

Abstract
Specialized variants of the constitutive 20S proteasome in the immune system like the immunoproteasomes and the thymoproteasome contain active site-bearing subunits which differ in their cleavage priorities and substrate binding pockets. The immunoproteasome plays a crucial role in antigen processing and for the differentiation of pro-inflammatory T helper cells which are involved in the pathogenesis of autoimmunity. Selective inhibitors of the immunoproteasome and constitutive proteasome have recently been generated which interfere with the development and progression of autoimmune diseases. Here we describe these inhibitors and their therapeutic potential as predicted from preclinical models.
Introduction
The ubiquitin-proteasome pathway is the major quality-control pathway for newly synthesized proteins in every eukaryotic cell. The 26S proteasome is a large (2.5 MDa), multi-subunit, ATP-dependent proteolytic complex that processively degrades proteins into peptides. It consists of a hollow cylindrical 20S proteolytic core and one or two 19S regulatory particles (RP). The 19S RP recognizes ubiquitylated substrates and prepares them for proteolysis, which occurs inside the 20S core. The 20S cores are hollow cylindrical structures comprising two pairs of 14 different polypeptides arranged in four stacked rings. Three subunits on each inner ring carry catalytic residues for the proteolytic sites (Figure 1). The catalytic residues of the chymotrypsin-like sites are located on β5 subunits and cleave peptide bonds predominantly after hydrophobic residues. The caspase-like sites are located on the β1 subunits which cleave peptide bonds after acidic residues. The third pair of sites is located on the β2 subunits. They cleave after basic residues and are referred to as trypsin-like sites. The lymphoid tissues contain immunoproteasomes, in which β5, β2, β1 subunits are replaced with β5i (LMP7), β2i (MECL1), β1i (LMP2) subunits, respectively (Figure 1). Moreover, epithelial cells in the cortex of the thymus express thymoproteasomes consisting of the active site subunits β5t, β2i and β1i. Here we describe the recent development of inhibitors for these tissue specific proteasome variants and how they can be exploited for immunomodulation in antigen processing and the therapy of autoimmune diseases.
Figure 1.
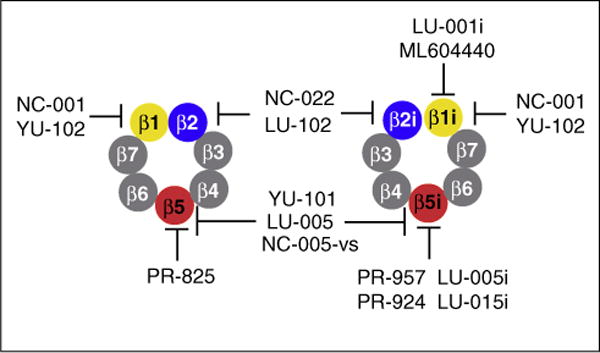
Subunits of the constitutive proteasome (left) and the immunoproteasome (right) with assignment of their respective inhibitors as mentioned in the text.
The majority of proteasome inhibitors inhibit multiple active sites
The inhibitors of proteolytic sites of the 20S core have served as indispensible tools to identify proteasome substrates and establish proteasome involvement in a variety of biochemical, immunological, and physiological processes for two decades (see [1••] for review). One of the earliest findings with these inhibitors was that transformed cells rapidly undergo apoptosis upon treatment with proteasome inhibitors, while much higher concentrations are needed to induce death of their non-transformed counterparts. These discoveries stimulated development of proteasome inhibitors as anti-neoplastic agents and, in 2003, bortezomib was approved by the FDA for the treatment of multiple myeloma. It took few additional years to realize that exquisite sensitivity of this particular cancer to proteasome inhibitors is caused by an exceptionally high load on the proteasome in myeloma cells, which secrete large amounts of immunoglobulins [2]. In 2012, another proteasome inhibitor, carfilzomib [3], has been approved for the treatment of multiple myeloma.
Because results of site-directed mutagenesis in yeast revealed that the chymotrypsin-like sites are most important in protein degradation, and because of the ability of hydrophobic peptides to enter cells, the various synthetic proteasome inhibitors were optimized according to their capacity to block the β5 sites, which cleave preferentially after hydrophobic residues [1••]. The primary target of all natural product inhibitors is also the chymotrypsin-like site. All compounds targeting chymotrypsin-like sites (e.g., MG-132, bortezomib, epoxomicin, clasto-lactacystin-β-lactone) effectively inhibit protein degradation inside the cells. However, none of these inhibitors can be considered chymotrypsin-like sites selective, and inhibition of protein degradation occurs at concentrations that co-inhibit either caspase-like or trypsin-like sites [4], raising the possibility that contribution of these two sites to protein degradation is larger than previously appreciated. This observation, together with the fact that bortezomib inhibits the caspase-like activity of the proteasome [4–6], stimulated the interest in the development of specific inhibitors of the individual active sites, to be used as tools to dissect contributions of individual sites to protein degradation, and to define their individual roles as targets for anti-neoplastic and anti-inflammatory agents.
Subunit-specific proteasome inhibitors
The great majority of site-specific proteasome inhibitors consist of an electrophilic trap that interacts with the catalytic threonine at the N-terminus of mature β-type subunits and a peptide moiety. A peptide epoxyketone YU-101 (Figure 2A) was the first highly potent and specific inhibitor of the chymotrypsin-like sites to be developed [7]. Carfilzomib is a derivative of this inhibitor [3]. It is the most specific inhibitor of the chymotrypsin-like sites among FDA-approved agents or agents undergoing clinical trials. NC-005 is even more specific than YU-101 and carfilzomib [8] while IPSI-001 inhibits both β1i and β5i [9]. Further efforts to improve selectivity have challenged the common dogma that active-site specificity is determined by the peptide portion of the inhibitor but not by the active-site electrophile. It was found that replacing epoxyketone in NC-005 (Figure 2A) with a vinyl sulfone moiety increases specificity for the chymotrypsin-like sites [10•]. Finally, utilization of fluorinated amino acids had led to another compound with improved specificity for the chymotrypsin-like activity, LU-005 (Figure 2A) [11].
Figure 2.
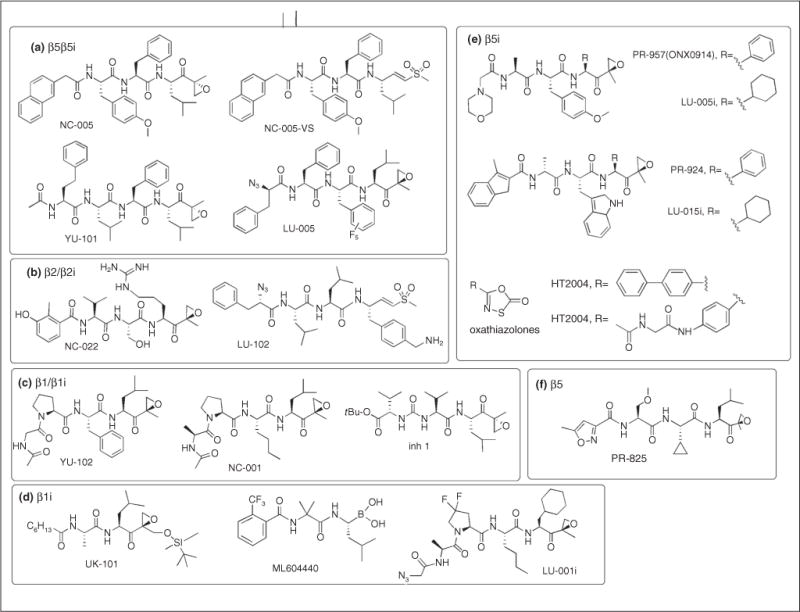
Structures of proteasome inhibitors mentioned in this review grouped according to their proteasome subunit specificities.
Systematic modification of the peptide epoxyketone scaffold led to the development of the β5-specific inhibitor PR-825 (Figure 2F), and two β5i inhibitors, PR-924 and PR-957 (later renamed ONX-0914, Figure 2E) [12•,13••]. Replacement of phenyl side chains in the P1 position in PR-924 and PR-957 with a cyclohexyl generated the highly selective β5i inhibitors LU-015i and LU-005i (Figure 2E) [14]. Oxathiazolones (Figure 2E) were first discovered as inhibitors of mycobacterial proteasomes but a recent study revealed that they show considerable selectivity toward β5i over β5 sites [15•]. The residues involved in the stabilization of the oxathiazolone covalent adducts with the mycobacterial proteasomes and the β5i site of human immunoproteasomes are similar. Oxathiazolones have a short half-life in aqueous solutions [15•], which may limit their in vivo activity. Lastly, a number of non-peptide inhibitors of β5i were identified recently by virtual screening [16].
YU-102 was the first inhibitor of caspase-like sites to be developed [17], followed by the more specific NC-001 and compound 1 [18] (Figure 2C). All three epoxyketones inhibit both the β1 and β1i sites. Replacement of the isopropyl side chain in the P1 position with a cyclohexyl moiety, and fluorine substitutions in the 3-postion of the P3-Pro residue converted NC-001 into the β1i-specific inhibitor LU-001i (Figure 2D). LU-001i is more β1i-specific than another peptide epoxyketone UK-101 (Figure 2D), which also inhibits β5i sites [14,19]. Dipeptide boronate ML604440 is a more potent and specific inhibitor of β1i sites [20]. All inhibitors are cell-permeable.
A bigger challenge has been the development of cell-permeable inhibitors of the trypsin-like sites. The first specific inhibitors of these sites were cell impermeable [21,22]. Another structural class, peptide vinyl esters [23], initially reported as cell-permeable inhibitors of the trypsin-like sites, did not have any inhibitory activity when re-synthesized by another group [10•]. Finally two cell-permeable peptide epoxyketones, NC-002 and NC-022 (Figure 2B), were discovered a few years ago [24]. They were followed by the more potent vinyl sulfone LU-102 (Figure 2B), which also has a better cell permeability [25••]. Just as with inhibitors of the chymotrypsin-like sites, these studies revealed that replacing the epoxyketone electrophile with vinyl sulfone dramatically increases the specificity of compounds for its primary target.
Most of these specific inhibitors were initially used to define the role of different active sites as drug targets in cancer. For example, it was found that increasing the specificity of compounds for the chymotrypsin-like sites dramatically decreases the cytotoxicity for HeLa cells [10•]. Inhibitors of trypsin-like and caspase-like sites, while not cytotoxic to cells when used as a single agent, sensitized multiple myeloma cells to inhibitors of the chymotrypsin-like sites, e.g. NC-005 and LU-005 [8]. Furthermore, inhibitors of the trypsin-like sites selectively sensitized myeloma cells to bortezomib and carfilzomib [24]. In other words, cytotoxicity was observed only when two types of active sites were inhibited. The lack of cytotoxic effect of site-specific inhibitors offers opportunities for using them for immunomodulation.
Selective inhibition of the immunoproteasome allows in vivo modulation of antigen processing and T cell generation
When cells are stimulated with interferon (IFN)-γ and tumor necrosis factor (TNF)-α, they produce vast amounts of the inducible β-type subunits β1i, β2i, and β5i. In such a pro-inflammatory environment virtually all newly synthesized 20S proteasomes incorporate these inducible subunits rather than their constitutive homologues β1, β2, and β5 to form immunoproteasomes. In mice infected with viruses, bacteria or fungi it has been shown that in the liver and other organs (but not the brain [26]) constitutive proteasomes are largely replaced by immunoproteasomes [27,28]. The proteasome is centrally involved in the generation of peptide ligands of major histocompatibility complex (MHC) class I molecules which are recognized by the antigen receptors of cytotoxic T lymphocytes (CTL) (Figure 3). Under stimulation with IFN-γ or TNF-α the cell surface expression of class I molecules is up-regulated approximately tenfold. The immunoproteasome is required to produce this surplus of class I peptide ligands needed because deletion of β5i alone or all three immunosubunits (β1i, β2i, β5i) together reduces class I surface expression in spleen, lymph nodes and thymus by approximately 50% [29]. Moreover, the treatment of cells or mice with the β5i selective inhibitor ONX-0914 (formerly called PR-957) leads to a similar reduction in MHC class I surface expression indicating that β5i is pivotal for the elevated generation of class I ligands [13••,30]. ONX-0914 exerts this effect in wild type but not in β5i−/− mice indicating an on target effect of this first selective inhibitor of the immunoproteasome.
Figure 3.
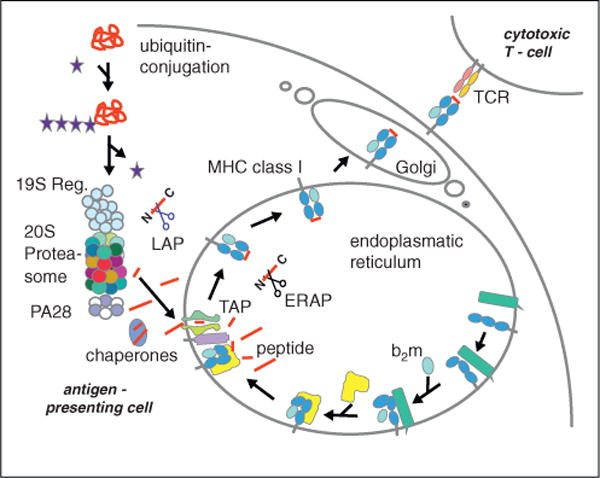
Antigen processing along the MHC class I pathway. Proteins synthesized in the cell are polyubiquitylated and degraded by the proteasome. Peptides produced are either of the ideal length for class I binding (8–9 amino acids) or are N-terminally extended precursors which can be cleaved by amino peptidases in the cytoplasm. The transporter associated with antigen processing (TAP) transports the peptides into the endoplasmic reticulum (ER) where they can be further trimmed by ER amino peptidase (ERAP)1/2. Peptides binding with high affinity to the MHC class I heavy chain/β2-microglobulin (b2m) complex induce a final folding and release from the ER lumenal chaperone calreticulin (in yellow) and the transmembrane chaperon calnexin (in green) to allow exit from the ER to the plasma membrane.
The most likely reason why immunoproteasomes produce more or better suited class I ligands is that compared to constitutive proteasomes they alter the cleavage preferences such that class I ligands with higher binding affinities for the peptide binding groove of class I molecules are generated. The caspase-like activity of β1, for example, produces weaker class I ligands than the chymotrypsin-like activity of β1i because their C-terminal anchor residues must possess hydrophobic or basic but not acidic side chains to enable tight binding into the peptide cleft of class I molecules [31]. Since β5 and β5i both exert a chymotrypsin-like activity, the requirement for this exchange was less obvious. However, the recently reported high resolution structures of mouse constitutive- and immunoproteasomes revealed that the S1 substrate pocket of β5i accommodates better bulky aliphatic and aromatic hydrophobic side chains [32••] which are most frequently found in peptides eluted from class I molecules. In spite of this valuable structural insight, the consequence of replacing β2 with β2i has remained elusive as they both harbor an equivalent trypsin-like activity [33,34] and no obvious differences between the peptide binding pockets of β2 and β2i have been noted. Accordingly, the generation of selective inhibitors for β2 and β2i will be a formidable task.
The effect of immunoproteasome deficiency or inhibition on the bulk supply of MHC class I ligands on immune defence is limited. Mice lacking the inducible subunits are immunocompetent except for an enhanced susceptibility to experimental infections with Toxoplasma gondii [35] and Listeria monocytogenes [36]. However, on the level of the unique epitope the presence of immunoproteasomes can be crucial either because the activity of β1i, β2i, or β5i is required for generating the epitope [37] or because epitope destruction through the constitutive subunits β1, β2, or β5 needs to be prevented by replacement with the respective immuno-subunits [20]. The important role of immunoproteasome subunits in defining the repertoire of peptides presented on MHC class I molecules has been underlined by the mass spectrometric analysis of peptides eluted from two class I molecules of splenocytes of wild type and β1i/β2i/β5i triply gene deleted mice [29]. The advent of subunit specific inhibitors has therefore enabled the pharmacological modulation of peptide antigen generation and presentation as well as the development and expansion of cognate T cells in vivo [13••,30]. Whether this potential of subunit selective proteasome inhibitors can be exploited for pharmacological intervention with infectious or autoimmune diseases remains to be determined.
Immunoproteasome subunits as novel targets for the suppression of autoimmunity
Accumulating evidence indicates that the immunoproteasome plays a crucial role for the pathogenesis of several autoimmune diseases and that this role is unrelated to the MHC class I antigen processing pathway. It was found that both, CD8+ CTLs and CD4+ T helper cells, need the immunoproteasome to survive in a pro-inflammatory environment [38]. Consequently, the inhibition of β5i, β2i, or β1i may be a means to ameliorate pro-inflammatory autoimmune diseases which rely on these cells. In fact, the treatment of mice with the β5i selective inhibitor ONX-0914 prevented the development or progression of experimental arthritis [13••], diabetes [13••], inflammatory bowel disease [39], systemic lupus erythematosus [40], and experimental autoimmune encephalomyelitis (EAE) [41••] in preclinical mouse models. Unexpectedly, β5i-deficient mice did not display the same protection profile as ONX-0914 treated wild type mice in each of these models. Both, β5i inhibition and deficiency protected mice from dextrane sulfate sodium (DSS) induced colitis indicating that β5i plays an essential role in disease development which cannot be sustained by β5 activity [39,42]. In the multiple sclerosis model EAE, in contrast, protection from disease progression and relapse was observed in ONX-0914 treated wild type but not in β5i−/− mice [41••]. However, the treatment of β5i−/− but not wild type mice with the β5-selective inhibitor PR-825 suppressed EAE. Since β5 completely occupies the β5/β5i position in the proteasome of β5i−/− mice, this result strongly suggests that inhibition with PR-825 suppressed the chymotrypsin-like activity in β5i−/− mice to a degree which blocked the pathogenic cells. This interpretation is consistent with high immunoproteasome expression in bone marrow-derived cells which cause the disease. The lesson learnt from these experiments is that β5i selective inhibitors may not only be of therapeutic potential for diseases which rely on a non-redundant function of β5i but for all pro-inflammatory diseases which rely on leukocytes expressing high levels of immunoproteasomes.
At first sight one may argue that more general inhibitors of the proteasomal chymotrypsin-like activity could be applied. However, immunoproteasomes are largely confined to sites of inflammation and leukocytes and therefore β5i selective therapy will block them and not the majority of tissues which express mainly constitutive proteasomes. Consistently, it has been shown that ONX-0914 is effective tenfold below its maximally tolerated dose while inhibitors of β5i and β5 like bortezomib or carfilzomib have to be applied at the maximally tolerated dose. The resulting adverse effects are acceptable for the treatment of neoplastic diseases like multiple myeloma or mantle cell lymphoma but are probably too severe for the long term treatment of chronic autoimmune diseases.
Conclusion
The selective inhibition of proteasome subunits is an interesting approach especially if specific subunits of the immunoproteasome but perhaps also of the thymoproteasome [37,43] are targeted. The high resolution structures of the 20S constitutive- and immunoproteasome have suggested that immunoproteasomes should be druggable and the first inhibitor of β5i has proven the principle in preclinical models of autoimmunity. Immunoproteasomes are expressed in lymphocytes, antigen presenting cells and innate immune cells and it will be important to elucidate which cells are primarily targeted in which type of disease. A similarly big challenge will be to find out how the immunoproteasome is mechanistically involved on a molecular level in the pathogenesis especially in those disease models which rely on a non-redundant function of the immunoproteasome. Hopefully site specific inhibitors of the immunoproteasome will help to identify selective substrates or cleavage activities of the immunoproteasome. Based on the reported preclinical results research teams in academia and pharmaceutical industry have intensified the search for selective proteasome inhibitors which will hopefully soon be subjected to clinical testing.
Acknowledgments
Work described in this article was funded by the German Research Foundation grants GR1517/12-1 and GR 1517/14-1, the Swiss National Science Foundation grant 31003A_138451, and the Konstanz Graduate School Chemical Biology (to MG), as well as by the Norris Cotton Cancer Center NCI core grant (P30CA023108) and by NCI grant 1R01-CA124634 (to AFK).
Footnotes
Conflict of interest
The authors have no financial conflict of interest.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1••.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. A comprehensive review of proteasome inhibitors including a historic overview of the field and coverage of earlier literature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cenci S, Oliva L, Cerruti F, Milan E, Bianchi G, Raule M, Mezghrani A, Pasqualetto E, Sitia R, Cascio P. Pivotal advance: protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J Leukoc Biol. 2012;92:921–931. doi: 10.1189/jlb.1011497. [DOI] [PubMed] [Google Scholar]
- 3.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67:6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 4.Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- 5.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods. 2005;2:357–362. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 6.Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, Ploegh HL, Kessler BM. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 2005;65:7896–7901. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]
- 7.Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide alpha’, beta’-epoxyketones. Chem Biol. 1999;6:811–822. doi: 10.1016/s1074-5521(99)80128-8. [DOI] [PubMed] [Google Scholar]
- 8.Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts RA, Amir O, Goddard AL, Pelphrey PM, et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem Biol. 2009;16:1278–1289. doi: 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood. 2009;113:4667–4676. doi: 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Screen M, Britton M, Downey SL, Verdoes M, Voges MJ, Blom AEM, Geurink PP, Risseeuw MDP, Florea BI, vanderLinden WA, et al. Nature of pharmacophore influences active site specificity of proteasome inhibitors. J Biol Chem. 2010;285:40125–40134. doi: 10.1074/jbc.M110.160606. Describes how the nature of electrophilic groups affects the specificity of peptide inhibitors and reports on development of NC-005-VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geurink PP, Liu N, Spaans MP, Downey SL, van den Nieuwendijk AM, van der Marel GA, Kisselev AF, Florea BI, Overkleeft HS. Incorporation of fluorinated phenylalanine generates highly specific inhibitor of proteasome’s chymotrypsin-like sites. J Med Chem. 2010;53:2319–2323. doi: 10.1021/jm9015685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009;114:3439–3447. doi: 10.1182/blood-2009-05-223677. Reports on the development of PR-924 (IPSI) and PR-825 (CPSI) as β5i and β5 selective inhibitors, respectively. [DOI] [PubMed] [Google Scholar]
- 13••.Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009;15:781–787. doi: 10.1038/nm.1978. Demonstrates that a β5i selective inhibitor modulates antigen presentation and suppresses cytokine production, experimental diabetes, and rheumatoid arthritis. [DOI] [PubMed] [Google Scholar]
- 14.de Bruin G, Huber EM, Xin BT, van Rooden EJ, Al-Ayed K, Kim KB, Kisselev AF, Driessen C, van der Stelt M, van der Marel GA, et al. Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J Med Chem. 2014;57:6197–6209. doi: 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- 15•.Fan H, Angelo NG, Warren JD, Nathan CF, Lin G. Oxathiazolones selectively inhibit the human immunoproteasome over the constitutive proteasome. ACS Med Chem Lett. 2014;5:405–410. doi: 10.1021/ml400531d. Describes β5i inhibition by oxathiazolones and the molecular basis of their selectivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasam V, Lee NR, Kim KB, Zhan CG. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg Med Chem Lett. 2014 doi: 10.1016/j.bmcl.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myung J, Kim KB, Lindsten K, Dantuma NP, Crews CM. Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol Cell. 2001;7:411–420. doi: 10.1016/s1097-2765(01)00188-5. [DOI] [PubMed] [Google Scholar]
- 18.van der Linden WA, Willems LI, Shabaneh TB, Li N, Ruben M, Florea BI, van der Marel GA, Kaiser M, Kisselev AF, Overkleeft HS. Discovery of a potent and highly beta1 specific proteasome inhibitor from a focused library of urea-containing peptide vinyl sulfones and peptide epoxyketones. Org Biomol Chem. 2012;10:181–194. doi: 10.1039/c1ob06554h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho YK, BargagnaMohan P, Wehenkel M, Mohan R, Kim KB. LMP2-specific inhibitors: chemical genetic tools for proteasome biology. Chem Biol. 2007;14:419–430. doi: 10.1016/j.chembiol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basler M, Lauer C, Moebius J, Weber R, Przybylski M, Kisselev AF, Tsu C, Groettrup M. Why the structure but not the activity of the immunoproteasome subunit LMP2 rescues antigen presentation. J Immunol. 2012;189:1868–1877. doi: 10.4049/jimmunol.1103592. [DOI] [PubMed] [Google Scholar]
- 21.Nazif T, Bogyo M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA. 2001;98:2967–2972. doi: 10.1073/pnas.061028898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loidl G, Groll M, Musiol HJ, Huber R, Moroder L. Bivalency as a principle for proteasome inhibition. Proc Natl Acad Sci USA. 1999;96:5418–5422. doi: 10.1073/pnas.96.10.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marastoni M, Baldisserotto A, Cellini S, Gavioli R, Tomatis R. Peptidyl vinyl ester derivatives: new class of selective inhibitors of proteasome trypsin-like activity. J Med Chem. 2005;48:5038–5042. doi: 10.1021/jm040905d. [DOI] [PubMed] [Google Scholar]
- 24.Mirabella AC, Pletnev AA, Downey SL, Florea BI, Shabaneh TB, Britton M, Verdoes M, Filippov DV, Overkleeft HS, Kisselev AF. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem Biol. 2011;18:608–618. doi: 10.1016/j.chembiol.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25••.Geurink PP, van der Linden WA, Mirabella AC, Gallastegui N, de Bruin G, Blom AE, Voges MJ, Mock ED, Florea BI, van der Marel GA, et al. Incorporation of non-natural amino acids improves cell permeability and potency of specific inhibitors of proteasome trypsin-like sites. J Med Chem. 2013;56:1262–1275. doi: 10.1021/jm3016987. Reports on the development of LU-102, the most potent cell-permeable inhibitor of trypsin-like sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kremer M, Henn A, Kolb C, Basler M, Moebius J, Guillaume B, Leist M, VandenEynde BJ, Groettrup M. Reduced immunoproteasome formation and accumulation of immunoproteasomal precursors in the brains of lymphocytic choriomeningitis virus-infected mice. J Immunol. 2010;185:5549–5560. doi: 10.4049/jimmunol.1001517. [DOI] [PubMed] [Google Scholar]
- 27.Khan S, van den Broek M, Schwarz K, de Giuli R, Diener PA, Groettrup M. Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. J Immunol. 2001;167:6859–6868. doi: 10.4049/jimmunol.167.12.6859. [DOI] [PubMed] [Google Scholar]
- 28.Barton LF, Cruz M, Rangwala R, Deepe GS, Monaco JJ. Regulation of immunoproteasome subunit expression in vivo following pathogenic fungal infection. J Immunol. 2002;169:3046–3052. doi: 10.4049/jimmunol.169.6.3046. [DOI] [PubMed] [Google Scholar]
- 29.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ, Valenzuela DM, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol. 2012;13:129–135. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basler M, Beck U, Kirk CJ, Groettrup M. The antiviral immune response in mice devoid of immunoproteasome activity. J Immunol. 2011;187:5548–5557. doi: 10.4049/jimmunol.1101064. [DOI] [PubMed] [Google Scholar]
- 31.Basler M, Kirk CJ, Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Curr Opin Immunol. 2013;25:74–80. doi: 10.1016/j.coi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 32••.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. 2012;148:727–738. doi: 10.1016/j.cell.2011.12.030. Reports on the high resolution X-ray crystallographic structures of mouse constitutive proteasome and immunoproteasome. [DOI] [PubMed] [Google Scholar]
- 33.Basler M, Moebius J, Elenich L, Groettrup M, Monaco JJ. An altered T cell repertoire in MECL-1-deficient mice. J Immunol. 2006;176:6665–6672. doi: 10.4049/jimmunol.176.11.6665. [DOI] [PubMed] [Google Scholar]
- 34.Salzmann U, Kral S, Braun B, Standera S, Schmidt M, Kloetzel PM, Sijts A. Mutational analysis of subunit i beta 2 (MECL-1) demonstrates conservation of cleavage specificity between yeast and mammalian proteasomes. FEBS Lett. 1999;454:11–15. doi: 10.1016/s0014-5793(99)00768-1. [DOI] [PubMed] [Google Scholar]
- 35.Tu L, Moriya C, Imai T, Ishida H, Tetsutani K, Duan XF, Murata S, Tanaka K, Shimokawa C, Hisaeda H, et al. Critical role for the immunoproteasome subunit LMP7 in the resistance of mice to Toxoplasma gondii infection. Eur J Immunol. 2009;39:3385–3394. doi: 10.1002/eji.200839117. [DOI] [PubMed] [Google Scholar]
- 36.Strehl B, Joeris T, Rieger M, Visekruna A, Textoris-Taube K, Kaufmann SH, Kloetzel PM, Kuckelkorn U, Steinhoff U. Immunoproteasomes are essential for clearance of Listeria monocytogenes in nonlymphoid tissues but not for induction of bacteria-specific CD8+ T cells. J Immunol. 2006;177:6238–6244. doi: 10.4049/jimmunol.177.9.6238. [DOI] [PubMed] [Google Scholar]
- 37.Kniepert A, Groettrup M. The unique functions of tissue-specific proteasomes. Trends Biochem Sci. 2013;39:17–24. doi: 10.1016/j.tibs.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Moebius J, van den Broek M, Groettrup M, Basler M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur J Immunol. 2010;40:3439–3449. doi: 10.1002/eji.201040620. [DOI] [PubMed] [Google Scholar]
- 39.Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol. 2010;185:634–641. doi: 10.4049/jimmunol.0903182. [DOI] [PubMed] [Google Scholar]
- 40.Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ, et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012;64:493–503. doi: 10.1002/art.33333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41••.Basler M, Mundt S, Muchamuel T, Moll C, Jiang J, Groettrup M, Kirk CJ. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med. 2014;6:226–238. doi: 10.1002/emmm.201303543. Shows suppression of autoimmune encephalomyelitis by β5i inhibition and that blocking the proteasomal chymotrypsin-like activity of pathogenic cells prevents the disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt N, Gonzalez E, Visekruna A, Kuhl AA, Loddenkemper C, Mollenkopf H, Kaufmann SHE, Steinhoff U, Joeris T. Targeting the proteasome: partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut. 2010;59:896–906. doi: 10.1136/gut.2009.203554. [DOI] [PubMed] [Google Scholar]
- 43.Takahama Y, Takada K, Murata S, Tanaka K. beta5t-containing thymoproteasome: specific expression in thymic cortical epithelial cells and role in positive selection of CD8+ T cells. Curr Opin Immunol. 2012;24:92–98. doi: 10.1016/j.coi.2012.01.006. [DOI] [PubMed] [Google Scholar]