalis in the rat brain. An experimental study. J. Comp. Neurol. 146: 303–334. [DOI] [PubMed] [Google Scholar]
- 155. De France, J.F. & Yoshihara H.. 1975. Fimbria input to the nucleus accumbens septi. Brain Res. 90: 159–163. [DOI] [PubMed] [Google Scholar]
- 156. Sesack, S.R. , Aoki C. & Pickel V.M.. 1994. Ultrastructural localization of D2 receptor‐like immunoreactivity in midbrain dopamine neurons and their striatal targets. J. Neurosci. 14: 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hara, Y. & Pickel V.M.. 2005. Overlapping intracellular and differential synaptic distributions of dopamine D1 and glutamate N‐methyl‐D‐aspartate receptors in rat nucleus accumbens. J. Comp. Neurol. 492: 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tarazi, F.I. et al 1998. Localization of dopamine receptor subtypes in corpus striatum and nucleus accumbens septi of rat brain: comparison of D1‐, D2‐, and D4‐like receptors. Neuroscience 83: 169–176. [DOI] [PubMed] [Google Scholar]
- 159. Richfield, E.K. , Penney J.B. & Young A.B.. 1989. Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience 30: 767–777. [DOI] [PubMed] [Google Scholar]
- 160. Beaulieu, J.M. & Gainetdinov R.R.. 2011. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63: 182–217. [DOI] [PubMed] [Google Scholar]
- 161. Goto, Y. & Grace A.A.. 2005. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal‐directed behavior. Nat. Neurosci. 8: 805–812. [DOI] [PubMed] [Google Scholar]
- 162. Dalley, J.W. , Cardinal R.N. & Robbins T.W.. 2004. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci. Biobehav. Rev. 28: 771–784. [DOI] [PubMed] [Google Scholar]
- 163. O'Donnell, P. & Grace A.A.. 1995. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J. Neurosci. 15: 3622–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Berke, J.D. et al 2004. Oscillatory entrainment of striatal neurons in freely moving rats. Neuron 43: 883–896. [DOI] [PubMed] [Google Scholar]
- 165. O'Keefe, J. & Dostrovsky J.. 1971. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely‐moving rat. Brain Res. 34: 171–175. [DOI] [PubMed] [Google Scholar]
- 166. Lavoie, A.M. & Mizumori S.J.. 1994. Spatial, movement‐ and reward‐sensitive discharge by medial ventral striatum neurons of rats. Brain Res. 638: 157–168. [DOI] [PubMed] [Google Scholar]
- 167. van der Meer, M.A. & Redish A.D.. 2011. Theta phase precession in rat ventral striatum links place and reward information. J. Neurosci. 31: 2843–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Lansink, C.S. et al 2008. Preferential reactivation of motivationally relevant information in the ventral striatum. J. Neurosci. 28: 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kudrimoti, H.S. , Barnes C.A. & McNaughton B.L.. 1999. Reactivation of hippocampal cell assemblies: effects of behavioral state, experience, and EEG dynamics. J. Neurosci. 19: 4090–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Murray, E.A. 2007. The amygdala, reward and emotion. Trends Cogn. Sci. 11: 489–497. [DOI] [PubMed] [Google Scholar]
- 171. Huang, Y.Y. , Kandel E.R. & Levine A.. 2008. Chronic nicotine exposure induces a long‐lasting and pathway‐specific facilitation of LTP in the amygdala. Learn. Mem. 15: 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Baxter, M.G. & Murray E.A.. 2002. The amygdala and reward. Nat. Rev. Neurosci. 3: 563–573. [DOI] [PubMed] [Google Scholar]
- 173. Holland, P.C. & Gallagher M.. 2004. Amygdala‐frontal interactions and reward expectancy. Curr. Opin. Neurobiol. 14: 148–155. [DOI] [PubMed] [Google Scholar]
- 174. Everitt, B.J. & Robbins T.W.. 2013. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci. Biobehav. Rev. 37: 1946–1954. [DOI] [PubMed] [Google Scholar]
- 175. Kutlu, M.G. et al 2013. Role of insular cortex D(1) and D(2) dopamine receptors in nicotine self‐administration in rats. Behav. Brain Res. 256: 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Naqvi, N.H. et al 2007. Damage to the insula disrupts addiction to cigarette smoking. Science 315: 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hollander, J.A. et al 2008. Insular hypocretin transmission regulates nicotine reward. Proc. Natl. Acad. Sci. U. S. A. 105: 19480–19485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Pushparaj, A. et al 2015. Involvement of the rostral agranular insular cortex in nicotine self‐administration in rats. Behav. Brain Res. 290: 77–83. [DOI] [PubMed] [Google Scholar]
- 179. Kandel, D. 1975. Stages in adolescent involvement in drug use. Science 190: 912–914. [DOI] [PubMed] [Google Scholar]
- 180. Yamaguchi, K. & Kandel D.B.. 1984. Patterns of drug use from adolescence to young adulthood: II. Sequences of progression. Am. J. Public Health 74: 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Doyon, W.M. et al 2013. Potential substrates for nicotine and alcohol interactions: a focus on the mesocorticolimbic dopamine system. Biochem. Pharmacol. 86: 1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Huang, Y.Y. et al 2014. D1/D5 receptors and histone deacetylation mediate the Gateway Effect of LTP in hippocampal dentate gyrus. Learn. Mem. 21: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yang, K. & Dani J.A.. 2014. Dopamine D1 and D5 receptors modulate spike timing‐dependent plasticity at medial perforant path to dentate granule cell synapses. J. Neurosci. 34: 15888–15897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Levine, A. et al 2011. Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine. Sci. Transl. Med. 3: 107ra109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Huang, Y.Y. et al 2013. Nicotine primes the effect of cocaine on the induction of LTP in the amygdala. Neuropharmacology 74: 126–134. [DOI] [PubMed] [Google Scholar]
- 186. Doyon, W.M. et al 2013. Nicotine decreases ethanol‐induced dopamine signaling and increases self‐administration via stress hormones. Neuron 79: 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Mathers, C.D. & Loncar D.. 2006. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 3: e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Dani, J. , Kosten T. & Benowitz N.. 2014. The pharmacology of nicotine and tobacco (chapter 12) In The ASAM Principles of Addiction Medicine. 5th ed Ries R.K., Fiellin D.A., Miller S.C. & Saitz R., Eds.: 201–216. Philadelphia, PA: Wolters Kluwer. [Google Scholar]
- 189. Alkondon, M. et al 1997. Neuronal nicotinic acetylcholine receptor activation modulates gamma‐aminobutyric acid release from CA1 neurons of rat hippocampal slices. J. Pharmacol. Exp. Ther. 283: 1396–1411. [PubMed] [Google Scholar]
- 190. Hadjiconstantinou, M. & Neff N.H.. 2011. Nicotine and endogenous opioids: neurochemical and pharmacological evidence. Neuropharmacology 60: 1209–1220. [DOI] [PubMed] [Google Scholar]
- 191. Kenny, P.J. 2011. Tobacco dependence, the insular cortex and the hypocretin connection. Pharmacol. Biochem. Behav. 97: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. McGehee, D.S. et al 1995. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269: 1692–1696. [DOI] [PubMed] [Google Scholar]
- 193. Gray, R. et al 1996. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383: 713–716. [DOI] [PubMed] [Google Scholar]
- 194. Role, L.W. & Berg D.K.. 1996. Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16: 1077–1085. [DOI] [PubMed] [Google Scholar]