

Abstract
Background. BST2/tetherin is an innate immune molecule with the unique ability to restrict the egress of human immunodeficiency virus (HIV) and other enveloped viruses, including Ebola virus (EBOV). Coincident with this discovery was the finding that the HIV Vpu protein down-regulates BST2 from the cell surface, thereby promoting viral release. Evidence suggests that the EBOV envelope glycoprotein (GP) also counteracts BST2, although the mechanism is unclear.
Results. We find that total levels of BST2 remain unchanged in the presence of GP, whereas surface BST2 is significantly reduced. GP is known to sterically mask surface receptors via its mucin domain. Our evaluation of mutant GP molecules indicate that masking of BST2 by GP is probably responsible for the apparent surface BST2 down-regulation; however, this masking does not explain the observed virus-like particle egress enhancement. We discovered that VP40 coimmunoprecipitates and colocalizes with BST2 in the absence but not in the presence of GP.
Conclusions. These results suggest that GP may overcome the BST2 restriction by blocking an interaction between VP40 and BST2. Furthermore, we have observed that GP may enhance BST2 incorporation into virus-like particles. Understanding this novel EBOV immune evasion strategy will provide valuable insights into the pathogenicity of this deadly pathogen.
Keywords: Ebola virus, HIV, BST2, tetherin, Ebola GP, Ebola VP40, viral egress, host immune restriction
BST2 is an interferon-inducible gene product that limits the release of human immunodeficiency virus (HIV) type 1 particles from infected cells [1, 2]. Work performed in our laboratory and others has demonstrated that the HIV-1 Vpu protein forms a complex with BST2 that results in the lysosomal degradation of BST2 [3–8]. BST2 also blocks the release of viral particles produced by other members of the Retroviridae family as well as particles produced by the Marburg and Ebola filoviruses [9]. It has been hypothesized that BST2 represents an innate immune mechanism that functions to limit the spread of many enveloped viruses, if not all.
Human Ebola virus (EBOV) infection results in highly transmissible hemorrhagic fevers with mortality rates approaching 90% [10]. The worst Ebola epidemic in history is presently occurring in West Africa, with no Food and Drug Administration–approved drugs or vaccines currently available. Continued research is therefore paramount in finding potential antiviral targets and to understanding the pathogenic mechanisms used by this deadly virus. EBOV and HIV are both enveloped RNA viruses that rely on their respective matrix proteins for viral particle assembly and function. EBOV VP40 and HIV Gag both interact with the endosomal sorting complexes required for transport system, which directs viral budding in a manner analogous to multivesicular body formation (recently reviewed in [11]). In addition, VP40 and Gag are each necessary and sufficient for the generation of virus-like particles (VLPs) that, similar to complete virions, will bud from the plasma membrane in a BST2-restricted manner.
Using these VLP systems, Kaletsky et al [12] have shown that among the 7 EBOV genes, only the envelope glycoprotein (GP) was capable of overcoming the inhibitory action of BST2. Although GP has been reported to coprecipitate with BST2 [12, 13], studies by Lopez et al [14] show that unlike Vpu, GP leads to neither the degradation nor the down-regulation of BST2 from the cell surface. Later, Lopez et al [15] found that GP does not appear to function by removing BST2 from lipid rifts, which have been implicated in viral assembly and budding of both HIV and EBOV (reviewed in [16]). Earlier work has shown that the release of both VP40- and Gag-based VLPs is restricted by BST2, and that the release of both types of VLPs is enhanced when either Vpu or GP is present. However, these are clearly accomplished by different means. For example, even in the absence of BST2, GP enhances the assembly and/or release of VP40-based VLPs, whereas Vpu does not [17].
The mechanism for this GP-dependent enhancement is not known, but it does not seem related to changes in particle morphology [18]. These results suggest that GP might have 2 distinct functions to enhance EBOV release, one involving BST2 antagonism, and the other promoting viral assembly regardless of BST2. Alternatively, the ability of GP to enhance viral assembly may only inadvertently allow EBOV to avoid BST2 restriction.
Although the data thus far support distinct mechanisms of action for GP and Vpu, it remains unclear how GP functions to overcome BST2. The majority of studies have thus far examined the EBOV GP-dependent release of heterologous HIV VLPs. We have instead chosen to investigate the EBOV GP-dependent release of homologous EBOV VP40 VLPs, with the expectation that a more physiological system would clarify matters. This article describes the data gathered using this approach, which indicates that Ebola GP uses a novel mechanism for evading BST2 restriction.
METHODS
Cells and Expression Constructs
Stable 293T-based cell lines expressing a hemagglutinin (HA) tagged BST2 (293T::BST2-HA) were generated as described elsewhere [19] and maintained in complete Dulbecco's modified Eagle medium. All EBOV protein expression constructs were derived from plasmids pCEboZVP40 and pCEboZGP, kindly provided by Yoshihiro Kawaoka (University of Wisconsin) [20]. VP40 and GP were subcloned into the mammalian expression vector pCAGGS-MCS. VP40 was NH2-terminally FLAG tagged to generate FLAG-VP40, and GP was COOH-terminally Myc tagged to generate GP-Myc. To create secreted GP (sGP)-Myc, 8 adenosine nucleotides we deleted from the RNA editing site (nucleotides 1270–1288) in GP-Myc [21]. Likewise, GPΔmucin-Myc was generated by deleting nucleotides 1324–1827, from our GP-Myc construct [22]. The green fluorescent protein (GFP) expression construct pQ100 was kindly provided by Jeffrey Vieira (University of Washington), and the pcDNA-Vphu [23] by Klaus Strebel at the National Institute of Allergy and Infectious Diseases. The HIV Gag/Pol construct pMDLg/pRRE (Addgene plasmid 12251) and pRSV-Rev (Addgene plasmid 12253) contributed by Didier Trono [24] were used for HIV-VLP production.
Flow Cytometry
For fluorescence-activated cell sorter (FACS) analysis of surface BST2, 293T::BST2-HA cells were cotransfected with pQ100 and the indicated viral protein expression vector (Lipofectamine 2000; Invitrogen). Forty-eight hours after transfection, cells were harvested and resuspended in FACS buffer (×1 phosphate-buffered saline plus 3% fetal bovine serum). Cells were stained with an anti-BST2 antibody (generated in house), washed 3 times, stained with a chicken anti-mouse Alexa Fluor 647 secondary antibody (Invitrogen), washed again 3 times, and finally analyzed on a FACSCalibur flow cytometer (BD).
Confocal Immunofluorescence
The 293T::BST2-HA and control cells were transfected as described above. After 48 hours, cells were fixed, washed 3 times, and permeabilized (phosphate-buffered saline, 0.2% saponin, 1% normal goat serum [NGS; Sigma]). Permeabilization buffer was used during all subsequent steps. For surface staining, cells were fixed, washed 3 times, blocked with 5% NGS, and then incubated with antibody in 1% NGS. Three primary antibodies were used: anti-BST2 polyclonal antibody from the National Institutes of Health (NIH) AIDS reagent program; anti-GP monoclonal antibody (mAb), kindly provided by Christopher Basler at Mount Sinai School of Medicine [25]; and biotinylated anti-FLAG mAb (Sigma). Cells were then washed 3 times in wash buffers (permeabilization or surface) and then incubated with secondary antibodies, which included Alexa Fluor 595–conjugated anti-rabbit (for BST2), Alexa Fluor 647–conjugated anti-mouse (for GP), and Alexa Fluor 488–conjugated streptavidin (for FLAG). Fluorescent images were obtained using a Leica DM IRBE confocal microscope.
Coimmunoprecipitations and Immunoblots
For VP40 coimmunoprecipitations (co-IPs), 293T::BST2-HA cells were transfected with plasmids expressing FLAG-VP40, GP, and GFP. After 40 hours, cells were lysed in 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) buffer (150 mmol/L sodium chloride [NaCl], 50 mmol/L Tris [pH 7.5–8], 1% CHAPS, cOmplete protease inhibitor [Roche]) and then centrifuged to pellet cell debris. Next, 100 µg of each lysate was precleared with control mouse immunoglobulin G beads (Sigma). Anti-FLAG beads (Sigma) were added to precleared supernatants, and samples were rocked at 4°C for 4 hours. Beads were then pelleted, washed 3 times, transferred to SigmaPrep spin columns, and eluted with NuPage lithium dodecyl sulfate (LDS) sample buffer (Invitrogen). GP co-IPs were handled similarly, with the following modifications. First, 150 µg of each lysate was precleared with immunoglobulin G2a antibody (Sigma) plus 20 µL of Trueblot anti-mouse beads (Rockland). An anti-Myc antibody (clone 9B11; Cell Signaling) was then added to precleared samples and incubated at 4°C for 4 hours, followed by the addition of 20 µL Trueblot anti-mouse beads for 1 hour. Eluted immunoprecipitation (IP) samples were boiled, separated on 4%–12% NuPage gradient gels (Invitrogen), transferred to polyvinylidene fluoride (PVDF; Millipore), and then analyzed for coimmunoprecipitating proteins.
Blots were first probed for both HA (clone 3F10 [horseradish peroxidase (HRP) conjugate]; Roche Applied Science) and BST2 (mAb generated in house, HRP conjugated). Blots were then stripped and sequentially probed for (1) GP-Myc, using either an anti-GP mAb (clone 254/3.12, kindly provided by Yoshihiro Kawaoka at the University of Wisconsin, Madison) or an anti-Myc mAb (clone 9B11; HRP conjugate; Cell Signaling); (2) FLAG-VP40, using an anti-FLAG mAb (clone M2 [HRP conjugate]; Sigma); and (3) either GFP (goat anti-GFP 600-103-215; Rockland) or actin (anti-actin mAb JLA20; EMD Millipore). Blots were developed using ECL Plus Substrate (Pierce/Thermo Fisher) and then imaged using either X-ray film (Kodak) or a G:BOX system (Syngene).
VLP Production
VP40-based VLPs were made by transfecting 293T::BST2::HA cells with plasmids expressing FLAG-VP40, GP-Myc, and GFP. After 24 hours, supernatants were replaced with 1.5 mL of virus production–serum-free medium (Invitrogen), and the cells were incubated for another 24 hours. Supernatants were harvested, centrifuged to remove cellular debris, layered onto 25% sucrose cushions, and centrifuged at 100 000g for 1.5 hours. Supernatants were discarded, and pellets were resuspended in NuPage LDS sample buffer. For immunoblot analysis, VLP samples were boiled, separated on 4%–12% NuPAGE gradient gels, transferred to PVDF, and then probed with anti-HA (BST2) and anti-FLAG (VP40) antibodies. The corresponding cell pellets were lysed in radioimmunoprecipitation assay buffer (150 mmol/L NaCl, 50 mmol/L Tris [pH 7.5–8], 1% octylphenoxypolyethoxyethanol, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, cOmplete protease inhibitor) and 2.5 µg of lysate was loaded per lane.
HIV-based VLPs were produced as described for VP40-based VLPs, with the following modifications. 293T::BST2-HA cells were transfected with pMDLg/pRRE plus pRSV-REV, along with plasmids expressing GFP and either pGP-Myc or pVphu. Purified HIV-based VLPs were separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis, the proteins were transferred to PVDF, and the membranes were probed with rabbit HIV-1SF2 p24 antiserum obtained through the NIH AIDS Reagent Program (No. 4250).
Lipid Raft Gradient Fractionation
293T::BST2-HA cells were transfected with the indicated constructs, lysed in Triton-X-100 buffer (25 mmol/L Tris [pH 7.4], 0.1% Triton X-100, 150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, and protease inhibitors), and then homogenized via 10 passages through a 26.5-gauge needle. Iodixanol (Optiprep; Sigma) step gradients were constructed as described elsewhere [26]; 200 µg of each lysate was equilibrated to 40% iodixanol and then overlaid with 30% iodixanol. Gradients were centrifuged at 215 000g for 3.5 hours at 4°C. After 500 µL fractions were collected and lysed in ×4 NuPage LDS sample buffer, 30 µL of each fraction was separated on 4%–12% gradient gels, transferred to PVDF, and immunoblotted for BST2. Blots were stripped and reprobed with an anti-flotillin mAb (610820; BD Biosciences).
RESULTS AND DISCUSSION
Total BST2 Levels Remain Unchanged in the Presence of GP, but Surface BST2 Levels Are Down-regulated
Because data have shown that HIV Vpu down-regulates both surface and intracellular BST2 levels, we began with experiments to determine whether EBOV GP functioned similarly. As shown in Figure 1A, total BST2 levels in 293T::BST2-HA cells were decreased in the presence of Vphu (humanized Vpu protein), but not GP. This lack of GP-induced BST2 degradation confirms data presented by Lopez et al [14]. In contrast, flow cytometry of these same cells showed a significant, GP-dependent decrease in surface BST2 levels (Figure 1B), similar to the level of down-regulation observed in the presence of Vphu. Note that GP has been shown to sterically block the antibody-based detection of other cell surface receptors, including major histocompatibility complex class 1 and β1-integrin [27, 28], and these activities have been mapped to GP's highly glycosylated mucin domain [22, 29, 30].
Figure 1.
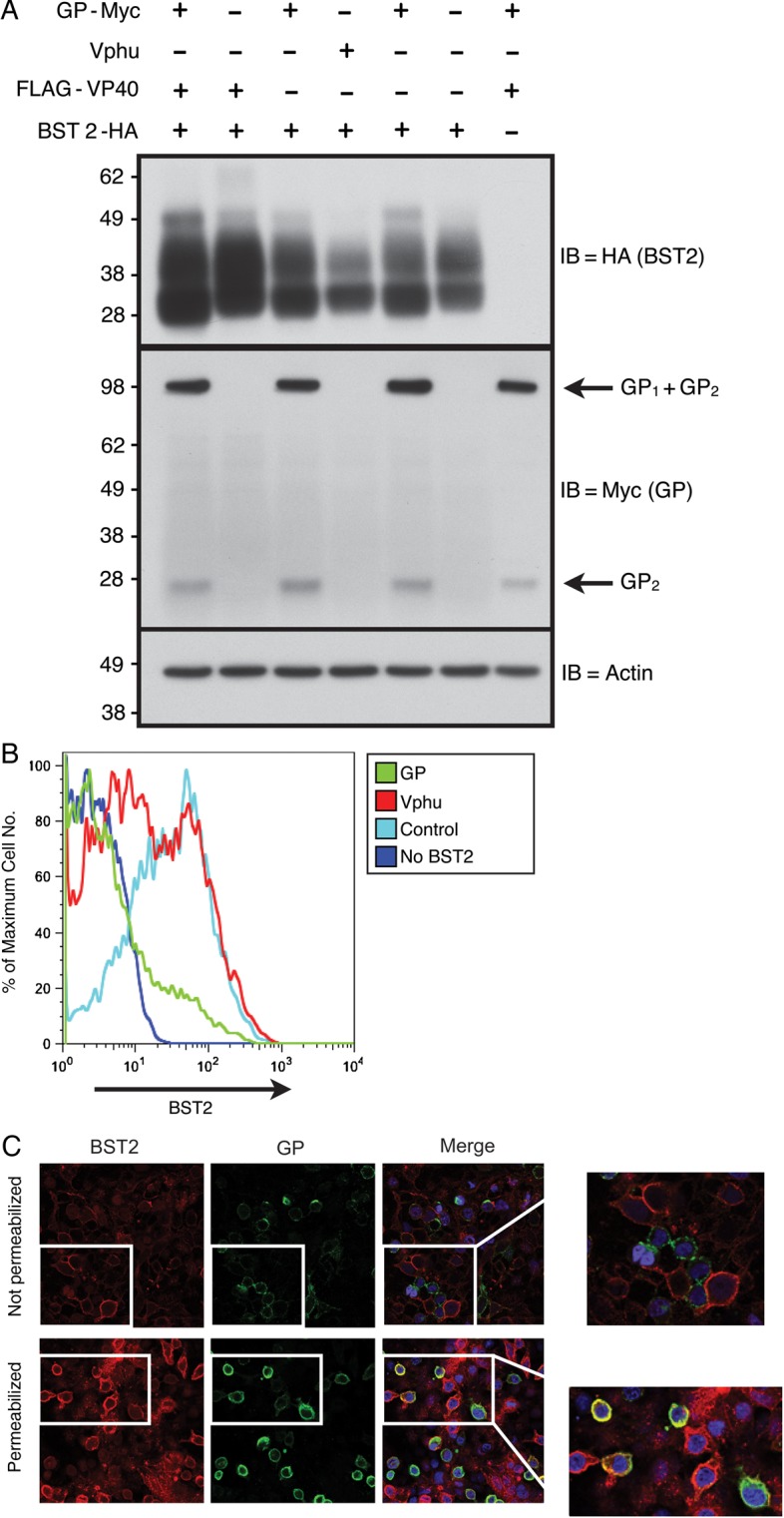
In the presence of glycoprotein (GP), total cellular BST2 levels remain unchanged, but BST2 surface levels seem to be down-regulated. A, Total BST2 levels. Stably transduced 293T cell lines expressing either the lentiviral vector only (293T::vector) or HA-tagged BST2 (293T::BST2-HA) were transfected with constructs expressing GP or Vphu. Protein lysates made from these cells were then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and probed for BST2 with an anti-HA antibody (immunoblot [IB] = BST2; top panel). The blot was sequentially stripped and reprobed with the indicated antibodies. B, Surface BST2 levels. 293T::vector or 293T::BST2-HA were cotransfected with an enhanced green fluorescent protein (EGFP)–expressing construct as well as constructs expressing either GP or Vphu. These cells were stained for BST2 without prior permeabilization and then subjected to flow cytometry. Histograms represent surface BST2 levels of EGFP-gated cells. C, BST2 and GP cellular localization. 293T::BST2-HA cells expressing GP were fixed and either surface stained (not permeabilized) or permeabilized with saponin for staining for BST2 (red), GP (green), and 4',6-diamidino-2-phenylindole (DAPI) (blue). Merged panel represents overlay of all 3 channels. Colocalization of BST2 and GP are indicated by yellow in the merged panel. A single z-section is shown. Abbreviation: HA, hemagglutinin.
We next performed confocal microscopy on cells that were either fixed and then stained for both surface BST2 and GP or were first permeabilized and then stained for intracellular BST2 and GP. As shown in Figure 1C (top panel), surface staining for BST2 and GP matched the flow cytometric data. Cells that stained positively for GP (green) were negative for BST2 staining (red), which is indicative of surface down-regulation. However, when the cells were permeabilized before staining, a significant amount of BST2 and GP was observed to colocalize at or near the cell surface (yellow), which suggests that BST2 down-regulation was not occurring. These disparate phenotypes were reminiscent of the ability of GP to sterically mask antibody epitopes of nearby surface proteins.
GP Mucin Domain is Responsible for the Apparent Down-regulation of Surface BST2 but not Necessary for Overcoming Block of VP40 VLP Release by BST2
We next sought to determine whether steric masking by GP's mucin domain could be responsible for the apparent BST2 down-regulation we observe and whether this masking phenotype might also function to block ability of BST2 to inhibit viral release. We generated plasmids expressing GPΔmucin (deleted for the mucin domain) and sGP (expressing inactive, secreted GP). The FACS data presented in Figure 2A confirm that the mucin domain is important for the observed down-regulation of surface BST2. Whereas wild-type GP decreased surface BST2 levels, neither GPΔmucin nor sGP displayed this activity. These data support a hypothesis in which GP does not promote the removal of BST2 from the cell surface but instead masks the antibody-dependent detection of BST2 at the cell surface.
Figure 2.
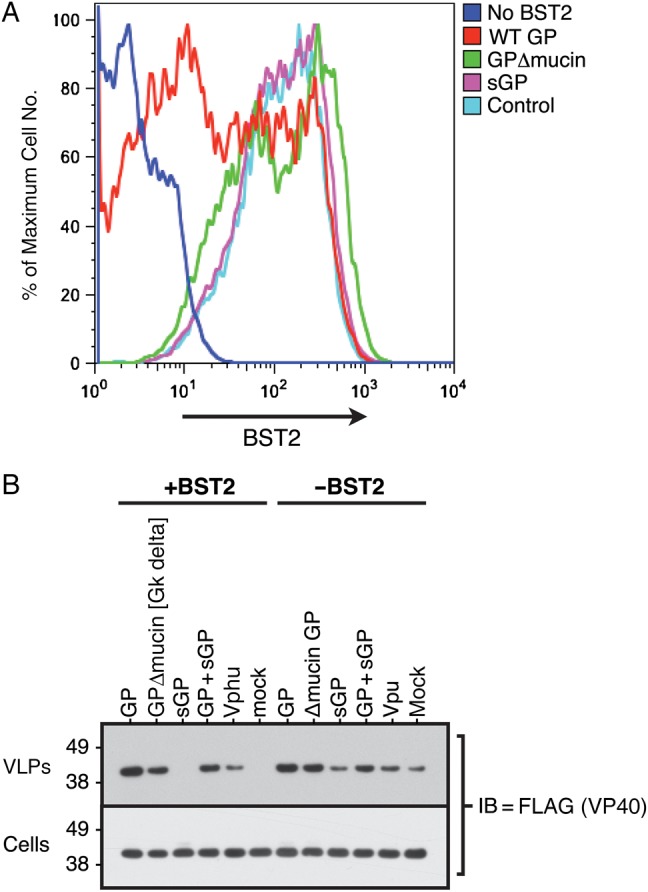
Although a glycoprotein (GP) molecule deleted for its mucin domain can still enhance virus-like particle (VLP) egress, it can no longer down-regulate cell surface BST2. 293T::vector or 293T::BST2-HA cells were cotransfected with both an enhanced green fluorescent protein (EGFP)–expressing construct and a construct expressing the indicated GP molecule. The cells were then stained for BST2, and flow cytometry was performed. A, Histograms represent the surface BST2 levels of EGFP-gated cells. B, VP40 VLP release. 293T::vector or 293T::BST2-HA cells were transfected with a construct expressing FLAG-VP40 along with a construct expressing the indicated GP or Vphu molecule. To quantitate VP40 levels, both total cell lysates and purified VLPs were separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis and FLAG immunoblots (IBs) were performed. Abbreviations: HA, hemagglutinin; sGP, secreted GP; WT, wild-type.
To determine whether this masking phenotype could also promote viral egress, we evaluated the ability of the GPΔmucin and sGP mutants to enhance the release of VP40-based VLPs in both the presence and absence of BST2. As shown in Figure 2B, in the absence of GP, VP40 VLPs were restricted by BST2, but the addition of GP significantly enhanced VLP release from cells regardless of BST2 expression. Interestingly, the mucin domain is clearly not important for these GP roles, because GPΔmucin increases VLP release as well as wild-type GP. Coupled with the fact that GPΔmucin could not down-regulate BST2, our data indicate that GP's steric masking function is separable from its ability to counteract BST2. Finally, as expected, sGP did not enhance VLP release, and Vphu was able to enhance VP40 VLP egress when BST2 was present. However, unlike GP, Vphu did not significantly enhance VLP release in the absence of BST2.
When Expressed Individually, GP and VP40 can Each Interact With BST2, but When Coexpressed, GP Prevents the Interaction Between BST2 and VP40
We next used IPs to examine interactions between BST2 and the EBOV matrix and envelope proteins. As shown in Figure 3A, BST2 coprecipitates with GP in both the presence and absence of VP40. In contrast, we do not observe VP40 specifically coimmunoprecipitating with GP, although some nonspecific VP40 binding is observed in the absence of GP. Consistent with the above IPs, when FLAG-VP40 was immunoprecipitated from these same lysates, GP was not recovered (Figure 3B). Interestingly, BST2 also coprecipitated with VP40. Although both the lower molecular weight endoplasmic reticulum (ER) form and the mature glycosylated forms of BST2 coprecipitated with VP40, the ER form also bound nonspecifically in the absence of VP40. Surprisingly, when GP was present, the higher molecular weight form of BST2 no longer interacted with VP40. Reciprocal BST2 IPs (Figure 3C) confirmed this phenotype. GPΔmucin similarly blocked the BST2-VP40 interaction, but sGP did not (Supplementary Figure 1). Unfortunately, because these proteins all associate with membranes, we cannot distinguish between direct protein interactions and precipitation of proteins within shared membranes or vesicles. Regardless, our data indicate that GP may act by preventing BST2 from interacting with VP40 and that only GP molecules with the ability to enhance VLP release can block the BST2-VP40 interaction.
Figure 3.

Glycoprotein (GP) and VP40 can each interact with BST2, but when all 3 are coexpressed, GP prevents the interaction between BST2 and VP40. 293T::vector or 293T::BST2-HA cells were transfected with a construct expressing GP-Myc and/or FLAG-VP40. An enhanced green fluorescent protein (EGFP)–expression construct was included as a transfection control. A, GP-Myc immunoprecipitations (IPs) were performed on cell lysates using an anti-GP antibody. Immune complexes were then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted for BST2 (IB = BST2 + HA). The blot was sequentially stripped and reprobed using the indicated antibodies. For comparison, 1/200 of the input cell lysate is shown. B, FLAG-VP40 IPs were produced on cell lysates using anti-FLAG–conjugated agarose beads. Immune complexes were then separated by SDS-PAGE and immunoblotted as described for Figure 3A. C, BST2-HA IPs were produced on cell lysates using anti-HA–conjugated agarose beads. Immune complexes were then separated by SDS-PAGE and immunoblotted for FLAG-VP40 (IB = FLAG). The blot was then sequentially stripped and reprobed for the indicated proteins. For comparison, 1/40 of the total lysate is shown. Abbreviation: HA, hemagglutinin.
VP40 and BST2 Colocalize Near the Cell Surface in the Absence but Not in the Presence of GP
To further evaluate the effect of GP on the interaction between VP40 and BST2, we used confocal microscopy to analyze the subcellular localization of these proteins. In agreement with the IP data, when GP is not expressed, VP40 and BST2 colocalize near the cell surface (Figure 4A, yellow overlay). In contrast, cells expressing GP (Figure 4B, blue arrows) showed reduced colocalization between VP40 and BST2. Cells that express low levels of GP (Figure 4B, white arrows) show considerable VP40 and BST2 colocalization in the overlay, indicating dose dependence. These data support the above co-IP data, and suggest that a spatial separation of these proteins occurs during VLP assembly and egress. It is possible that GP and BST2, both transmembrane proteins transported to the surface through the secretory system, may interact first in the ER or trans-Golgi network, and the BST2 trapped within this preformed protein complex is then unable to interact with VP40 at the cell surface. VP40 harbors neither a signal peptide nor a membrane-spanning domain and is therefore thought to associate with the inner leaf of the plasma membrane through hydrophobic domains present in its C-terminus [31]. Alternatively, the presence of GP during VLP assembly could modify VP40 or BST2 membrane localization to preclude their interaction.
Figure 4.
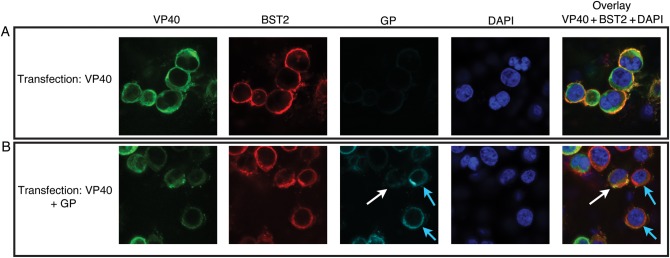
VP40 and BST2 colocalize near the cell surface in the absence of glycoprotein (GP) but not in its presence. 293T::BST2-HA cells were transfected with constructs expressing either VP40 alone (A) or VP40 and GP (B). The cells were then fixed and permeabilized with saponin before staining for VP40 (Alexa Fluor 488–conjugated antibody), BST2 (Alexa Fluor 595–conjugated antibody), GP (Alexa Fluor 647–conjugated antibody), and nuclei (DAPI). Each overlay is a merge of the green (VP40), red (BST2), and blue (DAPI) channels. Colocalization is indicated by yellow in the overlay. Confocal micrograph of a single z-section is shown. White arrows depict cells with low GP expression; blue arrows, cells with high GP expression. Abbreviations: DAPI, 4',6-diamidino-2-phenylindole; HA, hemagglutinin.
GP Does Not Displace BST2 From Lipid Rafts or VLPs
Both our co-IP and confocal microscopy data support a model in which GP prevents the interaction or colocalization between BST2 and VP40. Because BST2 is a lipid raft–associated protein, we speculated that GP might promote the displacement of BST2 from lipid rafts, thereby resulting in the observed phenotypes. We therefore performed an analysis to determine whether GP could affect the lipid raft association of either BST2 or VP40. As shown in Figure 5A, BST2 is enriched in the lipid raft fraction (fraction 2) in both the presence and absence of GP, demonstrating that GP does not alter the association of BST2with lipid rafts.
Figure 5.

Glycoprotein (GP) does not remove BST2 from lipid rafts or virus-like particles (VLPs). A, Lipid raft fractionation of BST2. 293T::BST2-HA cells expressing VP40 and/or GP were lysed in 0.1% Triton X-100 and then subjected to iodixanol (0%–40%) density gradient centrifugation. Fractions were collected from these gradients, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotted for BST2 (IB = BST2). The top blot was stripped and reprobed for the lipid raft marker flotillin (IB = flotillin). B, BST2 incorporation into VP40 VLPs. The 293T::BST2-HA or vector cells were transfected with a construct expressing FLAG-VP40 and/or a construct expressing GP-Myc. Cell lysates and purified VLPs were run on SDS-PAGE, and immunoblots for the indicated proteins were performed. Abbreviations: EGFP, enhanced green fluorescent protein; HA, hemagglutinin.
Several models suggest that BST2 tethers virions via homotypic interactions between BST2 that has been incorporated into virions and BST2 that remains at the infected cell surface [1, 32, 33]. We next investigated the possibility that GP might function by decreasing BST2 incorporation into VLPs. As shown in Figure 5B, in the presence of GP, a significant level of BST2 is detectable in VP40-based VLPs. In stark contrast, in the absence of GP, only low levels of BST2 were detected in VLPs. However, in the absence of GP, there also seems to be a concomitant decrease in VLP release. Thus, BST2 is indeed incorporated into VLPs in the presence of GP, suggesting that GP does not overtly prevent BST2 incorporation. However, the strong influence of GP on VLP release masks our ability to readily distinguish between increased BST2 incorporation into VLPs and an overall increase in released VLPs.
BST-2 Levels Are Increased in VLPs When GP is Present
To determine whether the increased BST2 that we observed in GP-containing, VP40-based VLPs was due to enhanced BST2 incorporation or merely to an overall increase in VLP production, we compared BST2 incorporation into VLPs in the presence of GP or Vphu. To determine whether this phenomenon was specific to VP40, we also assessed BST2 incorporation into GP-containing, HIV-based VLPs. As shown in Figure 6A, both GP and Vphu allow VP40 VLPs to overcome BST2 egress restriction, as measured by the increased levels of VP40-FLAG in VLPs. Interestingly, BST2 levels in these VLPs did not directly correlate with VP40 levels. Although Vphu promotes VP40/VLP release from cells, the levels of BST2 in those VLPs were significantly decreased. This is perhaps unsurprising, because Vpu is known to promote the destruction of BST2. However, BST2 levels in the Vphu-induced VP40 VLPs matched those from mock cells expressing no VP40 or Vphu. Because the levels of both VLP-associated BST2 and VLP-associated VP40 were significantly increased in the presence of GP, we cannot distinguish between increased BST2 incorporation per particle and increased VLP release. We speculate that the BST2 present in samples with no VP40 resides within secreted exosomes or microvesicles.
Figure 6.

BST2 levels are increased in virus-like particles (VLPs) when glycoprotein (GP) is present. A, BST2 incorporation into VP40 VLPs. The 293T::vector and 293T::BST2-HA cells were cotransfected with FLAG-VP40 and either GP, Vphu, or control expression constructs. An enhanced green fluorescent protein (EGFP)–expression construct was included as a transfection control. Cell lysates and purified VLPs were run on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblots (IB) for the indicated proteins were performed. B, BST2 incorporation into human immunodeficiency virus (HIV) VLPs. The 293T::vector and 293T::BST2-HA cells were transfected with the HIV Gag expression constructs and the indicated GP, Vphu, and control constructs. Cell lysates and purified VLPs were run on SDS-PAGE, and IB for the indicated proteins were performed. Abbreviations: HA, hemagglutinin; HIV, human immunodeficiency virus.
The presence of GP did not increase levels of BST2 in supernatants from mock cells indicating that the increased BST2 in VP40 VLPs is dependent on the presence of VP40. To determine whether this phenomenon is specific to VP40 or also occurs with VLPs derived from a heterogeneous matrix protein, we evaluated the release of HIV-based VLPs under similar conditions. As shown in Figure 6B, similar to what we observed for VP40-based VLPs, both GP and Vphu allowed HIV VLPs to overcome BST2-dependent restriction, as measured by increased p24 release. Correspondingly, although not to the same degree as for VP40 VLPs, we observed that BST2 levels in HIV VLPs increased when GP was expressed and decreased in the presence of Vphu. These data demonstrate that HIV VLPs exclude BST2 when either Vphu or no antagonists are present but incorporate it in the presence of GP. Our data indicate that the 2 viruses have evolved distinct mechanisms to circumvent BST2. We are currently evaluating other methods to determine whether this GP-dependent increase in VLP-associated BST2 is the result of more BST2 incorporated per particle or merely the result of enhanced particle release.
Summary
Our observation that GP prevents the interaction between VP40 and BST2 suggests that the matrix protein is more actively involved in BST2 tethering than previously thought. Earlier models predicted that because BST2 restricts such a wide variety of viruses, BST2 would not interact with specific viral proteins. Our observation that BST2 is both present at the cell surface and efficiently incorporated into VLPs from GP-expressing cells indicates either that BST2 restricts VP40-VLP release by an altogether different mechanism than HIV VLPs or that GP promotes BST2 incorporation into VLPs in a manner that prevents tethering.
The Marburg virus provides another example in which a viral matrix protein is required for tethering. A recent study [34] indicates that mouse-adapted Marburg VP40 is completely restricted by human BST2, but not mouse BST2, and that this is due to specific mutations in VP40 acquired during mouse adaptation. Another report, showing that the Lassa arenavirus is restricted by BST2 but does not seem to encode an antagonist, also highlights the potential importance of a matrix protein in BST2 restriction; in that study, Radoshitzky et al [35] evaluated whether EBOV GP and HIV Vpu could function as heterologous BST2 antagonists for Lassa Z VLP. Surprisingly, they found that only Vpu was able to enhance BST2-restricted Lassa Z release; EBOV GP did not counteract BST2 in the context of the Lassa Z matrix protein. Taking these findings together with our data, we conclude that (1) Vpu enhances VLP release through direct action on BST2, regardless of the matrix protein from which the VLPs are derived; and (2) the ability of GP to counteract BST2 is dependent on some feature common to both retroviral and filoviral matrix proteins.
We propose that evasion of BST2 restriction by EBOV depends on the ability of GP to block BST2 from interacting with VP40. Our future directions include the identification of those VP40 residues and/or domains that make contact with BST2, which should in turn shed light on how GP blocks this interaction. In an effort to determine how GP modifies the VLP composition, we also plan to conduct a detailed protein and lipid analysis of VP40 VLPs in the presence or absence of GP.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Lisa Clepper for her technical support and the following core facility directors for their technical assistance and expertise: Yibing Jia at the Molecular & Cellular Biology Core and Anda Cornea at the Imaging and Morphology Core of the Oregon National Primate Research Center and Daniel Cawley at the Vaccine and Gene Therapy Institute Monoclonal Antibody Core.
Financial support. This work was supported by the National Institutes of Health (grants 5 U54 A1081680-05 and 5 R01 AI090490-05).
Potential conflicts of interest. All authors: No potential conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Neil SJD, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008; 451:425–30. [DOI] [PubMed] [Google Scholar]
- 2.Van Damme N, Guatelli J. HIV-1 Vpu inhibits accumulation of the envelope glycoprotein within clathrin-coated, Gag-containing endosomes. Cell Microbiol 2008; 10:1040–57. [DOI] [PubMed] [Google Scholar]
- 3.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Früh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J Virol 2009; 83:7931–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goffinet C, Allespach I, Homann S, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009; 5:285–97. [DOI] [PubMed] [Google Scholar]
- 5.Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci 2009; 106:2868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. HIV-1 Vpu neutralizes the antiviral factor tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog 2009; 5:e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell RS, Katsura C, Skasko MA, et al. Vpu antagonizes BST-2–mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog 2009; 5:e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwabu Y, Fujita H, Kinomoto M, et al. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J Biol Chem 2009; 284:35060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jouvenet N, Neil SJD, Zhadina M, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 2009; 83:1837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez A, Geisbert TW, Feldmann H. Filoviridae: Marburg and Ebola Viruses. In: Knipe DM, Howley PM, eds. Fields virology, 5th ed. Vol 1. Philadelphia: Lippincott Williams & Wilkins, 2007: 1409–48. [Google Scholar]
- 11.Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe 2013; 14:232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci 2009; 106:2886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhl A, Banning C, Marzi A, et al. The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J Infect Dis 2011; 204(suppl 3):S850–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez LA, Yang SJ, Hauser H, et al. Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J Virol 2010; 84:7243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez LA, Yang SJ, Exline CM, Rengarajan S, Haworth KG, Cannon PM. Anti-tetherin activities of HIV-1 Vpu and Ebola virus glycoprotein do not involve removal of tetherin from lipid rafts. J Virol 2012; 86:5467–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suomalainen M. Lipid rafts and assembly of enveloped viruses. Traffic 2002; 3:705–9. [DOI] [PubMed] [Google Scholar]
- 17.Licata JM, Johnson RF, Han Z, Harty RN. Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol 2004; 78:7344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson RF, Bell P, Harty RN. Effect of Ebola virus proteins GP, NP and VP35 on VP40 VLP morphology. Virol J 2006; 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gustin JK, Douglas JL, Bai Y, Moses AV. Ubiquitination of BST-2 protein by HIV-1 Vpu protein does not require lysine, serine, or threonine residues within the BST-2 cytoplasmic domain. J Biol Chem 2012; 287:14837–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka Y. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol 2002; 76:4855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc Natl Acad Sci USA 1996; 93:3602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons G, Wool-Lewis RJ, Baribaud F, Netter RC, Bates P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J Virol 2002; 76:2518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen K-L, llano M, Akari H, et al. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 2004; 319:163–75. [DOI] [PubMed] [Google Scholar]
- 24.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998; 72:8463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez O, Valmas C, Basler CF. Ebola virus-like particle-induced activation of NF-κB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 2007; 364:342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krautkrämer E, Giese SI, Gasteier JE, Muranyi W, Fackler OT. Human immunodeficiency virus type 1 Nef activates p21-activated kinase via recruitment into lipid rafts. J Virol 2004; 78:4085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynard O, Borowiak M, Volchkova VA, Delpeut S, Mateo M, Volchkov VE. Ebolavirus glycoprotein GP masks both its own epitopes and the presence of cellular surface proteins. J Virol 2009; 83:9596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francica JR, Varela-Rohena A, Medvec A, Plesa G, Riley JL, Bates P. Steric shielding of surface epitopes and impaired immune recognition induced by the Ebola virus glycoprotein. PLoS Pathog 2010; 6:e1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takada A, Watanabe S, Ito H, Okazaki K, Kida H, Kawaoka Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology 2000; 278:20–6. [DOI] [PubMed] [Google Scholar]
- 30.Sullivan NJ, Peterson M, Yang ZY, et al. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J Virol 2005; 79:547–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J Virol 2001; 75:5205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Caballero D, Zang T, Ebrahimi A, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009; 139:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammonds J, Wang J-J, Yi H, Spearman P. Immunoelectron microscopic evidence for tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog 2010; 6:e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feagins AR, Basler CF. The VP40 protein of Marburg virus exhibits impaired budding and increased sensitivity to human tetherin following mouse adaptation. J Virol 2014; 88:14440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radoshitzky SR, Dong L, Chi X, et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J Virol 2010; 84:10569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.