

Abstract
Background:
Leptin, a 16 kDa peptide hormone synthesized and secreted specifically from white adipose cells protects neurons against amyloid β-induced toxicity, by increasing Apolipoprotein E (APO E)-dependent uptake of β amyloid into the cells, thereby, protect individuals from developing Alzheimer's disease (AD). The APO E ε4 allele is a known genetic risk factor for AD by accelerating onset. It is estimated that the lifetime risk of developing AD increases to 29% for carriers with one ε4 allele and 9% for those with no ε4 allele.
Objectives:
To determine the levels of serum leptin, cholesterol, low density lipoprotein (LDL-C), and high density lipoprotein (HDL-C) in the diagnosed cases of AD and the association of them with cognitive decline and Apolipoprotein E (APO E) genotypes in AD.
Materials and Methods:
Serum levels of serum leptin, cholesterol, LDL-C, and HDL-C along with APO E polymorphism were studied in 39 subjects with probable AD and 42 cognitive normal individuals.
Results:
AD group showed significantly lower levels of leptin (P = 0.00) as compared to control group. However, there was no significant difference in cholesterol, triglycerides, LDL-C, and HDL-C levels in AD and control groups. The frequency of ε4 allele in AD (38.5%) was found to be significantly higher than in control (10.3%). ε3 allele was more frequent than ε4 allele in AD and control group.
Keywords: Alzheimer's disease, APO E polymorphism, dementia, leptin, total cholesterol
Introduction
Dementia is an age-related disorder associated with elderly population.[1] In coming decades, increased prevalence of Alzheimer's disease (AD) may pose burden on society and healthcare services, given the rapid increase in aging population in developed and developing countries. Alzheimer's disease International (ADI) estimates that there are currently 30 million people with dementia in the world and will increase to be over 100 million by 2050.[2] It is a life course illness resulting from interaction of life-style risk factors with genetic, vascular, and other risk factors to affect risk of disease.[3] Studies show that mid life obesity, metabolic syndrome,[4] insulin resistance, and type 2 Diabetes[5] as well as atherogenic/ischemic diseases[6,7] have been associated with cognitive impairment and progression of AD by affecting myelin integrity and white matter connectivity. It is further supported by the fact that neuritic plaque and neurofibrillary tangle aggregates, hallmark of AD pathology are present in about 30% of cognitively normal elderly subjects.[8] Many prospective studies have shown association between overweight, obesity in middle age with decrease in cognitive function in general population.[9] However, underlying mechanism for this association has not been understood completely. Few studies in recent past have implicated the endocrine hormone leptin as a possible factor linking obesity and AD.[6] Leptin is a 16 kDa peptide hormone synthesized and secreted specifically from white adipose cells.[10] Leptin levels have been shown to be associated with anthropometric parameters such as the body mass index (BMI), total body fat, and subcutaneous fat. Leptin has a variety of important central and peripheral actions to regulate energy balance and bone metabolism that are mediated by specific cell surface leptin receptors.[11,12] Leptin has peripheral actions to stimulate vascular inflammation, oxidative stress, and vascular smooth muscle hypertrophy that may contribute to pathogenesis of type 2 diabetes mellitus, hypertension, atherosclerosis, and coronary heart disease.[12,13,14] Another emerging role for Leptin is the facilitation of memory processes. Leptin, after activation of Leptin receptors (LRp) increases N-methyl-D-aspartate (NMDA) receptor function, which in turn facilitates the induction of hippocampal long-term potentiation (LTP), partly through mitogen-activated protein kinase (MAPK).[15,16] It is supported by the studies showing additional function of leptin on brain, in CA1 region of the hippocampus, an area integral to learning and memory.[17] At this moment, it is difficult to say that at which stage of AD leptin acts and mechanism by which high circulating concentration of leptin protect individuals from developing AD, as leptin have been shown to modulate production by reducing the activity of β secretase, a protease that cleaves amyloid precursor protein (APP) as well as clearance of amyloid β in rodent models.[18,19] It also protects neurons against amyloid β-induced toxicity, by increasing Apolipoprotein E (APO E) dependent uptake of β amyloid into the cells.
The APO E ε4 allele is a known genetic risk factor for AD by accelerating onset.[20] It is estimated that the lifetime risk of developing AD increases to 29% for carriers with one ε4 allele and 9% for those with no ε4 allele.[21] APO E has a prominent role in the transport and metabolism of plasma cholesterol.[22] Carriers of the ε4 allele of the APO E gene (APO E ε4) have higher total and low density lipoprotein cholesterol levels than non carriers.[23] Many animal models of AD shows that high cholesterol levels increase β amyloid protein concentration and may increase APO E expression by which it increases the risk for AD, explaining the mechanism of action of APO E.[24,25] The uptake of APO E/Aβ complexes by neurons which is allele specific, serves as a mechanism for clearing Aβ from brain interstitium. APO E 3 allele appears to be more efficient in uptake of APO E/Aβ complex as compared to APO E 4.[26] Literature shows conflicting studies regarding influence of APO E genotype on the relationship between plasma cholesterol level and dementia risk.[27,28,29,30] It is well-established that the risk of AD and cognitive impairment increases in patients with pathological condition associated with high cholesterol levels, such as cardiovascular diseases.[31,32] Cholesterol might play a role in the biosynthesis of Amyloid β.[33] However, association between cholesterol levels and dementia are inconclusive. Few studies report high cholesterol level to be a significant risk factor for dementia, others have not confirmed this finding.[27,34]
In this hospital-based cross-sectional study, with the objectives to identify the biochemical risk factors in AD, we examined the levels of serum leptin, cholesterol, low density lipoprotein (LDL-C), and high density lipoprotein (HDL-C) in the diagnosed cases of AD. Also, attempt was made to explore the association of serum leptin, total cholesterol, LDL-C, and HDL-C with cognitive decline in AD and whether the levels of these can be determined by APO E polymorphism.
Materials and Methods
Study subjects
Subjects attending Neurobehavioral clinic in the Institute of Human Behavior & Allied Sciences from 2010 to 2012 were enrolled in this cross sectional study. Each participant underwent in person interview about general health and function at the time of their entry in the study, followed by assessment if medical history, physical, and neurological examination, and a neuropsychologic battery for cognition.
Of the 200 subjects undertaken for initial screening, 88 were diagnosed as probable AD. Out of 88 subjects, 12 were excluded as their APO E polymorphism study was not done and 16 subjects’ leptin level, lipid profile could not be obtained in the cross-sectional analysis (%). Thirty-nine subjects with probable AD (27 men and 12 women; mean age 66.90 years) and 42 cognitive normal individuals (22 men and 20 women; mean age 61.9 years) were involved in the cross-sectional analysis.
Patients were selected from outpatient only. All patients with history of cerebral stroke, epilepsy, head trauma, and moderate to severe depressive episodes were excluded from the study. Before enrolling subjects in the study, written informed consent was obtained. Participants who did not give consent for participation in the study were not included, in both case and control groups. Study was initiated after approval from the institutional ethics committee.
Diagnosis of AD
Dementia screening and diagnosis was done in two phases. In first phase, subjects were screened by the trained resident doctor using Mini-Mental State examination (MMSE). In second phase, screen positive subjects (MMSE <26) were subsequently assessed by a neurologist and underwent brain imaging. Diagnosis of probable AD was made according to the criteria of the National Institute of Neurological and Communicative Disorders Association-Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA).[35] The diagnosis was confirmed by cerebral magnetic resonance imaging (MRI) studies, which showed a generalized atrophy with selective temp-roparietal atrophy in all the patients. NIMHANS and PGI battery of neuropsychological tests for cognition were done. All the subjects underwent clinical examination along with routine biochemical, hormonal, and radiologic examinations, including glucose, cholesterol, triglycerides, LDL-C, HDL-C, leptin, and MRI. All the cases underwent genetic testing for APO E polymorphism.
Biochemical assessment
At baseline non-fasting serum samples were obtained from patients taking all the standard precautions. The specimen was centrifuged within 30 min of sample collection to separate the serum and examined for routine biochemistry including cholesterol, LDL-C, HDL-C. All the routine biochemical parameters were measured on fully automated Biochemistry Discrete Analyser AU 480 from Beckman Coulter. Specimen for evaluation of leptin was stored at −20°C until analysis. Serum leptin was measured by using sandwich immunoassay technique, using kit from DRG, DRG Instruments Gmbh, Germany. This test was performed on enzyme-linked immunosorbant assay (ELISA) Reader Ranskan Sprint from Ranbaxy Diagnostics.
APO E genotyping
Blood samples were obtained from all patients at the time of enrolment and genomic deoxyribonucleic acid (DNA) was isolated using a modification of the salting out procedure.[36] Two non-synonymous genetic variants, rs429358 and rs7412, which determine the three APO E ε isoforms were identified by sequencing 217bp exon 4 region of APO E gene. The 217bp fragment (reference sequence: GenBank AF261279)[37] was amplified using genomic DNA from each individual by forward (CCAAGGAGCTGCAGGCGGCGCA) and reverse (GCCCCGGCCTGGTACACTGCCA) primers. Amplification reaction was assembled in 25μl total volume containing 50 ng of genomic DNA using Taq polymerase (Thermo Scientific).
Following assembly, thermal cycling was performed with an initial denaturation at 94°C for 10 min followed by 35 cycles of denaturation at 94°C for 45 sec, primer annealing at 60°C for 45 sec, and primer extension at 72°C for 45 sec. After 35 cycles, a final extension was performed at 72°C for 10 min. PCR products were purified by cutting the specific product from a 1% agarose gel and isolating the product with GeneJET Gel Extraction Kit (Thermo Scientific). Purified PCR products were sequenced in 10 μl reactions with 25 ng of the sequencing primer on an ABI 3730xl DNA analyzer (Scigenom laboratory, India). Sequencing reactions were denatured for 10 sec at 9°6 C and subjected to 15 cycles at 96° C for 10 sec, 55° C for 5 sec, and 70° C for 1 min and 15 cycles at 96° C for 10 sec 70° C for 1 min. The results were analyzed using FinchTV V1.4.0 software [Geospiza, Inc].
Statistical analysis
Descriptive statistics (Mean & standard deviation) was used to describe the continuous variable under study. Independent t-test was applied to find out significant difference in average level between AD and control groups. The AD group patients were categorized in three sub-groups (mild, moderate, severe) on the basis of MMSE scores and mean of all the studied biochemical variables (serum leptin, cholesterol, triglycerides, LDL-C and HDL-C) were compared by analysis of variance (ANOVA) technique. Product-moment correlation method was applied to find out the strength and direction of relationship between MMSE score and biochemical variables among AD group. All analysis was carried out on Statistical Package of Social Sciences (SPSS) 18.0.
Results
Characteristics of study subjects
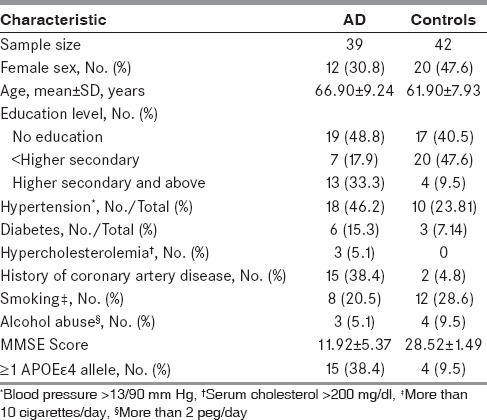
In the present case control cross-sectional study, 81 subjects, which included 39 AD and 42 control, were studied for serum levels of leptin, lipid profile, and APO E polymorphism. The average age in AD group was 66.90 ± 9.24 years, which was slightly but significantly higher than the controls (61.90 ± 7.93 years). The proportion of men in AD was more than control (27/12 & 22/20, respectively). which was not significant (P = 0.12). Table 1 describes the demographic characteristics of study population. It also describes the associated risk factors in AD and control group. AD group had more often history of hypertension (46.2%), diabetes (15.3%), hypercholesterolemia (5.1%), coronary artery disease (38.4%), and higher frequency of ε4 alleles (38.4%) compared with control group, whereas 28.6% and 9.5% subjects in control group had history of smoking and alcohol abuse, respectively, which was slightly higher than AD group. However, AD group had more uneducated subjects, i. e., cannot read and write (48.7%) than control group (40.5%). The case and control were not matched for age or education.
Table 1.
Baseline characteristics of the entire study sample
Allele and genotype distribution of the APO E gene in AD and control group
Table 2 shows genotype distribution of APO E gene in AD as compared to controls. The frequency of ε2, ε3, and ε4 was studied in 81 subjects. Allele ε2 was present in only one patient with AD as compared to 9 out of 42 control subjects. The frequency of ε4 allele in AD (38.5%) was found to be significantly higher than in control (9.5%). ε3 allele was more frequent than ε4 allele in AD and control group.
Table 2.
Allele and genotype distribution of the APO E gene in AD and control groups

The frequency of ε3ε3 genotype was highest in the AD group (61.5%), which was lower than control (69.0%), followed by ε3ε4 (25.6%) and ε4ε4 (10.3%), whereas ε4ε4 allele was absent in control group. In addition, ε2ε3 genotype was absent in AD, but present in 21.4% subjects in control group.
Serum leptin and cholesterol levels in AD
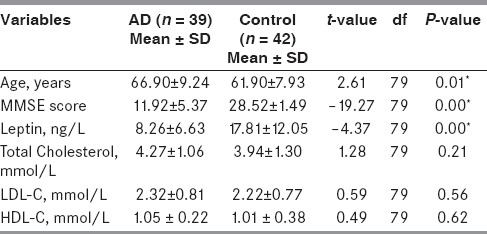
Table 3 describes the descriptive statistics (mean and SD) of age, MMSE score, and biochemical variables (serum leptin, cholesterol, LDL-C, and HDL-C levels). One-way ANOVA technique was applied to compare the levels of leptin, cholesterol, triglycerides, LDL-C, and HDL-C in AD and control groups. AD group showed significantly lower levels of leptin (P = 0.00) as compared to control group. There was no significant difference in cholesterol, LDL-C, and HDL-C levels in AD and control groups, but the mean value of these variables in serum was higher in AD group as compared to control.
Table 3.
Comparison of mean value of age, MMSE score, serum leptin, and lipids in AD and control groups
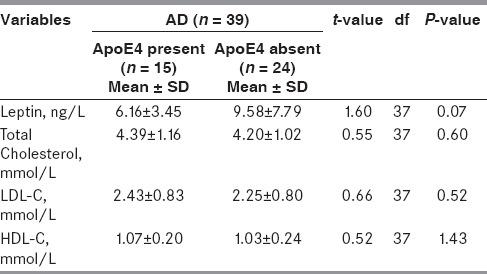
To assess the effect of APO E polymorphism on serum leptin, cholesterol, triglycerides, LDL-C, and HDL-C levels in AD and control group, analysis was stratified by APO E genotype (no ε4 vs. at least one ε4 allele). In subjects carrying at least one ε4 allele, serum leptin levels were slightly low as compared to subjects not carrying ε4 allele [Table 4]. However, this association was not significant (P = 0.07).
Table 4.
Comparison of serum leptin and lipids in AD group with/without ApoE ε4 allele
Association of leptin and lipid levels with cognitive decline in AD
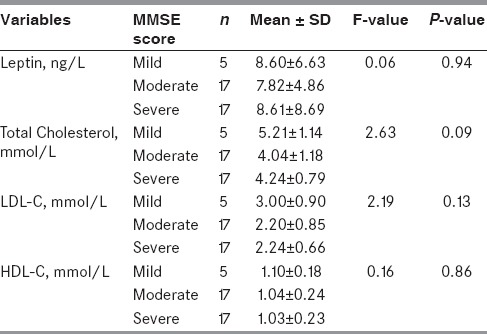
To explore the association of serum leptin, total cholesterol, triglycerides, LDL-C, and HDL-C levels with cognitive decline in AD, the AD group patients were categorized in three sub-groups- mild (19-24), moderate (11-18) and severe (0-10), and mean of all the studied biochemical variables (serum leptin, cholesterol, triglycerides, LDL-C, and HDL-C levels) were compared by analysis of variance (ANOVA) technique [Table 5]. Within subgroup no significant difference in mean value of serum leptin, cholesterol, triglycerides, LDL-C, and HDL-C levels was observed. The mean leptin level was almost comparable in all three MMSE score subgroups, whereas mean cholesterol level (155.88 ± 45.68 mg/dl)) was lowest in moderate MMSE score subgroup and highest in mild MMSE score subgroup (201 ± 44.16 mg/dl). Similarly, triglycerides, LDL-C, and HDL-C levels were highest in mild MMSE score subgroup. However, all these values were not significantly different.
Table 5.
Relation of MMSE score with serum leptin and lipids in AD group
Discussion
In the present study performed on small-sized sample taken from a tertiary care hospital, main findings are:
lower serum leptin levels were associated with a higher prevalence of AD,
APO E ε4 allele was raised in AD,
serum lipid levels were slightly but not significantly higher in AD as compared to controls, and
no significant association was found between low serum leptin and high serum cholesterol and AD prevalence in subjects carrying at least one APO E ε4 allele.
Our findings of reduced serum leptin levels and raised serum cholesterol and LDL-C are in agreement with other studies.[6,38,39,40] Lieb et al., 2012[6] in their landmark prospective study found association of higher baseline plasma leptin concentration with significantly lower risk of dementia and AD. During 12 year follow-up, 25% subjects developed AD in the lowest quartile of leptin as compared to 6% in highest quartile. They found correlation of circulating leptin levels with gray matter volume in brain, specifically in hippocampus. Their study suggests that higher leptin levels are associated with lower incidence of AD in non-obese individuals. Another prospective study by Holden et al., 2009[20] performed in normal elderly, a negative correlation observed between serum leptin levels and cognitive decline, further supported our findings. They observed that elders in the high leptin group had significantly less likelihood of cognitive decline. However, Teunissen et al., 2014[41] found no correlation between serum leptin levels and cognitive decline in AD patients, whereas Johnston et al., 2014[42] observed low serum leptin levels in cognitively impaired AD subjects. In vitro and in vivo studies have reported that leptin impacts Aβ homeostasis and tau phosphorylation.
It has also been suggested that abnormal neuronal AMP-dependent kinase (AMPK) leading to modification of metabolic pathways, modulated by leptin precede amyloid plaque and neurofibrillary tangle (NFT) development.[43] An abnormal AMPK activity could lead to abnormal lipid levels and membrane composition in neurons that could affect membrane fluidity, which may lead to Aβ production. Leptin, known to regulate the AMPK activity, can reduce tau phosphorylation. Taken together; leptin can influence these pathways, prior to amyloid deposition or tau phosphorylation. Further, leptin treatment can ameliorate AD pathology by reducing the levels of Aβ42 and phosphorylated tau.[44,45] These biochemical and pathological changes, hallmark of AD, after leptin treatment in animal models correlate with behavioral improvements. Leptin treatment helps in Aβ clearance by reducing β-secretase activity and increasing APO E-dependent Aβ uptake[44] and decreases the hyperphosphorylation of tau in a dose dependent and synergistic manner with insulin.[46] Hence it can be suggested that leptin not only reduces AD pathology but also improves cognitive functions[45] and leptin could serve as a therapy that increases learning and memory capacity in AD patients and reduces Aβ plaques in the brain, while helping maintain adequate metabolic control.[47] Our findings, as supported by these studies suggest that the risk of developing AD is significantly lower in persons with high leptin concentration, as leptin, besides having role in energy regulation, may be neuroprotective and mitigate the progression of AD.
Our second finding is that the frequency of ε4 allele in AD (38.5%) was significantly higher than in control (9.5%). Luthra et al., 2004[48] has reported 29.3% APO E ε4 allele frequency among AD in Indian population, which is almost similar to African American[49] (32.2%) and Japanese[49] (27.8%) population, but lower than Caucasian population[49] (36.7%). However, another study done in Indian population by Ganguly et al., 2000[50] reported 16.07% APO E ε4 frequency among AD patients in Indian population. As seen in these studies, APO E ε4 is associated with an increased risk of AD. But, many people with APO E ε4 allele did not develop AD and also, AD developed in its absence. Heterogeneity regarding the strength of risk of AD for carriers of APO E ε4 allele in the present study may be due to small sample size, genetic drift by founder effect, and/or different prevalence rates of AD. Also, other than selection bias, factors like age, sex, smoking, hypercholesterolemia, hyperglycemia are known to modify APO E-related risk in AD. Hence, neither APO E genotyping is recommended for routine clinical diagnosis nor can be used for predictive testing.[51] However, APO E genotyping may be applied to differentiate AD and non AD dementia. It can also play a vital role in sub grouping of both AD and non-AD dementia.[52]
Our third finding of slightly but not significantly raised serum lipid levels in AD as compared to controls is in agreement with longitudinal studies[53,54,55] showing a non-significant association between high cholesterol and HDL-C levels in midlife and the development of AD in the late life, whereas, Presecki et al., 2011[56] reported reduced serum cholesterol, LDL-C, and HDL-C levels in patients of AD. As published data available regarding blood lipid levels and incidence of AD are contradictory,[27,53,57] it is not possible to conclude whether cholesterol has a protective or neurotoxic role in the development of AD. Such varied results among studies may be due to lifestyle-related modifiable risk factors like diet, alcohol, exercise, smoking, in addition to age, gender, and different methods used for lipid determination. Another possible explanation, which needs to be considered, is that most of the data available come from the short-term cohort studies, with a time interval not more than 2-6 years before the diagnosis of AD, except one study undertaken 18 years before dementia was identified.[58] The long-term follow-up studies, undertaken in mid life as a starting point show results that are opposite to those of short-term follow-up studies done in older individuals.[56] Such contradictions may be explained by the fact that total cholesterol tends to increase with age in young- or middle-aged adults, but later decreases as individuals get older.[57] Although studies show high cholesterol levels at mid life as an independent risk factor for AD, this association was not observed in late-life.[59] However, no significant association was observed between low leptin and high cholesterol and AD prevalence in subjects having APO E ε4 allele. Also, there was no significant difference between APO E ε4 allele and non APO E ε4 allele for cholesterol levels. Though there were slightly raised leptin levels in AD patients with APO E ε4 allele, but they were not significant. Similar findings have been reported by Sabbagh et al., 2006,[60] which showed that APO E ε4 allele did not reveal any significant difference in the lipid profile except for LDL-C, whereas, Neeraj et al., 2012[40] reported significantly high levels of serum cholesterol and LDL-C cholesterol in APO E ε3/4 genotype in patients with AD as compared to controls. Studies done by Staito et al., 2003[61] shows that APO E ε4 isoforms have higher capacity to bind to VLDL receptor leading to raised levels of serum cholesterol and LDL-C. Hence, it can be concluded that high cholesterol serum cholesterol levels at midlife constitute a significant risk factor for AD later in life, independent to APO E ε4.[56] Lieb et al., 2009[6] also showed no effect modification by APO E ε4 allele for interaction of APO E and leptin for the models predicting all cause dementia.
The present study is not without limitations. First, subjects taken in the study were diagnosed cases of AD taken in late life, thus it is difficult to say whether these findings are applicable to all stages of AD and in other age-groups and will be unable to address the relationship between mid-life leptin and cholesterol and cognitive decline. Further, in the present study, leptin, and lipid levels were measured in serum and not in cerebrospinal fluid and only once in each participant. However, studies show that the levels of leptin in cerebrospinal fluid (CSF) are comparable to that of serum/plasma.[6] Also, confounding factors for leptin-like waist hip ratio, body mass index, and lipids like dietary habits especially dietary fats were not examined at the baseline examination.
Conclusions
Based on the findings of present study, it can be concluded that low levels of leptin may contribute to systemic and central nervous system abnormalities leading to AD, suggesting that a leptin may be considered as possible targets for the therapeutic intervention. This may be an attractive alternative to the drugs that are currently under development. Also, APO E genotyping might improve the specificity of the differential diagnosis of dementia and in addition to serum lipids may participate as an independent risk factor for AD.
Footnotes
Source of Support: Nil
Conflicts of Interest: None declared.
References
- 1.O’Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol Cell Neurosci. 2007;35:559–72. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Estimate of Worldwide Prevalence of Alzheimer's Disease. [Last dated on 2013 Jan 26]. Available from: http://www.emaxhealth.com/91/12843.html .
- 3.Peila R, Rodriguez BL, Launer LJ. Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 4.Yaffe K, Weston AL, Blackwell T, Krueger KA. The metabolic syndrome and development of cognitive impairment among older women. Arch Neurol. 2009;66:324–8. doi: 10.1001/archneurol.2008.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craft S. The role of metabolic disorders in Alzheimer's disease and vascular dementia: Two roads converged. Arch Neurol. 2009;66:300–5. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA. 2009;302:2565–72. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Une K, Takei YA, Tomita N, Asamura T, Ohrui T, Furukawa K, et al. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer's disease. Eur J Neurol. 2010;18:1006–9. doi: 10.1111/j.1468-1331.2010.03194.x. [DOI] [PubMed] [Google Scholar]
- 8.Sena A, Capela C, Nobrega C, Campos E, Pedrosa R, Ferret-Sena V. Plasma lipoproteins in brain inflammatory and neurodegenerative diseases. 2012. [Last accessed on 2015 Feb 25]. Available form: http://dx.doi.org/10.5772/51268 .
- 9.Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Jr, Yaffe K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ. 2005;330:1360. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human analogue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 11.Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: A review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–33. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 12.Seufert J. Leptin effects on pancreatic beta-cell gene expression and function. Diabetes. 2004;53:S152–8. doi: 10.2337/diabetes.53.2007.s152. [DOI] [PubMed] [Google Scholar]
- 13.Beltowski J. Leptin and atherosclerosis. Atherosclerosis. 2006;189:47–60. doi: 10.1016/j.atherosclerosis.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Beltowski J. Role of leptin in blood pressure regulation and arterial hypertension. J Hypertens. 2006;24:789–801. doi: 10.1097/01.hjh.0000222743.06584.66. [DOI] [PubMed] [Google Scholar]
- 15.Harvey J. Leptin regulation of neuronal excitability and cognitive function. Curr Opin Pharmacol. 2007;7:643–7. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvey J, Solovyova N, Irving A. Leptin and its role in hippocampal synaptic plasticity. Prog Lipid Res. 2006;45:369–78. doi: 10.1016/j.plipres.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey J. Leptin : The missing link in alzheimer's disease? Clin Chem. 2010;56:696–7. doi: 10.1373/clinchem.2010.144006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holden KF, Lindquist K, Tylavsky FA, Rosano C, Harris TB, Yaffe K. Health ABC study. Serum leptin level and cognition in the elderly: Findings from the Health ABC study. Neurobiol Aging. 2009;30:1483–9. doi: 10.1016/j.neurobiolaging.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR. Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology. 2004;62:1869–71. doi: 10.1212/01.wnl.0000125323.15458.3f. [DOI] [PubMed] [Google Scholar]
- 21.Seshadri S, Drachman DA, Lippa CF. Apolipoprotein E epsilon 4 allele and the lifetime risk of Alzheimer's disease. What physicians know, and what they should know. Arch Neurol. 1995;52:1074–9. doi: 10.1001/archneur.1995.00540350068018. [DOI] [PubMed] [Google Scholar]
- 22.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Reilly SL, Ferrell RE, Kottke BA, Kamboh MI, Sing CF. The gender-specific apolipoprotein E genotype influence on the distribution of lipid and apolipoproteins in the population of Rochester, MN. I Pleiotropic effects on means and variances. Am J Hum Genet. 1991;49:1155–66. [PMC free article] [PubMed] [Google Scholar]
- 24.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: The cholesterol connection. Nat Neurosci. 2003;6:345–51. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 25.Wu CW, Liao PC, Lin C, Kuo CJ, Chen ST, Chen HI, et al. Brain region-dependent increases in β-amyloid and apolipoprotein E levels in hypercholesterolemic rabbits. J Neural Transm. 2003;110:641–9. doi: 10.1007/s00702-002-0809-1. [DOI] [PubMed] [Google Scholar]
- 26.Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. 2004;18:1870–8. doi: 10.1096/fj.04-2572com. [DOI] [PubMed] [Google Scholar]
- 27.Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17:14–20. doi: 10.1159/000026149. [DOI] [PubMed] [Google Scholar]
- 28.Evans RM, Emsley CL, Gao S, Sahota A, Hall KS, Farlow MR, et al. Serum cholesterol, APOE genotype, and the risk of Alzheimer's disease: A population-based study of African Americans. Neurology. 2000;54:240–2. doi: 10.1212/wnl.54.1.240. [DOI] [PubMed] [Google Scholar]
- 29.Romas SN, Tang MX, Berglund L, Mayeux R. APOE genotype, plasma lipids, lipoproteins, and AD in community elderly. Neurology. 1999;53:517–21. doi: 10.1212/wnl.53.3.517. [DOI] [PubMed] [Google Scholar]
- 30.Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, et al. Apolipoprotein E epsilon 4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137:149–55. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 31.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet. 1997;349:151–4. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 32.Carmelli D, Swan GE, Reed T, Miller B, Wolf PA, Jarvik GP, et al. Midlife cardiovascular risk factors, ApoE, and cognitive decline in elderly male twins. Neurology. 1998;50:1580–5. doi: 10.1212/wnl.50.6.1580. [DOI] [PubMed] [Google Scholar]
- 33.Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport. 1999;10:1699–705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- 34.Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: A case-control study. Neurology. 1995;45:1092–6. doi: 10.1212/wnl.45.6.1092. [DOI] [PubMed] [Google Scholar]
- 35.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mrozek D, Małysiak-Mrozek B, Siążnik A. search GenBank: Interactive orchestration and ad-hoc choreography of Web services in the exploration of the biomedical resources of the National Center For Biotechnology Information. BMC Bioinformatics. 2013;14:73. doi: 10.1186/1471-2105-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonarek M, Barberger-Gateau P, Letenneur L, Deschamps V, Iron A, Dubroca B, et al. Relationships between cholesterol, apolipoprotein E polymorphism and dementia: A cross-sectional analysis from the PAQUID study. Neuroepidemiology. 2000;19:141–8. doi: 10.1159/000026249. [DOI] [PubMed] [Google Scholar]
- 39.Lesser GT, Haroutunian V, Purohit DP, Schnaider Beeri M, Schmeidler J, Honkanen L, et al. Serum lipids are related to Alzheimer's pathology in nursing home residents. Dement Geriatr Cogn Disord. 2009;27:42–9. doi: 10.1159/000189268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh NK, Chhillar N, Banerjee BD, Bala K, Mukherjee AK, Mustafa MD, et al. Gene-environment interaction in Alzheimer's disease. Am J Alzheimer's Dis Other Demen. 2012;27:496–503. doi: 10.1177/1533317512456067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teunissen CE, van der Flier WM, Scheltens P, Duits A, Wijnstok N, Nijpels G, et al. Serum leptin is not altered nor related to cognitive decline in alzheimer's disease. J Alzheimers Dis. 2014 doi: 10.3233/JAD-141503. [DOI] [PubMed] [Google Scholar]
- 42.Johnston JM, Hu WT, Fardo DW, Greco SJ, Perry G, Montine TJ, et al. Alzheimer's Disease Neuroimaging Initiative. Low plasma leptin in cognitively impaired ADNI subjects: Gender differences and diagnostic and therapeutic potential. Curr Alzheimer Res. 2014;11:165–74. doi: 10.2174/1567205010666131212114156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu X, McCorkle S, Wang M, Lee Y, Li J, Saha AK, et al. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: Prevention of diabetes and ectopic lipid deposition. Diabetologia. 2004;47:2012–21. doi: 10.1007/s00125-004-1570-9. [DOI] [PubMed] [Google Scholar]
- 44.Wallerstedt SM, Eriksson AL, Niklason A, Ohlsson C, Hedner T. Serum leptin and myocardial infarction in hypertension. Blood Press. 2004;13:243–6. doi: 10.1080/08037050410021405. [DOI] [PubMed] [Google Scholar]
- 45.van den Beld AW, Bots ML, Janssen JA, Pols HA, Lamberts SW, Grobbee DE. Endogenous hormones and carotid atherosclerosis in elderly men. Am J Epidemiol. 2003;157:25–31. doi: 10.1093/aje/kwf160. [DOI] [PubMed] [Google Scholar]
- 46.Greco SJ, Sarkar S, Johnston JM, Zhu X, Su B, Casadesus G, et al. Leptin reduces Alzheimer's disease-related tau phosphorylation in neuronal cells. Biochem Biophys Res Commun. 2008;376:536–41. doi: 10.1016/j.bbrc.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease response to therapeutic intervention. Circulation. 2008;117:3238–49. doi: 10.1161/CIRCULATIONAHA.107.741645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luthra K, Tripathi M, Grover R, Dwivedi M, Kumar A, Dey AB. Apolipoprotein E gene polymorphism in Indian patients with Alzheimer's disease and Indian patients with Alzheimer's disease and vascular dementia. Dement Geriatr Cogn Disord. 2004;17:132–5. doi: 10.1159/000076345. [DOI] [PubMed] [Google Scholar]
- 49.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex and ethnicity on the association between apolipoprotein E genotype and Alzheimer's disease. A meta analysis. For the APOE and Alzheimer's Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 50.Ganguli M, Chandra V, Kamboh MI, Johnston JM, Dodge HH, Thelma BK, et al. Apolipoprotein E polymorphism and Alzheimer's disease: The Indo-US cross-national dementia study. Arch Neurol. 2000;57:824–30. doi: 10.1001/archneur.57.6.824. [DOI] [PubMed] [Google Scholar]
- 51.Apolipoprotein E genotype in Alzheimer's disease. National Institute on Aging/Alzheimer's association Working Group. Lancet. 1996;347:1091–5. [PubMed] [Google Scholar]
- 52.Bang OY, Kwak YT, Joo IS, Huh K. Important link between dementia subtype and apolipoprotein E: A meta-analysis. Yonsei Med J. 2003;44:401–13. doi: 10.3349/ymj.2003.44.3.401. [DOI] [PubMed] [Google Scholar]
- 53.Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, et al. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain abeta 1-42 levels. Biochem Biophys Res Commun. 1998;252:711–5. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- 54.Tan ZS, Seshadri S, Beiser A, Wilson PW, Kiel DP, Tocco M, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: The Framingham Study. Arch Intern Med. 2003;163:1053–7. doi: 10.1001/archinte.163.9.1053. [DOI] [PubMed] [Google Scholar]
- 55.Li G, Shofer JB, Kukull WA, Peskind ER, Tsuang DW, Breitner JC, et al. Serum cholesterol and risk of Alzheimer disease: A community-based cohort study. Neurology. 2005;65:1045–50. doi: 10.1212/01.wnl.0000178989.87072.11. [DOI] [PubMed] [Google Scholar]
- 56.Presećki P, Mück-Seler D, Mimica N, Pivac N, Mustapić M, Stipcević T, et al. Serum lipid levels in patients with Alzheimer's disease. Coll Anthropol. 2011;35:115–20. [PubMed] [Google Scholar]
- 57.Mielke MM, Zandi PP, Sjögren M, Gustafson D, Ostling S, Steen B, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005;64:1689–95. doi: 10.1212/01.WNL.0000161870.78572.A5. [DOI] [PubMed] [Google Scholar]
- 58.Cauhan NB. Membrane dynamics, cholesterol homeostasis, and Alzheimer's disease. J Lipid Res. 2003;44:2019–29. doi: 10.1194/jlr.R300010-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Koudinov AR, Koudinova NV. Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. (233-40).J Nurol Sci. 2005:229–230. doi: 10.1016/j.jns.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 60.Sabbagh MN, Sandhu S, Kolody H, Lahti T, Silverberg NB, Sparks DL. Studies on the effect of the apolipoprotein E genotype on the lipid profile in Alzheimer's disease. Curr Alzheimer's Res. 2006;3:157–60. doi: 10.2174/156720506776383013. [DOI] [PubMed] [Google Scholar]
- 61.Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC, Lund-Katz S. Effects of polymorphism on the lipid interaction of human apolipoprotein E. J Boil Chem. 2003;278:40723–9. doi: 10.1074/jbc.M304814200. [DOI] [PubMed] [Google Scholar]