

Abstract
Ebola virus (EBOV) causes lethal disease in up to 90% of EBOV-infected humans. Among vaccines, only the vesicular stomatitis virus platform has been successful in providing postexposure protection in nonhuman primates. Here, we show that an adjuvanted human adenovirus serotype 5 (Ad5)–vectored vaccine (Ad5–Zaire EBOV glycoprotein) protected 67% (6 of 9) and 25% (1 of 4) of cynomolgus macaques when administered 30 minutes and 24 hours following EBOV challenge, respectively. The treatment also protected 33% of rhesus macaques (1 of 3) when given at 24 hours. The results highlight the utility of adjuvanted Ad5 vaccines for rapid immunization against EBOV
Keywords: Ebola, nonhuman primates, post-exposure, adenovirus, vaccine
To date, the outbreak of Ebola virus (EBOV) infection centered around Guinea, Sierra Leone, and Liberia during 2014–2015 has lasted approximately 1 year, with 9976 deaths from 24 282 cases [1]. Despite ongoing efforts to contain the outbreak via contact tracing, community education, and quarantine, as well as recent advancements in monoclonal antibody–based therapy against EBOV [2], the outbreak is still not under control and has been estimated by the World Health Organization to eventually cost almost $1 billion to fully contain [3]. This number highlights the importance of vaccine development as an important strategy to prevent new infections.
Advances over the past 15 years in EBOV vaccine development have resulted in several platforms that have successfully demonstrated protection in nonhuman primates (NHPs). These include EBOV-like particles, human parainfluenza virus type 3, rabies virus, adenovirus serotype 5 (Ad5) with and without a DNA prime, Ad26 prime with an Ad35 boost, Venezuelan equine encephalitis virus replicons, and vesicular stomatitis virus (VSV)–vectored vaccines [4]. To date, only the VSV-vectored vaccine has been successful at eliciting rapid protection in animals, as evidenced by the survival of 4 of 8 EBOV-infected NHPs that were given the vaccine 30 minutes after a lethal EBOV challenge [5].
Several compounds have also demonstrated postexposure efficacy in the past decade. In addition to antibody-based therapies, potential options include recombinant nematode anticoagulant protein c2 (rNAPc2), recombinant human activated protein C; encapsulated EBOV-specific small, interfering RNA; and positively charged phosphorodiamidate morpholino oligomers [6]. However, only vaccination is likely to be able to elicit robust immune responses capable of long-term protection against EBOV, and as such the development of a vaccine that is able to rapidly elicit protective immune responses after vaccination remains an important goal.
Ad5 vectors are a popular platform for experimental vaccines because they are well characterized and have been tested against a variety of infectious diseases, human conditions, and cancers. In EBOV studies, a single administration of Ad5–Zaire EBOV glycoprotein (ZGP) demonstrated sustained protection in NHPs that were challenged 21 weeks after immunization [7]. Preexisting immunity is a valid concern in sub-Saharan Africa because >90% of residents are seropositive for Ad5 antibodies [8], which may impact vaccine efficacy. However, studies have shown that airway delivery of Ad5-ZGP is still efficacious in NHPs under such circumstances [9]. In a previous study, we determined that Ad5-ZGP combined with an Ad5-interferon α (IFN-α) adjuvant elicited a synergistically elevated immune response, which resulted in rapid, postexposure protection against EBOV challenge in rodents [10]. The aim of this study was to assess the efficacy of Ad5-ZGP with various adjuvants in NHPs under postexposure conditions. Additionally, we evaluated specific cellular and humoral immune responses following vaccination/challenge and their relative importance to survival.
METHODS
The construction and production of the Ad5-ZGP vaccine was described previously [11]. Ad5–IFN-α was provided by Defyrus (Toronto, Canada), and the other products were obtained through commercial means. All humoral and cell-mediated immunity assays were performed as described in a previous publication [9]. All animal procedures were approved by the Institutional Animal Care Committee at the National Microbiology Laboratory of the Public Health Agency of Canada, according to the guidelines of the Canadian Council on Animal Care.
NHPs were challenged with a uniformly lethal dose of 1000 plaque-forming units of EBOV (Ebola virus H.sapiens-tc/COD/1995/Kikwit-9510621; GenBank accession number AY354458) and then treated at the indicated times with the corresponding regimens (Table 1). Blood specimens and oral, nasal, and rectal swab specimens were analyzed by reverse transcription quantitative polymerase chain reaction for the presence of EBOV after challenge, using methods described by a previous publication [2]. Levels of total ZGP-specific immunoglobulin G (IgG) antibody were assayed, and EBOV-specific cellular immune responses from NHP peripheral blood mononuclear cells were quantified by IFN-γ enzyme-linked immunosorbent spot analysis 14 or 15 days after infection.
Table 1.
Survival Outcomes of Cynomolgus and Rhesus Macaques Administered Adjuvanted Adenovirus Serotype 5 (Ad5)–Zaire Ebola Virus Glycoprotein (ZGP) After Ebola Virus Challenge
| Macaque ID | Macaque Type | Treatment | Treatment Initiation, Time After Exposure | Outcome |
|---|---|---|---|---|
| A1 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 days after infection | 30 min | Survived |
| A2 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 30 min | Survived |
| A3 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 30 min | Died 13 days after infection |
| A4 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection, along with 30 µg/kg rNAPc2 | 30 min | Died 11 days after infection |
| A5 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection, along with 30 µg/kg rNAPc2 | 30 min | Survived |
| A6 | Cynomolgus | One dose of 2 × 1010 IFU of Ad5-ZGP, followed by 3 × 109 PFU of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection, along with 30 µg/kg rNAPc2 | 30 min | Survived |
| A7 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 30 min | Survived |
| A8 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 30 min | Died 13 days after infection |
| A9 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 30 min | Survived |
| B1 | Cynomolgus | Phosphate-buffered saline | 30 min | Died 7 days after infection |
| B2 | Cynomolgus | Phosphate-buffered saline | 30 min | Died 7 days after infection |
| C1 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Died 7 days after infection |
| C2 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Died 7 days after infection |
| C3 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Died 9 days after infection |
| C4 | Cynomolgus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Survived |
| D1 | Cynomolgus | Phosphate-buffered saline | 24 h | Died 7 days after infection |
| D2 | Cynomolgus | Phosphate-buffered saline | 24 h | Died 7 days after infection |
| E1 | Rhesus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Died 11 days after infection |
| E2 | Rhesus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Died 17 days after infection |
| E3 | Rhesus | One dose of 4 × 109 IFU of Ad5-ZGP/kg, followed by 2 × 109 PFU/kg of Ad5–IFN-α intramuscularly; 0.44 µg/kg universal type I IFN was administered intramuscularly once daily on days 5–21 after infection | 24 h | Survived |
| F1 | Rhesus | Phosphate-buffered saline | 24 h | Died 8 days after infection |
| F2 | Rhesus | Phosphate-buffered saline | 24 h | Died 9 days after infection |
Abbreviations: ID, identification code; IFN, interferon; IFN-α, interferon α; IFU, infection-forming units; PFU, plaque-forming units; rNAPc2, recombinant nematode anticoagulant protein c2.
RESULTS
Six of 9 NHPs (67%) survived the challenge when treated 30 minutes after exposure, whereas 1 of 4 (25%) survived challenge when treated 24 hours after infection (Table 1). The 3 nonsurviving NHPs treated 30 minutes after challenge had a delayed time to death of 13, 11, and 13 days after infection, surviving nearly twice as long as the control animals, which died 7 days after infection. In contrast, the 3 nonsurviving cynomolgus macaques treated 24 hours after challenge died 7, 7, and 9 days after infection, which is similar to times of death for the control animals (Table 1). Nonsurviving, treated rhesus macaques died 11 and 16 days after infection, which is longer than the control animals (Table 1). Moribund NHPs displayed signs consistent with that of EBOV disease, including reduced activity, fever, lowered food and water intake, and early signs of subcutaneous hemorrhage, contributing to clinical scores above the ethical threshold for mandatory euthanasia (Supplementary Figure 1). Surviving NHPs had fully recovered from EBOV disease by 20 days after infection (Supplementary Figure 1).
In control animals and moribund animals treated 24 hours after challenge, viremia reached 104–108 genome equivalents/mL of blood. Oral, nasal, and rectal swab specimens also tested positive for EBOV RNA (Supplementary Table 1). In moribund animals treated 30 minutes after challenge, only viremia was detected upon euthanasia. In contrast, EBOV was not detected in blood specimens or in oral, nasal and rectal swab specimens from surviving NHPs (Supplementary Table 1).
All surviving cynomolgus macaques treated 30 minutes or 24 hours after EBOV challenge had elevated anti-ZGP IgG levels by 14 days after infection, and levels remained elevated until the termination of the experiment 28 days after infection (Figure 1A). ZGP-specific IgG was not detected in nonsurviving NHPs A4 and A8 but was detected in animal A3 by 7 days after infection (Figure 1A). For nonsurviving and control animals in the 24-hour treatment groups, ZGP-specific IgG was not detected, with the exception of animal E2 (Figure 1A and 1B). Surviving animals in the 24-hour treatment groups developed elevated anti-ZGP IgG levels by 14 days after infection, which remained elevated 28 days after infection (Figure 1A and 1B).
Figure 1.
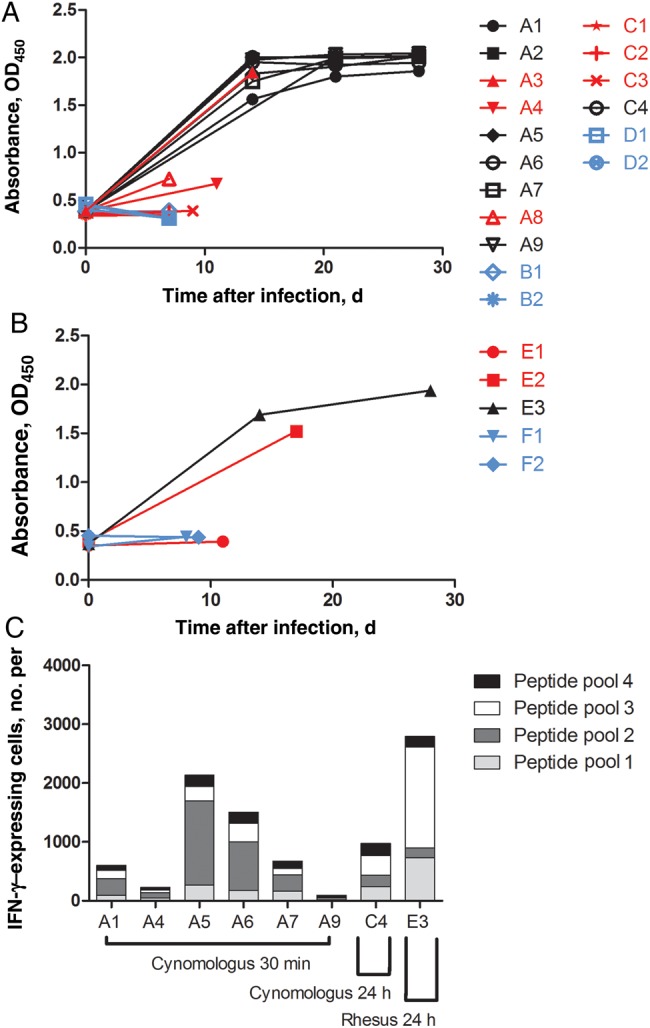
Adaptive immune response in cynomolgus and rhesus macaques following Ebola virus (EBOV) challenge and treatment. A and B, Zaire EBOV glycoprotein (ZGP)–specific immunoglobulin G (IgG) enzyme-linked immunosorbent assays were performed on serum specimens obtained from cynomolgus macaques (A) and rhesus macaques (B). Samples were assayed in triplicate, and absorbance values at 405 nm (OD405) from a 1:800 dilution are shown. C, Number of interferon γ (IFN-γ)–secreting cells per million cells in peripheral blood mononuclear cells obtained from surviving animals 14 or 15 days after infection, after stimulation by ZGP peptide pools, as measured by enzyme-linked immunosorbent spot assays. Red, black, and blue indicate treated nonsurvivors, treated survivors, and control animals, respectively.
The cumulative levels of IFN-γ produced in response to stimulation by various ZGP peptide pools varied between individual animals, ranging from 94 to 2134 IFN-γ–secreting lymphocytes per million cells for animals A1, A2, A5, A6, A7, and A9, which were treated 30 minutes after EBOV challenge (Figure 1C). The level of IFN-γ produced by animal C4 was 970 IFN-γ–secreting lymphocytes per million cells (Figure 1C). The rhesus macaque E3 produced 2796 IFN-γ–secreting lymphocytes per million cells (Figure 1C).
DISCUSSION
Untreated EBOV infections in humans result in mortality rates reaching as high as 90%. Ideally, the best postexposure treatments should be effective after the appearance of symptoms, and, owing to the inherent time needed to establish a robust adaptive immune response, adjuvanted Ad5-vectored vaccines cannot compare with the speed of the postexposure protection provided by antibody immunotherapy. However, the drawbacks with monoclonal antibodies include its large costs of production, the fact that filoviruses are difficult to neutralize completely, and antibody-mediated enhancement of EBOV entry into cells [12]. A rapidly effective vaccine may provide a viable alternative without the associated problems. Furthermore, the results of these studies show that the efficaciousness of an adjuvanted Ad5-ZGP regimen is at least similar to that of VSV-vectored vaccines and that the adjuvanted Ad5-ZGP regimen should be further investigated as an alternative vaccine capable of providing rapid protection to susceptible populations, including residents of EBOV-endemic areas, primary healthcare workers and researchers working in level 4 biocontainment facilities. We added the anticoagulant rNAPc2 to animals A4–A6 in a bid to enhance the adjuvanted Ad5-ZGP regimen. However, this treatment regimen was not superior in terms of survival (Table 1). In addition, administering adjuvanted Ad5-ZGP on the basis of animal weights did not alter the protective efficacy of the treatment regimens, compared with administering a fixed vaccine dose (Table 1).
A past report has established that total ZGP- and EBOV-specific IgG levels are reliable correlates of protection for Ad5-vectored vaccines, independent from the time of EBOV challenge, and that cellular immune responses also play a role in survival from EBOV infection [13]. Although ZGP-specific IgG was detected in 2 nonsurviving animals of this study, it may be that the level of antibodies produced and the strength of the adaptive immune response in general were not sufficient to alter the survival outcome, owing to advanced EBOV disease and varying levels of viremia to control between individual animals. Overall, these results reinforce the importance of the adaptive immune responses in survival from EBOV disease. The exact mechanisms of antibody-mediated protection remain unknown, but they are likely working in concert with the innate immune response through either antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, antibody-mediated phagocytosis, and the inhibition of virus entry or egress to keep EBOV viremia at manageable levels, whereas the cell-mediated immune response is responsible for clearing the virus from infected hosts.
NHP experiments are currently underway to investigate the postexposure efficacy of adjuvanted Ad5-ZGP delivery through the airway to address the issue of preexisting immunity. Furthermore, the current survival rate of 67% in the 30-minute treatment group combined, with rates of 25% and 33% among cynomolgus and rhesus macaques, respectively, in the 24-hour treatment group means that further improvements to the vaccination regimens are needed. We are currently addressing this by combining adjuvanted Ad5-ZGP with ZGP-specific monoclonal antibodies to investigate whether both candidates are able to work synergistically, potentially resulting in an Ad5-based vaccine with improved efficacy. In addition, this may also decrease the amount of monoclonal antibodies required to achieve full protection against EBOV in NHPs, thus yielding an affordable EBOV treatment with high potential for clinical and regulatory approval.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Alex Bello, Jason Gren, Shane Jones, Allen Grolla, and Jim Strong for their excellent technical assistance.
Financial support. This work was supported by the Public Health Agency of Canada; the Chemical, Biological, Radiological, or Nuclear Research and Technology Initiative (grants CRTI-06-0218RD and CRTI-09-453TD to G. P. K.); and the Canadian Institutes of Health Research (doctoral research award to G. W.).
Potential conflicts of interest. J. T. and J. E. were employees of Defyrus when these experiments were being planned and conducted and when the data were evaluated. J. T. and J. E. are inventors of the DEF201 technology that was assigned a patent by the US Patent and Trademark Office on 13 November 2012. Defyrus provided DEF201 at no cost or conditions to the Public Health Agency of Canada for these studies. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization. Ebola Situation Report – 11 March 2015, 2015. Available at: http://apps.who.int/ebola/current-situation/ebola-situation-report-11-march-2015. Accessed 12 March 2015.
- 2.Qiu X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014; 514:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler D, Morello L. Ebola by the numbers: The size, spread and cost of an outbreak. Nature 2014; 514:284–5. [DOI] [PubMed] [Google Scholar]
- 4.Marzi A, Feldmann H. Ebola virus vaccines: an overview of current approaches. Expert Rev Vaccines 2014; 13:521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldmann H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog 2007; 3:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong G, Qiu X, Olinger GG, Kobinger GP. Post-exposure therapy of filovirus infections. Trends Microbiol 2014; 22:456–63. [DOI] [PubMed] [Google Scholar]
- 7.Choi JH, Jonsson-Schmunk K, Qiu X, et al. A Single Dose Respiratory Recombinant Adenovirus-Based Vaccine Provides Long-Term Protection for Non-Human Primates from Lethal Ebola Infection. Mol Pharm 2014; 4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nwanegbo E, Vardas E, Gao W, et al. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol 2004; 11:351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson JS, Pillet S, Bello AJ, Kobinger GP. Airway delivery of an adenovirus-based Ebola virus vaccine bypasses existing immunity to homologous adenovirus in nonhuman primates. J Virol 2013; 87:3668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson JS, Wong G, Pillet S, et al. Evaluation of different strategies for post-exposure treatment of Ebola virus infection in rodents. J Bioterror Biodef 2011; S1:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson JS, Yao MK, Tran KN, et al. Enhanced protection against Ebola virus mediated by an improved adenovirus-based vaccine. PLoS One 2009; 4:e5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J Virol 2003; 77:7539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong G, Richardson JS, Pillet S, et al. Immune parameters correlate with protection against Ebola virus infection in rodents and nonhuman primates. Sci Transl Med 2012; 4:158ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.