

Abstract
Claudin-low tumors are a highly aggressive breast cancer subtype with no targeted treatments and a clinically documented resistance to chemotherapy. They are significantly enriched in cancer stem cells (CSCs), which makes claudin-low tumor models particularly attractive for studying CSC behavior and developing novel approaches to minimize CSC therapy resistance. One proposed mechanism by which CSCs arise is via an epithelial-mesenchymal transition (EMT), and reversal of this process may provide a potential therapeutic approach for increasing tumor chemosensitivity. Therefore, we investigated the role of known EMT regulators, miR-200 family of microRNAs in controlling the epithelial state, stem-like properties, and therapeutic response in an in vivo primary, syngeneic p53null claudin-low tumor model that is normally deficient in miR-200 expression. Using an inducible lentiviral approach, we expressed the miR-200c cluster in this model and found that it changed the epithelial state, and consequently, impeded CSC behavior in these mesenchymal tumors. Moreover, these state changes were accompanied by a decrease in proliferation and an increase in the differentiation status. miR-200c expression also forced a significant reorganization of tumor architecture, affecting important cellular processes involved in cell-cell contact, cell adhesion, and motility. Accordingly, induced miR200c expression significantly enhanced the chemosensitivity and decreased the metastatic potential of this p53null claudin-low tumor model. Collectively, our data suggest that miR-200c expression in claudin-low tumors offers a potential therapeutic application to disrupt the EMT program on multiple fronts in this mesenchymal tumor subtype, by altering tumor growth, chemosensitivity, and metastatic potential in vivo.
Keywords: miR-200c, claudin-low, EMT, CSCs
Introduction
Breast cancer is not a single disease, but rather a heterogeneous disease with multiple subtypes. The majority of claudin-low breast cancers are characterized as triple negative, with no amplification of HER2 or expression of estrogen and progesterone receptors, and very poor prognosis. A unique feature of these tumors is lack of cell-cell adherens and tight junction genes, such as E-cadherin and claudins (1). Loss of these genes is also characteristic of the epithelial-mesenchymal transition (EMT), a process that governs metastatic dissemination, during which cells lose cell-to-cell junctions, breach the basement membrane, and disseminate (2,3). Accordingly, in-depth characterization of claudin-low tumors revealed a significant overlap between their core genetic profiles and those of cells undergoing EMT (1,4,5). Notably, cells that undergo EMT often acquire properties associated with cancer stem cells (CSCs) (6). Consistently, claudin-low tumors are enriched in functional CSCs (7). Increasing evidence suggests that treatment resistance of cancers, including breast, is the result of residual CSC subpopulations that are capable of regenerating the epithelial components of a recurring tumor by intrinsic self-renewal mechanisms (8,9). Consequently, as claudin-low tumors exhibit properties associated with both EMT and CSC self-renewal, they are highly resistant to conventional radiation and chemotherapy (7,8).
Several transcription factors are known EMT inducers, including Zeb1/2 and Snail/Slug, (2,3). These factors bind to the promoter region of E-cadherin (Cdh1) and repress its expression. Downregulation of E-cadherin leads to loss of epithelial characteristics and a significant increase in expression of several mesenchymal markers, such as N-cadherin and vimentin. Importantly, E-cadherin causes decreased cell proliferation through contact inhibition (10), whereas loss of E-cadherin promotes metastasis by increasing cell motility (11).
Another central regulator of EMT is the microRNA-200 (miR-200) family, which consists of five members that are located in two gene clusters (miR-200c-141 and miR200a-b-429) (12). These miRNAs maintain cells in an epithelial state by forming a double-negative feedback loop with Zeb transcription factors (13). Pursuant to their role in EMT, miR-200 members also regulate self-renewal capabilities of both normal and breast cancer stem cells (14), and their expression is lost when cells transition into a stem-like state (15).
Previous studies demonstrated that constitutive overexpression of miR-200 reverses EMT in multiple cancer cell lines (16,17); however, these studies almost uniformly constitutively expressed miR-200, and although they uncovered many direct targets of miR-200 and provided insight into its biological functions, the effects of miR-200 expression in primary tumors in an in vivo context have not been fully characterized. Here, we show that induced expression of the miR-200c-141 cluster in an in vivo CSC-enriched claudin-low tumor model, decreased tumor growth and stem cell functionality, and resulted in loss of EMT features, accompanied by an increase in chemotherapeutic sensitivity. The model we utilize was developed by transplanting the p53null mammary tissue into wild-type recipient Balb/c mice (18). p53 is frequently mutated in human breast cancers, and confers poor prognosis and chemoresistance (19). Spontaneously arising p53null murine mammary tumors showed significant heterogeneity, recapitulating the properties of their human counterparts in subtype clustering (20). Derived murine claudin-low tumors exhibit a significant overlap in their gene expression profile with human claudin-low breast cancers, as well as the spindloid morphology, and are representative of human claudin-low cancer behavior, as illustrated by their pathology following serial transplantation, maintaining their mesenchymal properties and CSC enrichment (21,22). Therefore, they represents an improved syngeneic model to investigate agents that target the EMT pathway and CSC behavior, as we can monitor tumor response in orthotopic sites with an appropriate microenvironment and an intact immune system in a wild-type background, while employing the treatment regime potentially applicable in a clinical setting.
Results
miR-200c induction impairs tumor growth
MiR-200c suppresses progression of multiple tumor models propagated from established cell lines (16,23,24). However, most of these studies focused on reversal of EMT in vitro with constitutive expression of miR-200c prior to tumor establishment. Any potential clinical application would require miR-200c to be manipulated after tumor diagnosis. To determine the effects of miR-200c expression on established primary tumors in vivo, we used a doxycycline (DOX)-inducible lentiviral vector to express the miR-200c-141 cluster (miR-200c for simplicity) in our genetically-engineered transplantable p53null claudin-low tumor model (21,25). This vector allows for visualization of transduced cells by constitutive expression of GFP. Upon DOX administration, transduced cells upregulate both, miR-200c and RFP, allowing us to identify and isolate miR-200c-expressing cells. We confirmed the induction of RFP by fluorescence-activated cell sorting (FACS) and fluorescence microscopy (Figure 1A), and the corresponding induction of miR-200c by quantitative PCR (qPCR), as well as miR-141 (Supplemental Figure S1A). Importantly, the level of miR-200c expression achieved in DOX-treated tumors fell within the range of miR-200c expression observed in other p53null, non-claudin-low breast cancer subtypes (20) (Figure 1B).
Figure 1. miR-200c decelerates tumor growth.

(A) RFP was induced in tumors after DOX administration, as seen by FACS analysis and fluorescent microscopy. Scale bar represents 5mm. (B) miR-200c was induced to a physiological level, compared with its levels in different subtypes of p53null tumors (20), as shown by qPCR. (C) Tumor growth, as measured by the daily increase in tumor volume, was significantly lower in the DOX-treated group compared with untreated controls. (D) Tumor weight at the end of the treatment showed that the miR-200c-expressing tumors were much smaller than untreated controls. (E) The Ki67 proliferation marker was significantly decreased in miR-200c-induced tumors, revealing that the tumor growth deceleration was due, in part, to a lower proliferation rate. Scale bar represents 50μm. (F) Tumor latency increased with miR-200c upregulation. Equal numbers (5 000) of GFP+ and RFP+ cells were injected into the cleared fat pads of wild-type Balb/c recipient mice, and tumors were palpated every day to establish their latency, while maintaining vehicle or DOX treatment.
To determine the effect of miR-200c induction on overall tumor growth, tumor volume was measured daily for duration of DOX treatment. Vehicle-treated group exhibited a high average daily tumor growth (130 mm3 per day), whereas DOX treatment resulted in its significant reduction (50 mm3 per day) (Figure 1C). Correspondingly, tumor weight at the end of treatment was significantly lower in the miR-200c-expressing group (Figure 1D). Notably, DOX treatment did not cause a decrease in tumor growth of non-transduced claudin-low tumors, confirming that the observed growth inhibition was due to miR-200c induction (Supplementary Figure S1B). Immunofluorescent staining and subsequent quantification of Ki67 revealed that the growth defect is due to a significant decrease in the level of proliferation (Figure 1E). Moreover, injecting the same number of freshly isolated primary RFP+ (mir200c-induced) or GFP+ (mir200c-deficient) sorted cells into orthotopic sites of wild-type mice revealed a significant decrease in tumor latency in miR-200c-expressing group (Figure 1F). These data further confirm that cells with high expression of miR-200c exhibit decreased proliferation in vivo.
Induced expression of miR-200c reverses EMT in vivo
One hallmark of claudin-low tumor phenotype is the spindloid nature of tumor cells, underlining their EMT characteristics. After inducing expression of miR-200c, we observed a change in morphological properties that suggested a reversal of EMT, as hematoxylin and eosin (H&E) staining showed that tumor cells lost their mesenchymal features, and seemed to switch into a more organized epithelial phenotype (Figure 2A). Consistent with this phenotypic change, we observed a significant induction of E-cadherin mRNA and protein levels by qPCR and reverse phase protein array (RPPA) analyses, respectively (Figure 2B). Of note, RPPA showed no change in E-cadherin levels in DOX-treated tumors when they were transduced with a control lentivirus expressing a luciferase reporter rather than miR-200c, confirming that these changes were dependent on miR-200c expression.
Figure 2. miR-200c alters claudin-low tumor morphology and causes MET.

(A) Significant morphological changes were observed in tumors with induced miR-200c, as shown by standard H&E staining. Scale bars represents 50μm. (B) qPCR showed that E-cadherin mRNA was highly induced post-DOX. Reverse phase protein array (RPPA) analysis showed a significant upregulation of E-cadherin protein in miR-200c-induced tumors, but not in tumors containing luciferase (FF3). The two bands represent runs with two different validated antibodies, according to the RPPA core. (C) There was no change in mRNA levels of vimentin following DOX administration, as shown by qPCR. (D) Images of immunofluorescent staining show that vimentin protein expression was decreased in cells that were positive for E-cadherin staining. Scale bar is 50μm.
Surprisingly, no difference in vimentin mRNA levels was detected between DOX-treated and untreated tumors (Figure 2C). However, there was a decrease in vimentin protein levels in tumors following miR-200c induction, as shown by immunofluorescent staining (Figure 2D, separate images shown in Supplemental Figure S1C.). Specifically, vimentin was lost in cells that were positive for E-cadherin. To compare the cells expressing miR-200c to control cells, we sorted cells from primary tumors based on their GFP or RFP positivity. Although there was a significant difference in the mRNA levels of Zeb2 between the sorted populations by qPCR, no such difference was observed in Zeb1, Snai2, and N-cadherin (Figure 3A). However, RFP+ cells from DOX-treated group had significantly lower levels of these mesenchymal proteins (Figure 3B), implying that their expression is controlled at the level of translation, rather than transcription or mRNA turnover.
Figure 3. miR-200c upregulation leads to a decrease in protein expression of several mesenchymal markers.

(A) Other mesenchymal markers showed either a significant decrease (Zeb2), or no significant change (Zeb1, Snai2, and N-cad) by qPCR. (B) Western blots show a dramatic reduction in Zeb1 and N-cad, as well as Snai2 protein levels in DOX-treated cells. Protein was isolated from primary tumor cells that were sorted for GFP or RFP positivity to isolate untreated and DOX-treated cells, respectively.
Induced expression of miR-200c enhances differentiation of claudin-low tumors in vivo
Previous studies revealed low expression of basal and luminal lineage markers in claudin-low tumors, highlighting their undifferentiated state (22). To investigate whether reversal of EMT by miR-200c induction affected the differentiation profile of this claudin-low tumor model, we performed immunostaining for well-known keratin markers of basal and luminal lineages. Indeed, we found that DOX treatment led to an increase in expression of basal marker keratin-14 that co-localized with increased expression of luminal marker keratin-8 (K8), as seen by double-immunofluorescence staining (Figure 4A), as well as single staining shown in Supplemental Figure S1D. Additionally, upregulated K8 expression was observed primarily in cells with low vimentin expression, based on immunofluorescence analysis of sorted GFP+ and RFP+ cells (Supplementary Figure S1E).
Figure 4. miR-200c induction alters the differentiation state of claudin-low tumors.

(A) Images of immunofluorescent staining show that luminal and basal markers, specifically K8 and K14, were upregulated after miR-200c induction, suggesting a change in their differentiation profile. Scale bar represents 50μm. (B) Microarray analysis confirmed this enhanced differentiation by revealing a significant increase in the differentiation score of miR-200c-expressing primary tumors compared with our no dox treated tumors. Note that the miR-200c tumors had a differentiation score close to the basal tumors, and no longer fell into the claudin-low category. The classes of murine tumors are as described in detail previously (51), where Myc, Erbb2 and Neu tumors are of luminal subtype clustering, and p53null-basal, class 14 and C3-tag cluster into the basal subtype. (C) Microarray analysis also revealed a significant change in 1457 genes, with 562 being down-regulated, and 895 being up-regulated after treatment with DOX. The microarray analysis was performed on RNA extracted from GFP+ or RFP+ cells sorted from primary tumors, and the heatmap depicts genes with a 0% FDR. The analysis was consistent with a reversal of EMT, as many of the genes found to be downregulated are associated with the mesenchymal state, such as FoxC2, and also confirmed the induction of several keratin markers. (D) The graph indicates the top 12 most significantly enriched gene ontology terms in the 895 miR-200c upregulated gene set. Importantly, most of them were related to terms describing cell-to-cell adhesion and motility.
To further investigate the effects of miR-200c expression on the differentiation of claudin-low tumors, we performed a microarray analysis on sorted GFP+ and RFP+ primary cell populations and compared their overall differentiation scores. The transcriptional similarity of tumors to normal mammary cell populations, referred to as the differentiation score (1), was determined for our claudin-low tumor model, with and without miR200, and compared to the scores of multiple murine subtypes of breast cancer (21). Low differentiation scores indicate a similarity to adult mammary stem cells, medium scores a similarity to luminal progenitor cells, and higher scores a similarity to mature luminal cells (1). Consistent with observed upregulation of keratin expression, microarray analysis demonstrated a significant increase in the tumor differentiation score following miR-200c induction (Figure 4B). Interestingly, differentiation of the miR-200c-induced tumors was more similar to the differentiated basal tumors than to the undifferentiated claudin-low tumors from which they originated. Further analysis revealed significant changes in expression of 1457 genes between the two cell populations, with 562 being downregulated and 895 being upregulated in miR-200c-induced population (Figure 4C). Gene Ontology analysis of the 895 upregulated genes showed an enrichment for categories related to cell-cell adhesion and contact, consistent with the concept of an EMT reversal upon miR-200c expression (Figure 4D).
Functional cancer stem cells are diminished in miR-200c expressing tumors
Because EMT properties are linked to stem-like cell behavior and claudin-low tumors are highly enriched in CSCs, we sought to determine if reversal of EMT that occurred following miR-200c induction also altered CSC frequency in our model. Indeed, cells with miR-200c induction exhibited significant decreases in expression of the stem cell-associated genes Ezh2 and Bmi1 (Figure 5A), previously shown to be direct targets of miR-200c, and increased levels of the differentiation markers Elf5 and Gata3 (Figure 5B), in further support for the role of miR-200c in promoting a more differentiated cell state with a concomitant decrease in stem-like cell properties. Next, we analyzed tumor cells by FACS, using the cell surface antigens CD24 and CD29 to identify the CSC-enriched population (CD24high/CD29high) (22). FACS analysis revealed a change in overall profile and a decrease in the number of double-positive cells in tumors with miR-200c expression, suggesting a reduction in CSCs (Figure 5C).
Figure 5. Stem cell functionality is compromised after miR-200c induction.
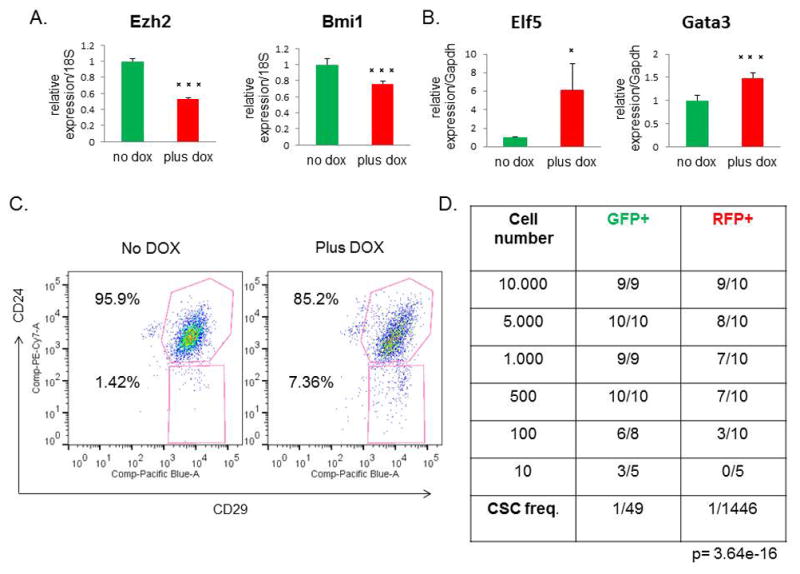
(A) There was a significant decrease in the stem cell markers Ezh2 and Bmi1 by qPCR post-DOX treatment. (B) Conversely, the mRNA levels of the luminal progenitor marker Elf5 and the mature luminal marker Gata3 were increased in RFP+ cells, compared with GFP+ cells. (C) A representative FACS profile for CD24+/CD29+ cells shows a change in the overall profile of miR-200c tumors, with the number of double-positive cells decreased relative to untreated controls. (D) Table depicts the results from the limiting dilution transplantation assay. As decreasing numbers of either GFP+ or RFP+ sorted cells were transplanted back into the recipient mouse, fewer tumors arose in each transplantation group. The RFP+ miR-200c-expressing cells showed a significantly reduced CSC frequency, as shown in the bottom row, confirming that miR-200c leads to a reduction in stem cell functionality.
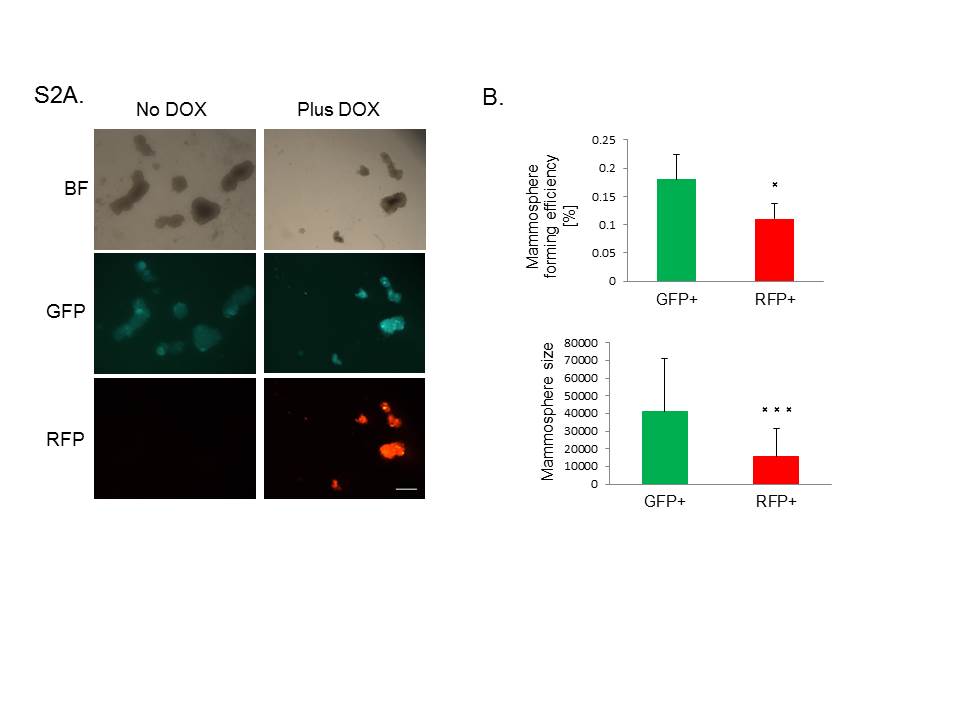
To validate the decrease in CSC frequency observed by FACS, we also performed functional assays to measure CSC activity. First, we evaluated the mammosphere forming efficiency after DOX administration. Notably, we found a significant decrease in the number and size of mammospheres formed in the miR-200c-induced group, demonstrating that miR-200c significantly impairs CSC functionality in claudin-low tumors (Supplementary Figure S2A–B). To confirm these findings in vivo, we performed a limiting dilution transplantation assay, which is the standard method to determine the in vivo repopulating ability, or CSC frequency of cells after transplantation. We injected decreasing numbers of GFP+ and RFP+ sorted cells into recipient mice, with continued DOX administration, and evaluated the number of resulting tumors. Consistent with the results of the mammosphere assay, we found that miR-200c expression significantly reduced CSC frequency and repopulation potential of primary tumor cells (Figure 5D). These data validate that stem cell functionality is severely impaired after miR-200c induction in claudin-low tumors.
Chemotherapy resistance is compromised in tumors expressing miR-200c
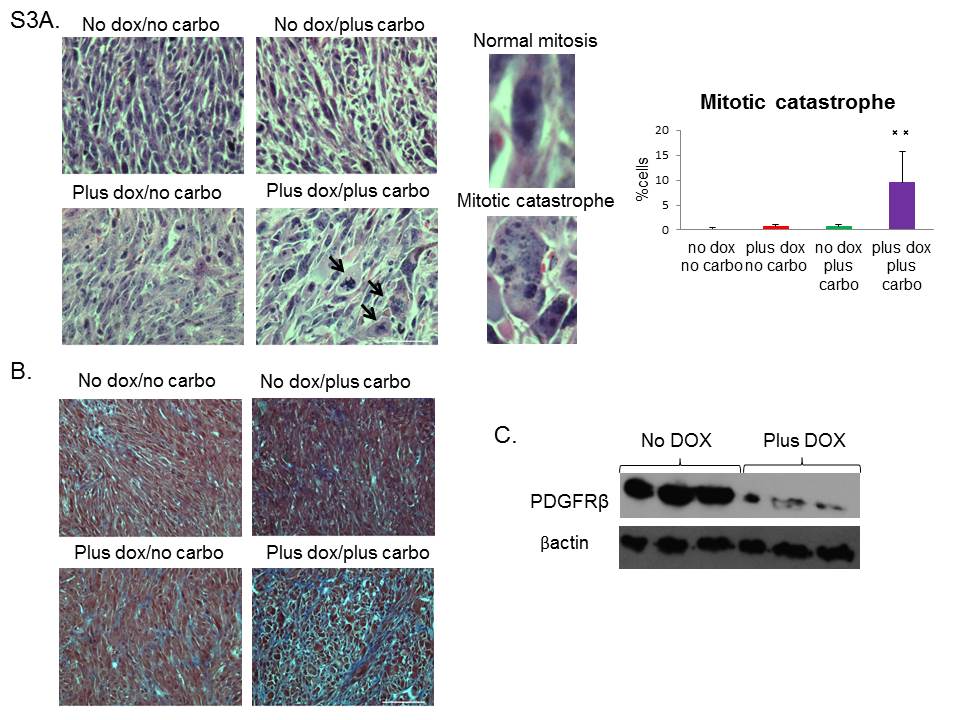
The claudin-low subtype of breast cancer is highly resistant to conventional chemotherapy, and a claudin-low signature was observed in residual chemotherapy-resistant breast cancer (7). In this study, miR-200c expression markedly decreased CSC functionality of these tumors, and enhanced their differentiation. Therefore, we evaluated whether it also sensitized tumors to chemotherapy drug carboplatin. To this end, we applied either combination or single agent treatment for a course of 10 days. Although DOX treatment alone led to a decrease in tumor growth, combination treatment with DOX and carboplatin caused complete tumor stasis, with a significantly lower growth rate than the other groups (Figure 6A). Additionally, the final change in tumor volume was significantly decreased in combination-treated group as compared to all other treatment groups (Figure 6B). As claudin-low tumors are resistant to single carboplatin treatment due to high percentage of CSCs (26), we hypothesized that combination of DOX and carboplatin led to the death of miR-200c-expressing cells, as these had diminished CSC functionality. Accordingly, we performed FACS analysis using Annexin V staining to determine the extent of apoptosis in treated tumors. After combination treatment, there was a 10-fold increase in the level of apoptosis compared with untreated mice. This observation was further strengthened by the fact that combination treatment eliminated the miR-200c-expressing cells, as seen by a decrease in the total number of RFP+ cells. Indeed, almost half of the RFP+ cells were Annexin V-positive and were, therefore, undergoing apoptosis (Figure 6C–D). Of note, miR-200c induction alone lead to an increase in cell death; however this phenotype is amplified by carboplatin addition. Further H&E analysis showed that these cells were undergoing mitotic catastrophe, as seen by a significantly higher number of giant, multinucleated cells (27) in tumors that had undergone combination treatment, compared to normal mitosis of vehicle treated tumors (Supplementary Figure S3A). In addition, combination treatment led to an increase in collagen deposition in tumors, as noted by Mason’s Trichrome staining (Supplemental Figure S3B), presumably due to the wound healing response that occurred in response to the high level of cell death. Taken together, our data suggest that re-expressing miR-200c in therapy-resistant tumors leads to chemosensitization by increasing their susceptibility to chemotherapy-induced cell death.
Figure 6. miR-200c sensitizes tumor cells to chemotherapy.

(A) DOX and carboplatin combination treatment caused tumor stasis, as seen by a significant difference in the growth rate of combination group, compared to all other treatment groups. Arrows point to the time of carboplatin injection. (B) There is a significant decrease in the average change in volume after treatment (n=20 for carboplatin+DOX group, n=15 for DOX only, n=8 for vehicle and carboplatin alone groups) (C–D) AnnexinV FACS analysis shows a significant increase in the number of cells undergoing early stages of apoptosis in the combination treatment group, as compared to all other groups. Sytox red staining represents dead cells, therefore cells positive only for AnnexinV were considered currently undergoing apoptosis. Analysis included only GFP+ or RFP+ cells, therefore AnnexinV positive cells are represented only through these gates, and quantified to the left.
MiR-200c induction decreases metastasis
Metastasis is strongly correlated with angiogenesis in primary tumors (28–30). Thus, considering that miR-200 members have been reported to control the levels of pro-angiogenic factors in several cancer models (31,32), we next investigated whether miR-200c induction affected pro-angiogenic factors in our model. Although we observed no change in VEGFR protein levels, we found a significant loss of a prominent metastasis-related protein PDGFRβ in RFP+ compared with GFP+ sorted primary tumor cells (Supplemental Figure S3C). Importantly, PDGFRβ expression correlates with breast cancer aggressiveness and metastasis in human patients, and coincides with EMT-associated metastasis in transgenic mice (33). Therefore, we asked if miR-200c induction and the corresponding downregulation of PDGFRβ correlated with a decreased metastatic potential of primary claudin-low tumor cells. To address the role of miR-200c in pulmonary colonization, we performed tail-vein injections of GFP+ sorted cells after transduction and started DOX treatment 16 hours post-injection. This allowed the cells to travel to the lungs prior to miR-200c induction. After 8 weeks of treatment, induction of miR-200c led to a significant decrease in the number of lungs containing any histologically detectable metastatic lesions (Figure 7A), and importantly, a decrease in the metastatic burden (Figure 7B). Representative images of whole lungs (Figure 7C), as well as H&E stained sections (Figure 7D), are shown. Collectively, our data suggest that miR-200c re-expression in claudin-low tumors may provide a novel therapeutic approach, as it reduces tumor growth, decreases the number of functional CSCs, sensitizes tumor cells to chemotherapy, and ultimately, decreases metastatic potential.
Figure 7. Metastatic potential is severely impaired following miR-200c induction.
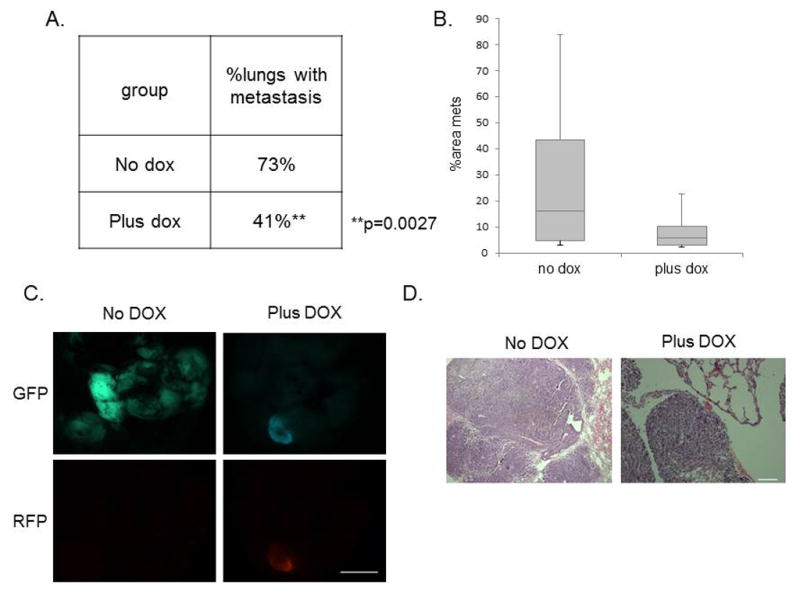
(A) Table shows that the percentage of tumor-bearing lungs was significantly reduced in the miR-200c-induced group. Data was analyzed using a Chi-squared test for two populations. (B) The average areas of metastasis were also lower in the miR-200c-induced group compared with the vehicle-treated group. (C) Fluorescent images of GFP and RFP expression shows metastasis present in the lung of both groups. Scale bar equals 5mm. (D) Representative H&E images of lungs are shown following 8 weeks of DOX treatment after tail-vein injection of 30 000 primary GFP+ cells. Scale bar equals 100μm.
Discussion
Given the central position of the miR-200 family in the process of EMT, restoration of its expression has a tremendous impact on mesenchymal and stem-like properties of cells (15,16,34,35). Although it was previously shown that miR-200c overexpression reduces mesenchymal features in several in vitro immortalized cell lines, this study for the first time utilized an in vivo primary syngeneic tumor model of breast cancer that has a gene expression signature which parallels those of human claudin-low tumors. Additionally, several studies confirmed that this model closely mimics human claudin-low tumors in their pathology and CSC enrichment (1,20,21). Here, we demonstrate that induced expression of the miR-200c cluster in vivo altered underlying CSC features of claudin-low tumors, making them more sensitive to chemotherapy and dampening their metastatic potential. These effects were achieved through EMT reversal, decreases in proliferation and stem cell functionality, and ultimately, an enhanced susceptibility to cell death.
First, we showed that induction of miR-200c pushed tumor cells into more organized epithelial-like structures and significantly reduced their proliferation. These changes were accompanied by an increase in differentiation score, suggesting a change in stem cell context. Since more differentiated tumors are thought to originate from cells that have more limited progenitor potential, they display less therapy resistance and tumor recurrence (9,36) than human and mouse CSCs (7,37,38). Indeed, our results conclusively showed that miR-200c-expressing tumors had a significant decrease in frequency, as well as functional self-renewing potential of CSCs, shown by limiting dilution transplantation, and that they do not transition into a more proliferative epithelial-like CSC state, as suggested by a recent study (39). Importantly, the change in CSCs observed upon miR-200c re-expression led to their heightened sensitivity to carboplatin. Accordingly, miR-200c-expressing cells also showed a significant reduction in expression of stem cell markers Ezh2 and Bmi1, shown previously to be direct targets of miR-200 (14,40,41). Interestingly, previous studies demonstrated that knockdown of these proteins decreases breast CSC frequency and increases their sensitivity to therapy (40,42,43). Our data support the concept that miR-200c, potentially via its regulation of Ezh2 and Bmi1, can influence functionality and frequency of CSCs in vivo, a finding that could be exploited to overcome therapy resistance manifested in CSCs-enriched tumors. Furthermore, significant loss of Zeb1 protein upon miR-200c re-expression suggests a potential decrease in DNA repair capacity (44), which may in turn increase the susceptibility of miR-200c-induced tumors to chemotherapy.
MiR-200c plays a central role in preventing EMT; thus, it is considered to be anti-metastatic within primary tumors. Nonetheless, there is considerable controversy regarding its role in promoting metastatic growth at the secondary tumor site. Some studies suggested that, although the promotion of EMT increases the intravasation potential of cancer cells, reversal of EMT, or MET, must occur at secondary sites for cells to be able to revert into the epithelial state in the distant organ and form macrometastases (17,45). However, these were performed with miR-200 over-expressing tumor cell lines prior to injecting them into the tail vein, or from the orthotopic site, where they grew with high miR-200c expression. Another study showed that high miR-200c levels in lung tissue correlate with higher metastatic burden and poorer prognosis (46). However, this result was observed only in luminal subtypes of breast cancer, whereas basal-like breast cancer exhibited worse survival with decreased miR-200c expression (31). In support of these data, high miR-200 levels were also found to be anti-metastatic in triple-negative breast cancer cells transplanted into an orthotopic site of mouse mammary glands (47). Unfortunately, our p53null claudin-low model is not suitable for investigation of metastasis from the orthotopic site, as it is not feasible to detect distant metastasis prior to sacrifice of the animals. Alternatively, we resected the primary tumors early during tumor progression, but after several months no metastases were observed in either of the groups, vehicle or DOX treated. An immune T-cell response to claudin-low tumors in syngeneic mice may inhibit intravasation in this model. However, our tail-vein injection studies suggest that even if intravasation did occur, the growth of macrometastases in the secondary site would be impeded by miR-200c, as we compared the extravasation and colonization potential of primary claudin-low tumor cells with and without miR-200c expression. These studies indicated that miR-200c induction not only inhibits primary tumor growth, but also decreases the metastatic potential of claudin-low tumors, most likely by decreasing colonization. Additional studies should be performed to study the role of miR-200c in cell survival in the bloodstream and extravasation into the lungs in this model.
Multiple RNAi therapeutics are currently being tested in clinical trials, and several groups have already reported in vivo microRNA delivery using nanoparticles with very promising results (48). Hence, our studies offer a potential target of microRNA delivery for clinical treatment of an aggressive subtype of breast cancer that is otherwise highly resistant to conventional radiation and chemotherapy. Success of these therapeutic approaches will depend upon improvements in both efficiency and specificity of these delivery systems. In this study, we demonstrate that miR-200c has therapeutic effects in an in vivo model of claudin-low breast cancer, which should provide the basis for clinical applications in miR-based drug development to treat this subtype of breast cancer. Finally, the detailed characterization of this transplantable in vivo model provides the foundation for future studies in which the interplay of EMT and the immune system can be investigated in therapeutic response and metastasis, something that has not been feasible to date using the current cell line and xenograft models.
Materials and Methods
Lentiviral vector construction
The doxycycline inducible vector was a kind gift from Dr. Thomas Westbrook at Baylor College of Medicine (25). The shRNA sequence was excised and replaced with the human pre-miR-200c-141 sequence, kindly provided by Dr. Gregory Goodall, University of Adelaide, Australia.
Lentiviral infection, tumor digestion and transplantation
Claudin-low tumors from the p53null mouse tumor bank (20) were digested into single cells according to our laboratory protocol (see below), transduced with the lentivirus containing the insert at an MOI of 20, as previously described (49), and injected into the cleared fat pad of wild-type Balb/c recipient mice. Detailed protocols can be found at the following website addresses: https://mediasrc.bcm.edu/documents/2014/bc/tumorpropagationfreezingprotocol.pdf, and https://mediasrc.bcm.edu/documents/2014/70/lentiviralproductionandtitering.pdf. Tumors were harvested after they reached approximately 1000 mm3 and again digested into single cells. Cells were analyzed and sorted for GFP positivity. Ten thousand GFP+ cells were injected into the cleared fat pads of host mice. Upon tumor palpability, the mice were randomized into treatment groups. GFP+ tumors were frozen in DMEM media containing 10% DMSO and 10% FBS for future transplants. Treated tumors were digested and sorted for GFP+ and RFP+ cells. Sorted cells were washed in PBS and either flash frozen in liquid nitrogen for RNA or protein isolation, or injected back into host mice at different dilutions for the limiting dilution transplantation assay. Limiting dilution analysis was performed using the ELDA program (50). All cell injections and surgeries were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal protocols were reviewed and approved by the Animal Protocol Review Committee at Baylor College of Medicine. Balb/c mice were obtained from Harlan Laboratories.
Metastasis assay
30 000 GFP+ cells were injected into the tail veins of 8 to 10-week-old Balb/c mice and DOX treatment was initiated 16 hours post-injection. The lungs were collected after 8 weeks of treatment, and fixed in 4% PFA overnight for histological assays.
Doxycycline and carboplatin treatment
Doxycycline (Clontech 631311) was administered to mice in water, using 5% sucrose as the vehicle. 2 mg/ml doxycycline solution was freshly prepared twice a week. Carboplatin (MP Biomedicals 198873) was resuspended in 0.9% NaCl and i.p. injected into mice once a week, at a concentration of 50 mg/kg.
Immunostaining
Tumors were fixed in 4% PFA overnight, embedded into paraffin, and sectioned into 5 μm sections. Tumor sections were subjected to H&E, or immunofluorescence staining, https://mediasrc.bcm.edu/documents/2014/78/immunofluorescencegeneralprotocol.pdf, as per the protocol, and probed overnight at 4°C with antibodies for E-cadherin (Cell Signaling 3195), keratin-14 (Covance PRB-155P), keratin-8 (DSHB Troma-1-c), Ki67 (Vector labs VP-K451), vimentin (Cell Signaling 5741), Zeb1 (Cell Signaling 3396), N-cadherin (Invitrogen 333900), Snai2 (Cell Signaling 9585), all diluted according to manufacturer’s instructions.
RNA isolation and quantitative PCR
RNA was isolated using Qiagen microRNeasy kit (217084). cDNA was synthesized either using a High Capacity RNA to cDNA kit (Applied Biosystems 4387406), or a microRNA kit (Applied Biosystems 4366597). qPCR was performed with Taqman assays (Cdh1 Mm01247357_m1, Zeb1 Mm00495564_m1, Zeb2 Mm00497193_m1, Vimentin Mm01333430_m1, N-cadherin Mm01162497_m1, Snai2 Mm00441531_m1, Ezh2 Mm00468464_m1, Bmi1 Mm03053308_m1, Gata3 Mm00484683_m1, Elf5 Mm00468732_m1) on a Step One Plus real-time PCR machine.
Western blot and RPPA analysis
Flash frozen tumor pieces or sorted cells were homogenized in RIPA buffer with added protease and phosphatase inhibitors (Roche) using a Polytron tissue homogenizer or vigorous pipetting. Protein concentrations were determined using a Bradford assay. Equal amounts of proteins were loaded onto a BioRad Pre-Cast gel, run at 100V, and subsequently transferred onto a PVDF membrane. Blocking was performed in 5% non-fat dry milk diluted in TBS/T. Primary antibodies were diluted in 5% BSA in TBS/T and incubated overnight at 4°C. Protein bands were visualized on autoradiography films. RPPA analysis was performed in the core of Dr. Gordon Mills at MD Anderson Cancer Center.
Flow Cytometry
Single cells were labeled with antibodies for CD24-PeCy7 and CD29-PacBlue, or Annexin V-PacBlue (BD Pharmingen), based on the manufacturer’s data sheet. Lineage cells were excluded using the BD Pharmingen lineage cocktail. Dead cells were excluded by staining with Sytox Red (1:1000).
Microarray analysis
Microarray was performed and analyzed as previously described (21). GEO accession number is GSE62230.
Statistical Analysis
Each experiment was repeated a minimum of three times, with n≥3 biological replicates in each group, unless otherwise noted. Two-tailed paired Student’s t-test was applied when only 2 groups were compared. Bars represent mean ± s.d. of ≥ three mice per group (*p<0.05, **p<0.01, ***p<0.001). One-way ANOVA analysis with Tukey p-value adjustment was applied to multiple group comparisons.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
These studies were supported by grants CA148761 (JMR and CP) from the National Cancer Institute and RP130485 (JMR) and RP140102 (JK) from CPRIT. This project was supported by the following core facilities and their funding: Biostatistics and Informatics (P30-CA125123) and Cytometry and Cell Sorting Core at Baylor College of Medicine (P30 AI036211, P30 CA125123, and S10 RR024574). RPPA arrays were performed at MD Anderson Cancer Center RPPA Core Facility – Functional Proteomics. The authors would like to thank Drs. Kevin Roarty Amy Shore, and Michael Toneff for helpful discussions and paper editing, Dr. Jason Herschkowitz for help and advice, and Ms. Shirley Small for animal husbandry.
Footnotes
Authors declare no conflict of interest.
References
- 1.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010 Jan;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Hennessy BT, Gonzalez-Angulo A-M, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee J-S, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–54. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–5. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:253–60. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- 9.Visvader JE, Lindeman GJ. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell. 2012;10:717–28. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Kim N-G, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108:11930–5. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–54. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 12.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c Links Breast Cancer Stem Cells with Normal Stem Cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim Y-Y, Wright Ja, Attema JL, Gregory Pa, Bert AG, Smith E, et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J Cell Sci. 2013;126:2256–66. doi: 10.1242/jcs.122275. [DOI] [PubMed] [Google Scholar]
- 16.Hurteau GJ, Carlson JA, Roos E, Brock GJ. Stable expression of miR-200c alone is sufficient to regulate TCF8 (ZEB1) and restore E-cadherin expression. Cell Cycle. 2009;8:2064–9. doi: 10.4161/cc.8.13.8883. [DOI] [PubMed] [Google Scholar]
- 17.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–8. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerry D, Kittrell F, Kuperwasser C, Laucirica R, Dickinson E, Bonilla P, et al. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–8. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 19.Network C genome AR. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci. 2012;109:2778–83. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013;14:R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008 Jun 15;68(12):4674–82. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13:R45. doi: 10.1186/bcr2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jurmeister S, Baumann M, Balwierz A, Keklikoglou I, Ward A, Uhlmann S, et al. MicroRNA-200c Represses Migration and Invasion of Breast Cancer Cells by Targeting Actin-Regulatory Proteins FHOD1 and PPM1F. Mol Cell Biol. 2012;32:633–51. doi: 10.1128/MCB.06212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:3665–70. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Usary J, Zhao W, Darr D, Roberts PJ, Liu M, Balletta L, et al. Predicting drug responsiveness in human cancers using genetically engineered mice. Clin Cancer Res. 2013;19:4889–99. doi: 10.1158/1078-0432.CCR-13-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 28.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 29.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 30.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 31.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun. 2013;4:2427. doi: 10.1038/ncomms3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roybal JD, Zang Y, Ahn Y-H, Yang Y, Gibbons DL, Baird BN, et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011;9:25–35. doi: 10.1158/1541-7786.MCR-10-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, et al. Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest. 2006;116:1561–70. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 35.Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–22. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 37.Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surg Oncol. 2010;19:27–32. doi: 10.1016/j.suronc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer Lett. 2013;341:56–62. doi: 10.1016/j.canlet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014;2:78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui J, Cheng Y, Zhang P, Sun M, Gao F, Liu C, et al. Down regulation of miR200c promotes radiation-induced thymic lymphoma by targeting BMI1. J Cell Biochem. 2014;115:1033–42. doi: 10.1002/jcb.24754. [DOI] [PubMed] [Google Scholar]
- 41.Kopp F, Oak PS, Wagner E, Roidl A. miR-200c Sensitizes Breast Cancer Cells to Doxorubicin Treatment by Decreasing TrkB and Bmi1 Expression. PLoS One. 2012:7. doi: 10.1371/journal.pone.0050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang CJ, Yang JY, Xia W, Chen C, Te Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-??-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin J, Zheng G, Jia X, Zhang Z, Zhang W, Song Y, et al. A Bmi1-miRNAs Cross-Talk Modulates Chemotherapy Response to 5-Fluorouracil in Breast Cancer Cells. PLoS One. 2013:8. doi: 10.1371/journal.pone.0073268. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014 Aug 3; doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009:4. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gravgaard KH, Lyng MB, Laenkholm AV, Søkilde R, Nielsen BS, Litman T, et al. The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res Treat. 2012;134:207–17. doi: 10.1007/s10549-012-1969-9. [DOI] [PubMed] [Google Scholar]
- 47.Humphries B, Wang Z, Oom AL, Fisher T, Tan D, Cui Y, et al. MicroRNA-200b targets protein kinase Cα and suppresses triple-negative breast cancer metastasis. Carcinogenesis. 2014 Jun 12;:1–10. doi: 10.1093/carcin/bgu133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu SY, Lopez-Berestein G, Calin Ga, Sood AK. RNAi Therapies: Drugging the Undruggable. Sci Transl Med. 2014;6:240ps7. doi: 10.1126/scitranslmed.3008362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welm BE, Dijkgraaf GJP, Bledau AS, Welm AL, Werb Z. Lentiviral Transduction of Mammary Stem Cells for Analysis of Gene Function during Development and Cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 51.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.