

Abstract
The balance and interplay between pro-death and pro-survival members of the B-cell lymphoma-2 (Bcl-2) family proteins play key roles in regulation of the mitochondrial pathway of programmed cell death. Recent NMR and biochemical studies have revealed that binding of the proapoptotic BH3-only protein PUMA induces significant unfolding of antiapoptotic Bcl-xL at the interface, which in turn disrupts the Bcl-xL/p53 interaction to activate apoptosis. However, the molecular mechanism of such regulated unfolding of Bcl-xL is not fully understood. Analysis of the existing Protein Data Bank structures of Bcl-xL in both bound and unbound states reveal substantial intrinsic heterogeneity at its BH3-only protein binding interface. Large-scale atomistic simulations were performed in explicit solvent for six representative structures to further investigate the intrinsic conformational dynamics of Bcl-xL. The results support that the BH3-only protein binding interface of Bcl-xL is much more dynamic compared to the rest of the protein, both unbound and when bound to various BH3-only proteins. Such intrinsic interfacial conformational dynamics likely provides a physical basis that allows Bcl-xL to respond sensitively to detailed biophysical properties of the ligand. The ability of Bcl-xL to retain or even enhance dynamics at the interface in bound states could further facilitate the regulation of its interactions with various BH3-only proteins such as through posttranslational modifications.
Introduction
Intrinsically disordered proteins (IDPs) frequently play crucial roles in cell signaling and regulation and are associated with numerous human diseases (1–7). Intrinsic conformational disorder of IDPs may offer many potential functional advantages, such as larger binding surface areas, inducibility by posttranslational modifications, and structural plasticity for binding multiple partners (2,7). Attesting to the fundamental importance of intrinsic disorder in biology, sequence analysis has revealed that over one-third of eukaryotic proteins contain long disordered segments or domains (8). Intensive efforts have been focused on characterizing the conformational properties of unbound IDPs and understanding how these properties may support facile and robust binding to specific targets (9,10). The ability of many regulatory IDPs to undergo coupled binding and folding transitions upon specific binding, in particular, has attracted much attention (9,11). It is also increasingly recognized that substantial conformational heterogeneity, and sometimes full disorder of the entire binding domain, may persist in the bound states of IDPs (12–15). Furthermore, examples have started to emerge in recent years where cellular signaling and regulation are achieved via regulated unfolding of proteins (16). Regulated unfolding could be local or global, and may be driven by a wide range of signaling stimuli, including posttranslational modifications (17), ligand or protein binding (18–20), changes in environmental conditions such as pH (21,22), and mechanical stress (23,24). Together, these examples illustrate a fascinatingly broad and versatile use of protein conformational disorder in cellular signaling and regulation.
A particularly intriguing example of regulated folding involves the binding of intrinsically disordered PUMA to protein Bcl-xL (18). Bcl-xL is an antiapoptotic member of the Bcl-2 family proteins, which are critical regulators of the mitochondrial pathway of programmed cell death (25). Interactions and balance between proapoptotic and antiapoptotic members of the Bcl-2 family proteins underlie the regulatory network that controls the switch between life and death of the cell (26). Misregulation of the Bcl-2 family proteins is frequently involved in cancers (25). Bcl-xL inhibits the proapoptotic function of cytoplasmic tumor suppressor p53 by sequestering it into inactive complexes (27,28). It also protects cells from programmed death by interactions with numerous proapoptotic BH3-only Bcl-2 family proteins, including BID, BIM, BAD, PUMA, BIK, HRK, BMF, and NOXA (29–31). All BH3-only Bcl-2 family proteins except BID are IDPs (32). Upon binding to Bcl-xL, the BH3 domain of PUMA folds into a single helix (18) (see Fig. 1B). Intriguingly, PUMA binding also leads to local unfolding of Bcl-xL (18), mainly in the α2 and α3 segments near the BH3-only protein binding interface (see Fig. 1B), which in turn disrupts the interactions with cytosolic p53 and releases the inhibition of the proapoptotic function of p53. The apparently unique ability of PUMA binding to drive drastic local unfolding of Bcl-xL appears consistent with the fact that PUMA is the only BH3-only protein that can efficiently release p53 from the inactive complex with Bcl-xL (33). However, the molecular basis of regulated unfolding in the Bcl-xL/PUMA interaction is not fully understood.
Figure 1.

(A–F) Representative experimental structures of the BH3-only protein binding interface of Bcl-xL. Bcl-xL is colored in gray except for residues 98–120, which are colored in red. The BH3-only protein binding partners (PUMA, BIM, and BAD) are shown in blue. Additional details of these PDBs are provided in Table 1. The cluster IDs are shown in parenthesis (see Table S2 for details). To see this figure in color, go online.
PUMA contains a unique Tryptophan at position 71 among BH3-only proteins. Of importance, its π-stacking interaction with Bcl-xL His-113 has been shown to be necessary for the observed regulated unfolding of Bcl-xL (18) (see Fig. 1B). Mutation of PUMA Trp-71 to Ala largely suppresses Bcl-xL partial unfolding in the bound state, and further abolishes the ability of PUMA binding to release cytosolic p53 from Bcl-xL inhibition (18). These observations have led to the conclusion that the π-stacking interaction between PUMA Trp-71 and Bcl-xL His-113 drives local unfolding in the adjacent α2 and α3 segments of Bcl-xL. However, the π-stacking interaction itself does not appear to be thermodynamically important, because the W71A mutant and wild-type PUMA BH3 domain bind to Bcl-xL with similar high affinities (18). It appears unusual that a single specific interaction between Bcl-xL His-113 and PUMA Trp-71, located on the edge of the binding interface, could fully account for the dramatic conformational changes observed for Bcl-xL. Curiously, the BH3 domain of BAD also contains a Tryptophan at position 70 (PUMA numbering), which is in position to potentially make a similar π-stacking interaction with Bcl-xL His-113 (see Fig. 1F), but it does not lead to similar local unfolding upon binding (18) (we note that α3 does become sufficiently distorted in Protein Data Bank (PDB):1G5J such that it is not fully assigned to the helical state in the secondary structure analysis; see Figs. 1F and 2). In this work, we analyze all existing PDB structures of Bcl-xL both in the apo form and in complex with various small molecule and peptide ligands, and perform extensive molecular dynamics (MD) simulations for six selected Bcl-xL apo structures and complexes to characterize the inherent conformational dynamics of Bcl-xL. The results together demonstrate that the BH3-only protein binding interface of Bcl-xL is much more dynamic than currently recognized. Such intrinsic interfacial conformational dynamics could provide a physical basis that enables Bcl-xL to respond sensitively to the nature of the bound ligand and/or environmental conditions, thus allowing facile unfolding upon specific binding of appropriate ligands such as PUMA.
Figure 2.

Secondary structures of Bcl-xL in 49 PDB entries in 2M04 numbering scheme. Helical residues are colored in red and disordered/loop residues in gray. White regions mark residues absent in the PDB structure. The PDB entries are ordered and colored based on the nature of the bound ligands: p53 (green), an α-β-foldamer homologs of the BIM BH3 domain (magenta), BH3-only peptides/proteins (blue), small molecules (red), and unbound (black). Note that some structures contain single or double mutations in Bcl-xL (see Table S1 for details). To see this figure in color, go online.
Materials and Methods
A total of 45 PDB entries were identified that contain either unbound Bcl-xL monomer or its complex with small molecule or peptide ligands (as of September 2014) (18,28,34–54). Four domain-swapped dimers of Bcl-xL, namely, 3FDL, 2YQ6, 2YQ7, and 4A1U, are also included, where α1 is involved in intermolecular interactions and adopts a very different configuration (e.g., see Fig. S1 in the Supporting Material). Information about all PDB entries included in the current analysis is provided in Table S1. For structural analysis, all entries were preprocessed to remove extra atoms that do not belong to Bcl-xL or its primary ligand (if present). The residue numbering of Bcl-xL was modified in all PDB entries to match the one used in 2M04 (Bcl-xL/PUMA) for convenience of comparison. For PDB entries containing multiple models/chains, only chain A of model 1 was included in the analysis. Clustering analysis was performed using the fixed radius clustering algorithm as implemented in the MMTSB/cluster.pl tool (with -kclust option) (55).
Atomistic MD simulations in explicit solvent were performed to further characterize the structure and dynamics of Bcl-xL in both bound and unbound states. These simulations were initiated from six representative PDB structures. Two were based on the NMR and x-ray structures of unbound Bcl-xL, namely, 2M03 (18) and 1R2D (36). The other four simulations were based on the complex structures of Bcl-xL with various BH3-only protein ligands, including 1BXL for Bcl-xL/BAK (43), 1G5J for Bcl-xL/BAD (53), 2M04 for Bcl-xL/PUMA (18), and 3FDL for Bcl-xL/BIM (52). We note that the long and presumably disordered loop between α1 and α2 is absent in all original PDB structures noted previously. To our best knowledge, this loop is not involved in the complex formation between BH3-only peptides and Bcl-xL, and thus omitted in the current simulations. A caveat is that the potential impacts of the disordered loop on the structure and dynamics of the rest of the protein will not be captured. All structures were solvated using TIP3P water molecules, with proper counter ions added to neutralize the whole system. The final solvated systems are illustrated in Fig. S2. They consist of 40,740 to 97,371 atoms with cubic box dimensions ranging from ∼73 to 98 Å. See Table 1, which provides a summary of the residue ranges, total atom numbers, box sizes, and total simulation times of all simulations.
Table 1.
Summary of All Six Simulations
| PDB | Proteins | Bcl-xL Residuesa | Water No. | Na+ No. | Total Atom No. | Initial Box Size (Å) | Simulation Time (ns) |
|---|---|---|---|---|---|---|---|
| 1R2D | Bcl-xL | 1–27, 82–196 | 12,824 | 6 | 40,740 | 73.0 | 730 |
| 2M03 | Bcl-xL | 1–44, 85–200 | 13,660 | 12 | 43,498 | 74.8 | 570 |
| 2M04 | Bcl-xL/PUMA | 1–44, 85–200 | 18,237 | 14 | 57,654 | 82.4 | 460 |
| 1G5J | Bcl-xL/BAD | 1–44, 85–211 | 14,233 | 10 | 45,836 | 76.5 | 580 |
| 3FDL | Bcl-xL/BIM | 1–26, 83–194 | 17,922 | 5 | 56,421 | 81.6 | 450 |
| 1BXL | Bcl-xL/BAK | 1–44, 85–217 | 31,439 | 12 | 97,371 | 98.1 | 310 |
Residue numbering is based on that of 2M04.
Each solvated system was energy minimized using steepest descent and adopted basis Newton-Raphson methods, followed by a short equilibration simulation of 100 ps using CHARMM (56,57) with a small harmonic restraint imposed on protein heavy atoms to slowly relax the system. After that, unrestrained MD production simulations were performed using NAMD (58). CHARMM36/CMAP force field was used to model proteins, water, and ions (59–61). These simulations were carried out under constant temperature (298 K) and constant pressure (1 bar), and periodic boundary conditions were imposed. Nonbonded interactions were truncated at a distance of 13 Å, with a smooth switching function starting from 12 Å. The pair list was updated every 10 steps with a pair list distance of 15 Å. Long-range electrostatic interactions were treated with the particle mesh Ewald method (62) with a grid spacing of ∼1 Å. Lengths of all hydrogen-related bonds were kept constant using the SHAKE algorithm (63), and a time step of 2 fs was used to integrate the equations of motion. The total production simulation lengths range from 310 ns (for 1BXL) to 730 ns (for 1R2D) (see Table 1). All analysis was performed using CHARMM and in-house scripts, and molecular illustrations were prepared using the visual molecular dynamics software (64).
Results and Discussion
Analysis of existing PDB structures of Bcl-xL
We first analyzed existing PDB structures of Bcl-xL in both bound and unbound states to examine the conformational flexibility of the BH3-only protein binding interface of Bcl-xL. Bcl-xL structure consists of eight helices (α1–8) connected by loops of different lengths (Fig. 1A). As summarized in Fig. 2, most helices are consistently present in all PDB structures except α3. Interestingly, PUMA does not appear to be the only ligand that can drive partial unfolding in the α2/α3 region of Bcl-xL. Instead, α3 appears to be unfolded in many complexes, such as 3ZK6, 3ZLN, 3ZLO, 2O2N, 1YSG, 1YSI, 1BXL, 1G5J, 3PL7, and 4A1U. These complexes involve both small molecule and peptide ligands, none of them contain analogous π-stacking interactions to the one between PUMA Trp-71 and Bcl-xL His-113 that has been postulated to be critical in driving partial unfolding in Bcl-xL (except potentially for 1G5J; see Fig. 1F). Even for the structures where α3 is not fully unfolded, the length and position of α3 can vary significantly among different PDB entries. For example, in 2LPC, α3 consists of 10 residues (residues 102–111), whereas in 1PQ1, α3 is only composed of 4 residues (residues 108–111). Other regions near the BH3-only protein binding interface, including both C-terminus of α2 and N-terminus of α4, also display substantial variance among different PDB structures. In particular, the C-terminus of α2 becomes disordered in two apo structures of Bcl-xL (1LXL and 2ME9), similar to what was observed in the Bcl-xL/PUMA complex (2M04).
Variations in the Bcl-xL PDB structures can be quantified by calculating the root mean-square fluctuation (RMSF) profiles. RMSF quantifies the magnitude of atomic positional fluctuation around the mean, and has been shown to correlate strongly with order parameters derived from NMR relaxation analysis (65). For this, all structures were first aligned using Cα atoms in the core region, which was identified as regions with minimal secondary and tertiary structure variations and included residues 85–98, 123–127, 140–156, and 162–175. We note that RMSF profiles calculated with structures aligned using the whole protein are not qualitatively different (e.g., see Fig. S3). Bcl-xL in domain swapped-dimers contains a dramatically different pose of α1 (see Fig. S1). Inclusion of the four domain-swapped dimer PDBs thus leads to artificially high RMSF values in α1 (Fig. 3, black trace). Excluding these four PDBs yields a RMSF profile that more accurately reflects the structural variations within the Bcl-xL monomer in bound and unbound states (Fig. 3, red trace). Clearly, the BH3-only protein binding interface of Bcl-xL is highly variant. The RMSF values exceed 3.5 Å in regions near α3, and are much higher than most loops except the long one between α1 and α2 (which is disordered and not resolved in most structures). Of importance, these variations among PDB structures seem to reflect the intrinsic conformational dynamics of Bcl-xL. For example, the RMSF profile converted from the B-factors of a representative x-ray crystal structure of unbound Bcl-xL (1R2G), shown as the green trace in Fig. 3, is highly consistent with that derived from all PDBs (red trace), which also suggest that regions near α3 is much more dynamic than the rest of the protein. Similar observations can be made on the RMSF profile calculated from the 20-member NMR ensemble of 2M04 (Fig. S4). The notion that the BH3-only protein binding interface of Bcl-xL is intrinsically more dynamic is further supported by sequence analysis by PrDOS (66), which predicts elevated disorder propensities near α3/4 compared to the other helical regions (Fig. S5).
Figure 3.

RMSF profiles calculated using all 49 Bcl-xL PDB structures (black trace) or 45 Bcl-xL PDB structures excluding 2YQ6, 2YQ7, 3FDL, and 4A1U (red trace). The four PDBs excluded form domain-swapped dimers with distinct α1 configurations (see Fig. S1). The green trace is converted from the B-factors of one representative PDB structure (1R2G). Only Cα atoms of residue 4–23 and 85–194 were used to calculate RMSF because these residues are present in all PDB entries. All structures were aligned using Cα atoms in the core region (residues 85–98, 123–127, 140–156, and 162–175) before RMSF calculation. To see this figure in color, go online.
Clustering analysis was performed to analyze the conformational distribution of the BH3-only protein binding interface of Bcl-xL. For this, all structures were also first aligned using the highly conserved and minimally varying core region, and then clustered based on mutual Cα root mean-square deviation (RMSD) of residues 98–120. The clustering led to many small clusters with a cutoff radius of 2.0 Å, confirming the significant level of conformational heterogeneity at the interface. A total of seven clusters were obtained when the clustering cutoff was set to 3.0 Å. The results are summarized in Table S2 and Fig. S6. Most unbound structures of Bcl-xL were assigned to the same cluster (cluster 1), whereas structures in complexes populate the other six clusters. The six PDB structures selected for MD simulations are representative of the four most populated clusters, with 2M04 representing the case with the most dramatic local unfolding of Bcl-xL.
Stability and fluctuation of simulation trajectories
MD simulations were performed in explicit solvent to further investigate the conformational dynamics of Bcl-xL in both unbound and bound states. For this, we focus on two representative structures of unbound Bcl-xL, namely 1R2D (by x-ray crystallography) (36) and 2M03 (by NMR) (18), and four Bcl-xL complexes that involve various BH3-only protein ligands as summarized in Table 1. The complexes were selected based on the varying degrees of Bcl-xL structural disorder observed at the binding interface (see Figs. 1 and 2). For example, Bcl-xL undergoes substantial unfolding in both α2 and α3 upon PUMA binding (2M04), but remains similarly structured when bound with BIM (3FDL). The helical structures in α3 appear to be sufficiently distorted both Bcl-xL/BAD (1G5J) and Bcl-xL/BAK (1BXL) complexes to be assigned as coil using the standard DSSP classification (67). Interestingly, α2 of Bcl-xL is extended by an extra turn upon BAK binding (Fig. 1C). The Bcl-xL/BIM complex (3FDL) was simulated without its domain-swapped partner for the sake of computational efficiency.
All six structures were stable during the simulations that lasted 310 to 730 ns. As shown in Fig. 4, the core region (see Fig. 3 caption for definition) deviated no greater than ∼2 Å from their corresponding initial conformations in all simulations. Without its domain-swapped partner, packing of α1 is very unstable during the initial stages of the Bcl-xL/BIM simulation (the 3FDL trace), leading to large fluctuations in the overall RMSD (Fig. 4A). During the simulation, α1 moves quickly toward the rest of the protein within the first 20 ns, and undergoes additional major conformational transitions around 170 and 250 ns (see Figs. S4 and S5). However, the final configuration of α1 in the 3FDL simulation remains quite different from the typical configuration observed in Bcl-xL monomer structures (see Fig. S9), which likely reflects limitations in conformational sampling with the 450 ns production run in explicit solvent. Moderate overall RMSD values of the 2M04 and 1R2D trajectories mainly arise from the unfolded BH3-only protein binding interface and fluctuations in the packing of α1, respectively. The structures of the bound BH3-only peptides were also very stable, except that PUMA helix becomes unfolded at the C-terminus (see Figs. S9 and S10). Interestingly, PUMA C-terminal spontaneous unfolding has also been implicated when bound to another Bcl-2 protein Mcl-1 and was shown to facilitate its dissociation from Mcl-1 (68).
Figure 4.

Evolution of Bcl-xL backbone heavy atom RMSD for (A) the whole protein and (B) core region during six simulations initiated from various PDB structures. See Fig. 3 caption for the definition of the core region. To see this figure in color, go online.
Once establishing the stabilities of all PDB structures during the simulations, we quantify the conformational dynamics of Bcl-xL by performing RMSF analysis. For this, all conformations in each trajectory were first aligned using Cα atoms in the core region. The resulting RMSF profiles, shown in Fig. 5, are highly similar to those extracted from existing PDB structures (Fig. 3). The implication is that variations among PDB structures indeed reflect the intrinsic conformational dynamics of Bcl-xL. The BH3-only protein binding interface (e.g., residues 98–120) is the most flexible region in all simulations, except that the α1 segment is artificially more dynamic in the 3FDL trajectory due to the absence of the domain-swapped partner. We note that the α3 region shows significantly larger fluctuations compared with other helices even for structures with well-formed α3 helix (e.g., 2M03, blue trace). Interestingly, binding of various BH3-only peptides do not appear to suppress the interfacial dynamics in general. Instead, the interface tends to become even more dynamic upon binding, e.g., comparing 1BXL (black trace) and 1G5J (red trace) versus 1R2D (green trace). Enhancement in interfacial dynamics is particularly dramatic in the case of Bcl-xL/PUMA complex due to complete local unfolding (cyan trace).
Figure 5.

RMSF of Bcl-xL derived from all six simulations. Only Cα residues 1–26 and 85–194 are shown because this is the common region among all simulation systems. The core region is marked with ×. RMSF profiles are consecutively shifted by 1.0 Å along the y axis for clarity. To see this figure in color, go online.
Conformational dynamics of the BH3-only protein binding interface
To further examine the details of conformational fluctuation at the BH3-only protein binding interface of Bcl-xL, we first focus on the stability and fluctuation at the secondary structure level. The results, summarized in Figs. 6 and S11, reveal that the α3 segment can undergo spontaneous helix-coil changes in both bound and unbound states. For example, for the Bcl-xL/PUMA complex, α3 is completely unfolded in the original PDB structure (2M04), but starts to sample short helical structures after ∼300 ns of simulations. The average residue helicity in the α3 segment calculated from the second half of the 2M04 simulation trajectory reach ∼0.3 (Fig. 7). Similar observation can be also made in simulations initiated from the 1G5J and 1BXL structures, except that the reformation of short helixes in the α3 segment occurred much earlier during the simulations. The neighboring region of α3, including both the N-terminus of α4 and C-terminus of α2, is dynamic as well. For example, α4 N-terminus of the unbound Bcl-xL became unfolded after ∼350 ns in the trajectory initiated from 2M03, but was extended after 75 ns in the trajectory initiated from 2M04. The averaged residue helicity profiles calculated from these trajectories, shown in Figs. 7 and S12, again illustrate the intrinsic dynamics and conformational heterogeneity of the BH3-only protein binding interface of Bcl-xL.
Figure 6.

Evolution of secondary structures near the BH3-only protein binding interface of Bcl-xL during six sets of simulations. The coloring scheme is the same as in Fig. 2. See Fig. S11 for the results for all Bcl-xL residues. To see this figure in color, go online.
Figure 7.
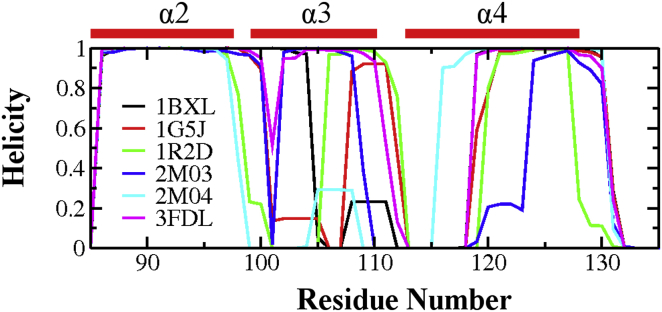
Average residue helicity profiles of Bcl-xL calculated from the second half of the simulation trajectories. Only residues 85–135 were shown here for clarity. See Fig. S12 for the full residue helicity profiles. To see this figure in color, go online.
Conformations of the BH3-only protein binding interface sampled during the atomistic simulations can be further visualized using the principal component analysis. For this, all snapshots from both the PDB set and six simulation trajectories were first aligned using the core region (see Fig. 3 caption for definition), and the Cα positions of residues 98–120 were then analyzed. The first two major components derived from the set of 49 PDB structures were used to project all PDB structures and MD snapshots. The results, summarized in Fig. 8, show that PDB structures assigned to different clusters nicely segregate on the two-dimensional projection as expected. All six atomistic simulations appear to mainly sample local conformational spaces centered around their corresponding initial conformations. The only exception is the 2M04 trajectory. With a completely unfolded α3, the interfacial structure of 2M04 quickly deviates from the initial conformation (marked by the blue ⊗ in Fig. 8B), and mainly sample a moderately large space projected to overlap with clusters 3 and 4 (orange and purple ×s). We note that the 2MO4 ensemble is quite heterogeneous but most of its members (particularly model 1 used to initiate the simulation) do not overlap with the final conformational space sampled (Fig. S13). The limited conformational sampling could suggest that the interfacial structures of Bcl-xL is restricted to different conformational subspaces depending on the binding partner, but is also likely a direct consequence of the finite simulation times in explicit solvent. Particularly for unbound Bcl-xL (2M03 and 1R2D trajectories), spontaneous unwinding and reforming of interfacial helices and changes in interfacial tertiary structures likely occur at timescales beyond those accessed by the current simulations. A more advanced sampling technique might be required to reliably probe the conformational space accessible to Bcl-xL in bound and unbound states (69–71).
Figure 8.

(A–F) Projection of six independent simulation trajectories on the first two major components derived from the set of 49 PDB structures. The projected locations of all PDB structures are marked using ×, except that the starting structure of each simulation is marked with ⊗ and pointed by arrows. Clusters 1 through 7 are colored in red, black, orange, purple, cyan, blue, and green, respectively. The cluster numbers of the PDB structures used to initiate the simulations are also indicated. To see this figure in color, go online.
Conclusion
It has been increasingly recognized that regulated unfolding of proteins is widely involved in cellular signaling and transduction (16). Bcl-xL, in particular, has been recently shown to undergo dramatic local unfolding upon binding and folding of the intrinsically disordered PUMA protein (18). There is an important need to understand the molecular principles of how such binding-induced folding and unfolding may be achieved with high reliability and efficiency to be viable in cellular signaling and regulation. Critical analysis of existing PDB structures of Bcl-xL in bound and unbound states has revealed that its BH3-only binding interface is intrinsically more dynamic than the rest of the protein. In particular, the specific π-stacking interaction between PUMA Trp-71 and Bcl-xL His-113 is clearly required for Bcl-xL interfacial unfolding in the case of PUMA (18), but does not appear necessary for Bcl-xL partial unfolding itself. Intrinsic dynamics at the BH3-only protein binding interface of Bcl-xL has been further confirmed by atomistic simulations of six representative bound and unbound structures in explicit solvent, even though these simulations are apparently insufficient to sample large-scale spontaneous conformational fluctuations in explicit solvent. Together, the current study supports that the BH3-only protein binding interface of Bcl-xL is highly dynamic and poised to adopt alternative conformations in response to ligand binding, as well as changes in solution conditions and posttranslational modifications. Such intrinsic interfacial conformational plasticity is likely the main physical basis of regulated unfolding observed in the Bcl-xL/PUMA interaction. With the ability to interact with numerous proapoptotic proteins including p53, Bcl-xL has been targeted by small molecules for cancer therapy (72). The dynamic nature of its BH3-only binding interface should be a critical consideration in such rational drug design and optimization efforts.
Author Contributions
X.L., A.B., and J.C. designed research; X.L. and A.B. performed research; X.L., A.B., and J.C. analyzed data; X.L. and J.C. wrote the article.
Acknowledgments
This work was supported by the National Science Foundation (MCB 0952514) and Johnson Cancer Research Center. Computer resources were used at Extreme Science and Engineering Discovery Environment (XSEDE) facilities (TG-MCB140210). Part of computing for this project was performed on the Beocat Research Cluster at Kansas State University, which is funded in part by NSF grants CNS-1006860, EPS-1006860, and EPS-0919443. This work is contribution number 15-291-J from the Kansas Agricultural Experiment Station.
Editor: Rohit Pappu.
Footnotes
Thirteen figures and two tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(15)00778-X.
Supporting Material
References
- 1.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 2.Uversky V.N., Oldfield C.J., Dunker A.K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 3.Smock R.G., Gierasch L.M. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilser V.J., Thompson E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai C.J., Ma B., Nussinov R. Structured disorder and conformational selection. Proteins. 2001;44:418–427. doi: 10.1002/prot.1107. [DOI] [PubMed] [Google Scholar]
- 6.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J. Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 2012;524:123–131. doi: 10.1016/j.abb.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 8.Zhang C., Ma J. Folding helical proteins in explicit solvent using dihedral-biased tempering. Proc. Natl. Acad. Sci. USA. 2012;109:8139–8144. doi: 10.1073/pnas.1112143109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyle N., Das R.K., Pappu R.V. A quantitative measure for protein conformational heterogeneity. J. Chem. Phys. 2013;139:121907. doi: 10.1063/1.4812791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H.X. Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 2012;37:43–48. doi: 10.1016/j.tibs.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 13.McDowell C., Chen J., Chen J. Potential conformational heterogeneity of p53 bound to S100B(betabeta) J. Mol. Biol. 2013;425:999–1010. doi: 10.1016/j.jmb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Mittag T., Orlicky S., Forman-Kay J.D. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. USA. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittag T., Marsh J., Forman-Kay J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitrea D.M., Kriwacki R.W. Regulated unfolding of proteins in signaling. FEBS Lett. 2013;587:1081–1088. doi: 10.1016/j.febslet.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimmler M., Wang Y., Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 18.Follis A.V., Chipuk J.E., Kriwacki R.W. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013;9:163–168. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denisov A.Y., Madiraju M.S., Gehring K. Solution structure of human BCL-w: modulation of ligand binding by the C-terminal helix. J. Biol. Chem. 2003;278:21124–21128. doi: 10.1074/jbc.M301798200. [DOI] [PubMed] [Google Scholar]
- 20.Hinds M.G., Lackmann M., Day C.L. The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J. 2003;22:1497–1507. doi: 10.1093/emboj/cdg144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardwell J.C.A., Jakob U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012;37:517–525. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foit L., George J.S., Bardwell J.C. Chaperone activation by unfolding. Proc. Natl. Acad. Sci. USA. 2013;110:E1254–E1262. doi: 10.1073/pnas.1222458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J., Zolkiewska A. Force-induced unfolding simulations of the human Notch1 negative regulatory region: possible roles of the heterodimerization domain in mechanosensing. PLoS One. 2011;6:e22837. doi: 10.1371/journal.pone.0022837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephenson N.L., Avis J.M. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc. Natl. Acad. Sci. USA. 2012;109:E2757–E2765. doi: 10.1073/pnas.1205788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yip K.W., Reed J.C. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 26.Petros A.M., Olejniczak E.T., Fesik S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Green D.R., Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Follis A.V., Llambi F., Kriwacki R.W. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat. Struct. Mol. Biol. 2014;21:535–543. doi: 10.1038/nsmb.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michels J., Kepp O., Galluzzi L. Functions of BCL-X L at the interface between cell death and metabolism. Int. J. Cell Biol. 2013;2013:705294. doi: 10.1155/2013/705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chipuk J.E., Moldoveanu T., Green D.R. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chipuk J.E., Green D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinds M.G., Smits C., Day C.L. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 33.Chipuk J.E., Bouchier-Hayes L., Green D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 34.Oltersdorf T., Elmore S.W., Rosenberg S.H. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 35.Priyadarshi A., Roy A., Hwang K.Y. Structural insights into mouse anti-apoptotic Bcl-xl reveal affinity for Beclin 1 and gossypol. Biochem. Biophys. Res. Commun. 2010;394:515–521. doi: 10.1016/j.bbrc.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Manion M.K., O’Neill J.W., Hockenbery D.M. Bcl-XL mutations suppress cellular sensitivity to antimycin A. J. Biol. Chem. 2004;279:2159–2165. doi: 10.1074/jbc.M306021200. [DOI] [PubMed] [Google Scholar]
- 37.Aritomi M., Kunishima N., Morikawa K. Crystal structure of rat Bcl-xL. Implications for the function of the Bcl-2 protein family. J. Biol. Chem. 1997;272:27886–27892. doi: 10.1074/jbc.272.44.27886. [DOI] [PubMed] [Google Scholar]
- 38.Liu X., Dai S., Kappler J.W. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19:341–352. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- 39.Feng Y., Zhang L., Liu D. A conserved hydrophobic core at Bcl-xL mediates its structural stability and binding affinity with BH3-domain peptide of pro-apoptotic protein. Arch. Biochem. Biophys. 2009;484:46–54. doi: 10.1016/j.abb.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Muchmore S.W., Sattler M., Fesik S.W. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 41.Lessene G., Czabotar P.E., Watson K.G. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 42.Feng W., Huang S., Zhang M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007;372:223–235. doi: 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 43.Sattler M., Liang H., Fesik S.W. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 44.Boersma M.D., Haase H.S., Gellman S.H. Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J. Am. Chem. Soc. 2012;134:315–323. doi: 10.1021/ja207148m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou H., Chen J., Wang S. Design of Bcl-2 and Bcl-xL inhibitors with subnanomolar binding affinities based upon a new scaffold. J. Med. Chem. 2012;55:4664–4682. doi: 10.1021/jm300178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee E., Czabotar P., Fairlie W. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14:1711–1713. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- 47.Bruncko M., Oost T.K., Elmore S.W. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 48.Schroeder G.M., Wei D., Borzilleri R.M. Pyrazole and pyrimidine phenylacylsulfonamides as dual Bcl-2/Bcl-xL antagonists. Bioorg. Med. Chem. Lett. 2012;22:3951–3956. doi: 10.1016/j.bmcl.2012.04.106. [DOI] [PubMed] [Google Scholar]
- 49.Sleebs B.E., Czabotar P.E., Baell J.B. Quinazoline sulfonamides as dual binders of the proteins B-cell lymphoma 2 and B-cell lymphoma extra long with potent proapoptotic cell-based activity. J. Med. Chem. 2011;54:1914–1926. doi: 10.1021/jm101596e. [DOI] [PubMed] [Google Scholar]
- 50.Wysoczanski P., Mart R.J., Allemann R.K. NMR solution structure of a photoswitchable apoptosis activating Bak peptide bound to Bcl-xL. J. Am. Chem. Soc. 2012;134:7644–7647. doi: 10.1021/ja302390a. [DOI] [PubMed] [Google Scholar]
- 51.Okamoto T., Zobel K., Czabotar P.E. Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem. Biol. 2013;8:297–302. doi: 10.1021/cb3005403. [DOI] [PubMed] [Google Scholar]
- 52.Lee E.F., Sadowsky J.D., Fairlie W.D. High-resolution structural characterization of a helical alpha/beta-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew. Chem. Int. Ed. Engl. 2009;48:4318–4322. doi: 10.1002/anie.200805761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petros A.M., Nettesheim D.G., Fesik S.W. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czabotar P.E., Lee E.F., Colman P.M. Mutation to Bax beyond the BH3 domain disrupts interactions with pro-survival proteins and promotes apoptosis. J. Biol. Chem. 2011;286:7123–7131. doi: 10.1074/jbc.M110.161281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feig M., Karanicolas J., Brooks C.L., 3rd MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 56.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM - a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 57.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 60.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 61.Best R.B., Zhu X., Mackerell A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Darden T., York D., Pedersen L. Particle mesh Ewald - an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 63.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical-integration of Cartesian equations of motion of a system with constraints - molecular-dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 64.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 65.Berjanskii M., Wishart D.S. NMR: prediction of protein flexibility. Nat. Protoc. 2006;1:683–688. doi: 10.1038/nprot.2006.108. [DOI] [PubMed] [Google Scholar]
- 66.Ishida T., Kinoshita K. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics. 2008;24:1344–1348. doi: 10.1093/bioinformatics/btn195. [DOI] [PubMed] [Google Scholar]
- 67.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 68.Rogers J.M., Oleinikovas V., Clarke J. Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. USA. 2014;111:15420–15425. doi: 10.1073/pnas.1409122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lane T.J., Shukla D., Pande V.S. To milliseconds and beyond: challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013;23:58–65. doi: 10.1016/j.sbi.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang W., Chen J. Accelerate sampling in atomistic energy landscapes using topology-based coarse-grained models. J. Chem. Theory Comput. 2014;10:918–923. doi: 10.1021/ct500031v. [DOI] [PubMed] [Google Scholar]
- 71.Zuckerman D.M. Equilibrium sampling in biomolecular simulations. Annu. Rev. Biophys. 2011;40:41–62. doi: 10.1146/annurev-biophys-042910-155255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jeng P.S., Cheng E.H. Cancer therapeutics: pulling the plug on BCL-X(L) Nat. Chem. Biol. 2013;9:351–352. doi: 10.1038/nchembio.1256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.