

Abstract
Cancer stem cells can escape therapeutic killing by adopting a quiescent or dormant state. The reversibility of this condition provides the potential for later recurrence or relapse, potentially many years later. We describe the genomics of a rare case of childhood BCR-ABL1-positive, B-cell precursor acute lymphoblastic leukemia that relapsed, with an acute myeloblastic leukemia immunophenotype, 22 years after the initial diagnosis, sustained remission and presumed cure. The primary and relapsed leukemias shared the identical BCR-ABL1 fusion genomic sequence and two identical immunoglobulin gene rearrangements, indicating that the relapse was a derivative of the founding clone. All other mutational changes (single-nucleotide variant and copy number alterations) were distinct in diagnostic or relapse samples. These data provide unambiguous evidence that leukemia-propagating cells, most probably pre-leukemic stem cells, can remain covert and silent but potentially reactivatable for more than two decades.
Introduction
Recurrence or relapse of cancer many years1, 2, 3 or occasionally decades4 after an initial diagnosis has been frequently recorded. These observations raise difficult issues related to presumptions of cure, risk assessment and monitoring of residual disease. A plausible mechanism for persistent, covert cancer cells during and after treatment is provided by the observation that some cancer stem cells can adapt a reversible quiescent or dormant state in which they are relatively resistant to radiation and chemotherapy.5, 6, 7, 8 However, the assumption is usually made that late recurring cancer is a derivative of the original clone at diagnosis, evidence for which is very limited, with the exception of some acute leukemias where physiological rearrangement of immunoglobulin genes (IGH/IGK) provide clone-specific markers.9, 10, 11
Materials and methods
Targeted capture libraries, cloning and sequencing of gene fusions
A cell line, MR-87, was established from the original leukemic cells of the 4-year-old patient and it showed the same immunophenotype and karyotype of the diagnostic leukemic cells. These cells were also shown to express the p190 BCR-ABL1 protein.12 Illumina paired-end libraries (Illumina, San Diego, CA, USA) covering the entire genomic regions of the BCR, ABL1 and IKZF1 genes were prepared from the MR87 cell line DNA (diagnosis) using the Agilent SureSelectXT 2 Custom (1–499 kb) DNA bait library (Agilent Technologies, Santa Clara, CA, USA). The custom libraries were sequenced on a HiSeq2500 (Illumina) to a coverage depth of 99 ×. Casava software (v1.8, Illumina) was used to make base calls and demultiplex the sequencing data and the genomic fusion breakpoints of BCR-ABL1 and IKZF1 were roughly determined using Burrows–Wheeler Aligner and Breakdancer software (The Genome Institute, St Louis, MO, USA). The BCR-ABL1 breakpoint fusion was predicted based on the location of read pairs that mapped to the fusion partners and the average fragment size of the capture library (320 bp). Using GRCh37.p13, the predicted breakpoint region in BCR was at chr22:23533568–23533950 (intron 1) and the breakpoint region in ABL1 was expected at chr9:133608500–133608811 (intron 1). A large deletion in IKZF1 (~50 kb) was observed between regions chr7:50412887–50463541. PCR primers were then designed to span the putative breakpoints using Primer3 plus (www.primer3plus.com/). Primers used for cloning the BCR-ABL1 fusion were: 5′-GTCAAAGCATTTTCCCCTGC-3′ and 5′-TCTTGATACTGGGTTGGCTGC-3′, and for the IKZF1 deletion were: 5′-GTCCTGGGTTTAAGCTTCAGTTCTCTGCCT-3′ and 5′-GGGTTGATAAGGAGGGTTTTGTGTCCCAGT-3′. Patient-specific gene fusions were amplified using AccuPrime Taq DNA Polymerase High Fidelity (Life Technologies, Carlsbad, CA, USA) and PCR products sequenced using BigDye Terminator v1.1 and an ABI-3730xl Genetic Analyzer (Applied Biosystems, Warrington, UK). Sequences were aligned by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Screening for IG and TCR gene rearrangements
DNA was extracted from diagnostic (MR87) and relapse cells (peripheral blood and bone marrow). PCR amplification of immunoglobulin (IG) heavy-chain variable–diversity–joining (IGH V(D)J; complete and incomplete), IGκ variable–joining (IGK-VJ), IGK Vκ-deleting element (Kde), intron recombination signal sequence–Kde, IGλ (IGL), T-cell receptor-β (TCRB), TCRγ (TCRG) and TCRδ gene rearrangements were performed using primers and conditions recommended by the BIOMED-2 Consortium.13 The 6-carboxyfluorescein labelled products were analysed using an ABI 3500 Genetic Analyzer (Applied Biosystems), clonality was assessed by GeneScan software (Applied Biosystems) and the results were interpreted in accordance with the EuroClonality/BIOMED-2 guidelines.14 PCR products were cloned into pCR2.1 (Life Technologies), sequenced and analysed as above. Junction analyses were performed using IgBLAST (www.ncbi.nlm.nih.gov/igblast/) and the ImMunoGeneTics database (www.imgt.org/).
Genome-wide copy number analysis
Single-nucleotide polymorphism array analysis was carried out using Affymetrix SNP 6.0 arrays (Affymetrix, Santa Clara, CA, USA), according to the manufacturer's instructions, on DNA extracted using standard methods from the diagnostic cell line (MR87) and from relapse bone marrow. Genotyping and generation of quality control data were performed in Genotyping Console v4.1.4 software (Affymetrix). The sample files were scrutinised for copy number alteration (CNAs) by visual inspection and by using the Partek Genomics Suite 6.6 software (Partek, St Louis, MO, USA). Copy number was determined by normalisation to Partek-distributed baseline files, which comprise 270 Hapmap files, using a genomic segmentation algorithm.
Whole exome sequencing
Exome capture was performed using the Agilent SureSelect Human All Exon V5 kit as per the manufacturer's instructions (Agilent) and sequenced by Illumina paired-end sequencing (protocol v1.2). Briefly, DNA was sheared by fragmentation (Covaris, Woburn, MA, USA), purified using Agencourt AMPure XP beads (Beckman Coulter, Pasadena, CA, USA) and the resulting fragments analysed on an Agilent 2100 Bioanalyzer. Fragment ends were repaired and adaptors ligated to the fragments, and the library was purified using beads. After amplification and hybridisation with biotinylated RNA baits, bound genomic DNA was purified with streptavidin-coated magnetic Dynabeads (Life Technologies) and re-amplified to include barcoding tags before finally pooling for sequencing on an Illumina HiSeq 2000.
Exome analysis was completed in Oxford Gene Technology's (Begbroke, UK) exome pipeline. Briefly, reads were aligned to the human genome reference sequence GRCh37 using Burrows–Wheeler Aligner 0.6.2.15 Local realignment was performed around indels with the Genome Analysis Toolkit (GATK v1.6) IndelRealigner.16 Optical and PCR duplicates were marked in BAM files using Picard 1.107 (http://picard.sourceforge.net). Original HiSeq base quality scores were recalibrated using GATK TableRecalibration and per-sample variants called with GATK UnifiedGenotyper (Broad Institute, Cambridge, MA, USA). Indels and single-nucleotide variants (SNVs) were hard filtered according to the Broad Institute best-practice guidelines, to eliminate false-positive cells.
Copy number variants, somatic SNV and somatic indels were identified between presentation and relapse samples using VarScan2.17 Variant annotation was performed with a modified version of Ensembl Variant Effect Predictor.18
Results
Clinical and haematological features of case
A brief case report of the patient was already published19 and is summarised as follows. A 4-year-old boy was diagnosed as having precursor B-cell acute lymphoblastic leukemia (ALL) but with a mixed lympho-myeloid phenotype: positive for myeloperoxidase, CD13+, CD10+ and CD19+. Cytogenetics on leukemic cells showed 46,XY,9p−,t(9q+22q−), indicating Ph+ pre-B-ALL. Reverse transcriptase-PCR confirmed the presence of the minor breakpoint (p190) BCR-ABL1 fusion.
The patient was treated with chemotherapy and achieved complete remission. Eight weeks after the diagnosis, he developed a central nervous system relapse, which was successfully treated with cranial irradiation and intrathecal drug administration. Three months after the diagnosis, he received a bone marrow transplant (BMT) from his human leukocyte antigen-identical (non-twin) brother when in the second complete remission. BMT transplantation was successful and no major complications were observed.
At the age of 25 years (20 years after BMT transplantation), the patient presented with general fatigue. His white blood cell count was 16.7 × 109/l with 7% blasts and his bone marrow aspirates showed leukemic cells with a myeloid immunophenotype positive for CD13 and CD33, and negative for CD10 and CD19. The leukemic cell karyotype was 46,XY,t(9;22)(q34;q11) × 2 plus other complex abnormalities. He was tentatively diagnosed as having a relapse of the initial Ph+ pre-B-ALL and received intensive chemotherapy resulting in complete remission. He underwent the second BMT transplant from a human leukocyte antigen-identical unrelated donor but had bone marrow relapse 35 weeks after the second BMT transplant and subsequently died of the disease.
Diagnostic and late relapse clones share an identical BCR-ABL1 fusion sequence
The putative breakpoint regions of the BCR and ABL1 genes were identified by targeted whole-genome sequencing of DNA isolated from the cell line (MR87) derived from patient cells at diagnosis. PCR primers were designed 5′ to the putative breakpoint in BCR and 3′ to that in ABL1, and the patient-specific BCR-ABL1 gene fusion was amplified, cloned and sequenced. The breakpoint detected in the BCR gene occurred within intron 1 at GRCh37.p13 position ch22:23533768 and within intron 1 of the ABL1 gene at position ch9:133608599 (Figure 1). The breaks in both BCR and ABL1 are therefore outside the recognised cluster regions described for Ph+ leukemia.20 The same set of PCR primers were next used to interrogate DNA from the peripheral blood and bone marrow at relapse and an identical sized fusion product was obtained. Cloning and sequencing of the relapse fusion products proved the BCR-ABL1 fusion sequence to be identical to that present at diagnosis (Figure 1a).
Figure 1.

Diagnostic and late relapse clones share an identical BCR-ABL1 fusion sequence with a discordant intra-gene deletion of IKZF1. (a) Upper panel: PCR primers that span the BCR-ABL1 breakpoint identified in diagnostic material (MR87, CL) were used to interrogate DNA isolated at relapse (bone marrow (BM) and peripheral blood (PB)). An identical product is seen in all patient samples but not in leukemia controls. Lower panel: DNA sequence of the BCR-ABL1 fusion and comparison with wild-type BCR and ABL1 gene sequences (GRCh37.p13). The DNA sequence was identical in both diagnostic and relapse samples. (b) Upper panel: DNA fusion sequence (GRCh37.p13) of the IKZF1 deletion at diagnosis reveals a 50-kb deletion between introns 2 and 7. Lower panel: the deletion/fusion product present at diagnosis (MR87 and CL) is not observed at relapse (BM and PB) or in DNA from leukemia controls.
Genome-wide copy number analysis
SNP 6.0 analysis on DNA isolated from the diagnostic cell line showed the following recurrent leukemia CNA: deletion of MTAP, CDKN2A/B, PAX5, 6q14.1–6q16.1 and IKZF1. In addition, amplification of MDM2 was noted. Relapse material was discordant for the diagnostic CNA drivers (Table 1); however, copy number loss of 9p was demonstrated (including loss of the same genes deleted at diagnosis: CDKN2A/B, MTAP and PAX5) but results clearly demonstrated that this 9p deletion was a re-iterative event with distinct breakpoints to the diagnostic sample (Supplementary Tables S1 and S2, and Supplementary Figures S1A–F). Potential drivers newly acquired in the relapse material also included deletion of the majority of chromosome 21q, gain of chromosome 20 and deletion of 8p (Supplementary Table S2).
Table 1. Summary of ‘driver' CNAs present at diagnosis versus relapse detected by SNP analysis.
| Chr | Start | End | Cytoband | CNA loss/gain | Diagnosis | Relapse | Genes (max three listed) | |
|---|---|---|---|---|---|---|---|---|
| 1 | chr6 | 78095353 | 93349377 | 6q14.1–6q16.1 | Loss | Present | Absent | IRAK1BP1, PHIP, SYNCRIP |
| 2 | chr7 | 50389071 | 50477011 | 7p12.2 | Loss | Present | Absent | IKZF1 |
| 3 | chr9 | 16523946 | 21901150 | 9p22.3–9p21.3 | Loss | Present | Absent | MLLT3, MTAP, FOCAD |
| 4 | chr9 | 21901150 | 22347406 | 9p21.3 | Homozygous Loss | Present | Absent | CDKN2A, CDKN2B |
| 5 | chr9 | 36412035 | 38493465 | 9p13.2–9p13.1 | Loss | Present | Absent | MELK, PAX5, FRMPD1 |
| 6 | chr12 | 68970862 | 69406845 | 12q15 | Gain | Present | Absent | RAP1B, NUP107, MDM2 |
| 7 | chr 7 | 50389060 | 50486601 | 7p12.2 | Loss | Absent | Present | IKZF1 |
| 8 | chr8 | 31254 | 43078119 | 8p23.3–8p11.21 | Loss | Absent | Present | ZNF596, FBXO25, DLGAP2 |
| 9 | chr9 | 6988784 | 21926959 | 9p24.1–9p21.3 | Loss | Absent | Present | MPDZ, MLLT3, MTAP |
| 10 | chr9 | 21926959 | 22200408 | 9p21.3 | Heterozygous Loss | Absent | Present | CDKN2A, CDKN2B |
| 11 | chr9 | 22200408 | 132070825 | 9p21.3–9q34.11 | Loss | Absent | Present | MOB3B, LINGO2, PAX5 |
| 12 | chr20 | 61305 | 62956154 | 20p13–20q13.33 | Gain | Absent | Present | DEFB125, DEFB126, TOP1 |
| 13 | chr21 | 15939316 | 48096958 | 21q11.2–21q22.3 | Loss | Absent | Present | CHODL,CXADR,NCAM2 |
Abbreviations: CNA, copy number alteration; SNP, single-nucleotide polymorphism.
Based on NCBI37/hg19 assembly.
Given that deletions in the tumour suppressor gene IKZF1 are considered a driving force of leukemogenesis, we used targeted sequencing of diagnostic DNA to design PCR primers that spanned the putative boundaries of the 50 kb IKZF1 intra-gene deletion. Subsequent PCR produced an ~4 kb amplification product that was further cloned and sequenced (Figure 1b). The 5′-breakpoint was determined at GRCh37.p13 position 7:50412893 and the 3′-breakpoint at position 7:50463650, with loss of 50 757 bp of DNA and the random insertion of 3 nucleotides (Figure 1b). Using the same primer set, we could not detect a deletion in the IKZF1 gene by conventional PCR or sensitive quantitative PCR in the peripheral blood or bone marrow at relapse (Figure 1b and Supplementary Figures S2 and S5). These data indicate that some genes (IKZF1 and CDKN2A) were subject to reiterated CNA in diagnosis and relapse but no CNA was preserved from diagnosis to relapse.
Clonality of immunoglobulin gene rearrangements at diagnosis and relapse
Screening for clonal IG and TCR gene rearrangements to assess clonality was performed on both the diagnostic and relapse DNA using multiplex PCR reactions and ABI GeneScan profiling. Clonal rearrangements were identified in both IGH VDJ (FR1 and 2) and IGL VJK reactions (Figure 2 and Supplementary Figure S3) with weaker clonal rearrangements observed in TCRBB/C and in TRG1 (data not shown). A V(N)JK light-chain rearrangement was shown to be identical between diagnosis and relapse (Figure 2), and the two major IGH V(N)D(N)J peaks identified at 294 and 335 bp at diagnosis were similarly shown to have identical sequences to the respective minor peaks observed at relapse (Supplementary Figure S3). However, the two major peaks identified in relapse at 330 and 341 bp were not detected in diagnostic material by conventional or quantitative PCR (Supplementary Figure S4). One interpretation of these data is that the ‘founder' IGH rearrangement present in the diagnostic samples underwent further rearrangement in relapse. Taken together, these data further suggest that the diagnostic and relapse clones may have arisen from a pre-leukemic progenitor cell already partially committed to the B-cell lineage. However, the myeloid or acute myeloblastic leukemia immunophenotype seen in relapse indicates that the leukemia was essentially ‘mixed lympho-myeloid' and may have arisen in a cell with some myeloid differentiation capacity despite the clonal IGH rearrangements present in the bulk cell progeny.
Figure 2.

Clonality of immunoglobulin light chain gene rearrangements at diagnosis and relapse. Upper panel: PCR amplifications of IGλ (IGL V(N)J) rearrangements at diagnosis and relapse were assessed by GeneScan software. Lower panel: DNA sequence analysis of the major V(N)J rearrangement identified at diagnosis shows an identical sequence to that observed at relapse. N region insertion is shown in bold italics.
Whole exome sequencing analysis
We performed whole exome sequencing on patient DNAs isolated at diagnosis, remission (germline) and relapse. All possible cross-comparisons between these three time points were assessed in the data analyses. In terms of somatic alterations, at diagnosis we identified, before filtering, a total of 2189 SNVs and 648 insertions and/or deletions. At relapse we identified 7320 SNVs and 1567 indels.
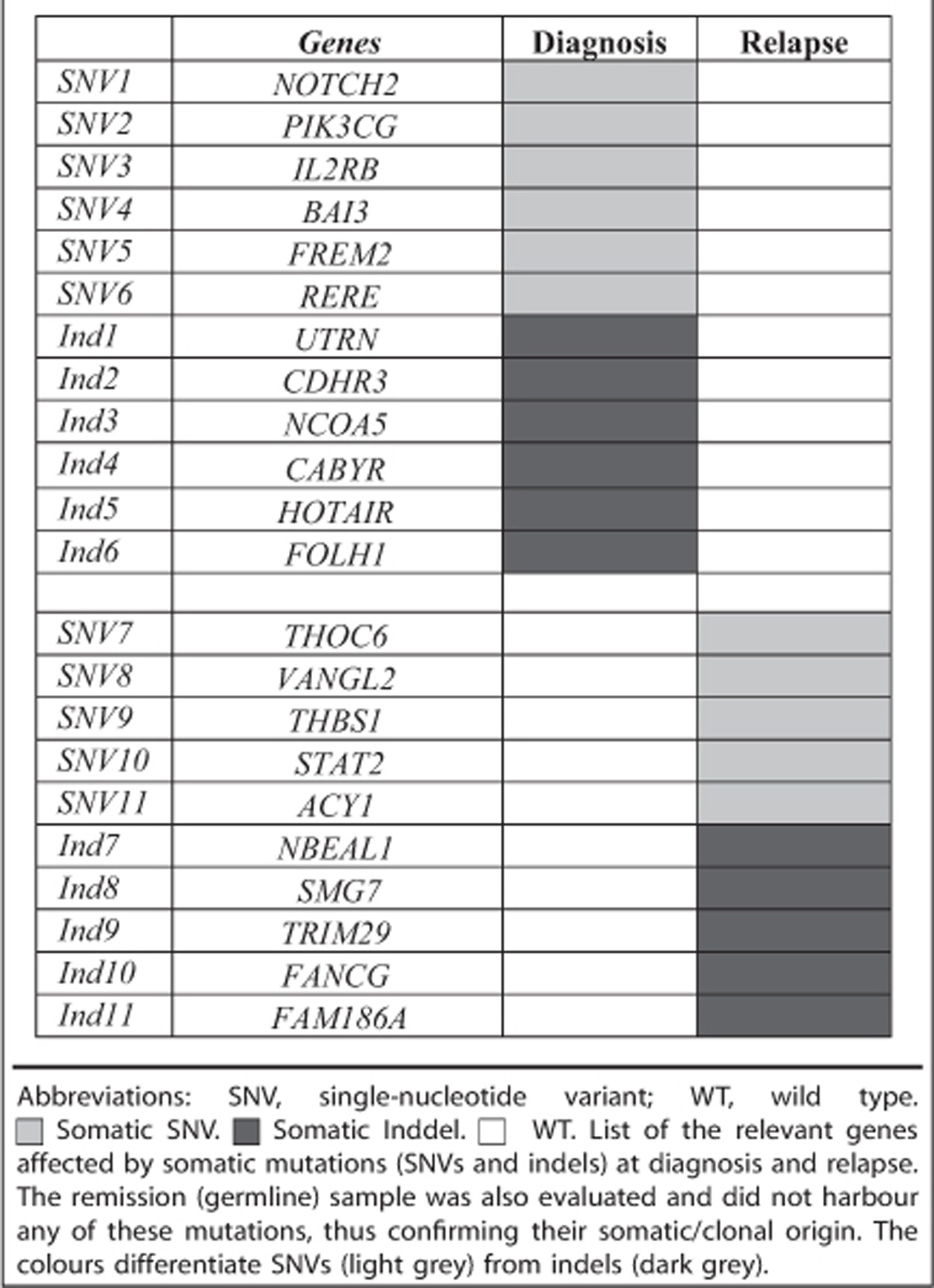
In further analyses, we highlighted relevant functional alterations. In the diagnostic sample, after filtering the data by read depth (between 30–170 ×), coding areas only and SNVs predicted to alter protein structure and deleterious/possibly damaging at the protein level (Ensembl Variant Effect Predictor), we detected 92 somatic SNVs and 59 indels. We selected those genes with functions known to be associated with cancer of which there were 12—SNVs: NOTCH2, PIK3CG, IL2RB, BAI3, FREM2 and RERE; indels: UTRN, CDHR3, NCOA5, CABYR, HOTAIR and FOLH1 (Table 2). In the relapse sample after a similar filtration, we identified 156 SNVs and 46 indels, and identified 10 potential ‘driver' genes—SNVs: THOC6, VANGL2, THBS1, STAT2 and ACY1; indels: NBEAL1, SMG7, TRIM29, FANCG and FAM186A (Table 2). All 22 genes have previously been shown to have a relevant role in tumourigenesis or have potential to be a ‘driver' of leukemogenesis. We confirmed selected heterozygous point mutations or indels by Sanger sequencing, that is, NOTCH2, HOTAIR, STAT2 and FANCG (data not shown). None were shared between the diagnostic and relapse samples, and were absent in remission (control, constitutive DNA).
Table 2. Principal mutations detected by whole exome sequencing of the patient DNA at diagnosis and relapse.
Discussion
The identity of shared and clone-specific genotypic sequences in this patient's diagnostic and very late relapse leukemia cell population provides unambiguous evidence that the relapse derived, after 22 years, from descendent progeny of the original founder clone (Figure 3). Late relapses derived from the founder diagnostic clones in ALL have been described before,9, 10 but this is the longest dormancy interval recorded with the possible exception of a case relapsing after 34 years in which the genetic evidence was very limited.21 It is striking that although the BCR-ABL1 fusion gene was identical in the paired diagnostic/relapse samples, all other genetic abnormalities detected by the single-nucleotide polymorphism arrays as CNA or by exomic sequencing as SNVs were distinctive, although the same gene was in some reiteratively mutated (for example, CDKN2A and IKZF1). Reiterative CNA have been reported before in ALL22, 23 and the predominant mutational mechanism for these structural changes appears to be driven by the lymphoid recombinases RAG1/2.24 SNV in ALL have a different mutational mechanism involving APOBECs.24 It is unclear whether the predominance of CNA as recurrent changes in ALL is a reflection of the relative activity of these different mutational mechanisms, the prevalence of different selective pressures or differential functional impacts of CNA versus SNV on cellular fitness.
Figure 3.

Model for the pre-leukemic origins of very late relapse. CNA, SNV and indel numbers—distinctive mutations found in diagnostic and relapse clones (numbers 1–13 refer to aberrations noted in Tables 1 and 2).
ALLs have multiple, genetically distinct stem cells at diagnosis.22 Our interpretation of the genomic data on this patient is that the long term surviving stem cells that spawned very late relapse derived from stem cells of a minor clone at diagnosis and most likely from a pre-leukemic clone that harboured a founder BCR-ABL1 lesion but not other secondary genetic changes (Figure 3). Evidence for such pre-malignant clones in ALL with BCR-ABL or other founder lesions have been provided by comparative genetics of monozygotic twins with discordant ALL.25, 26, 27 Sharing of identical or clonotypic BCR-ABL1 genomic fusions in monozygotic twins with concordant or discordant ALL but discordance of other genetic changes27 suggests that the BCR-ABL1 fusion in such cases is an early or likely founder or initiation event spawning a pre-leukemic clone. Limited comparative genetics had previously suggested that some late relapses in ALL might be spawned by persistent pre-leukemic clones.28, 29 Immunophenotypically or genetically defined pre-leukemic cells have previously been shown to preferentially survive chemotherapy in ALL30 and acute myeloblastic leukemia.31 Recently, Zhang et al.32 reported a relapse after 17 years in a case of acute promyelocytic leukaemia. The comparative genetics in this case was also compatible with the relapse originating from pre-leukemic stem cells.
Many mechanisms have been proposed to explain protracted clinical dormancy of cancer including balanced proliferation, cell death, non-angiogenic phenotypes, negative signalling within stromal niches maintaining cells out of cycle and immune surveillance.6, 7, 33 Whatever the prevailing restraints, a late recurrence derived from the original clone requires that cells with self-renewal or stem cell potential survive to re-establish disease. In this respect, the recognised therapeutic resistance of quiescent cancer stem cells and residence in specialised bone marrow niches33 provides a basis for their survival in a dormant state, as we assume occurred in our patient.
Adopting dormancy as a survival strategy is not unique to cancer stem cells. Normal blood stem cells fluctuate between proliferative and quiescent or out of cycle phases.34 Bacteria ‘hunker down' or adopt a non-proliferative state when confronted with stressful conditions.35 The capacity of cancer stem cells to avoid lethal therapy by switching to a dormant state can be seen as a legacy of evolutionary programming of protective mechanisms for essential normal stem cells.
The case reported here reflects an extremely rare occurrence and does not conflict with the suggestion that cure in childhood ALL can be operationally defined by remission of 4 years post cessation of treatment when the risk of relapse is <1%.10, 36
Although many very late (>20 years) recurrences or relapses of cancer have been recorded, the assumption that this reflects re-awakening of the original clone or one of its subclones requires genetic scrutiny. Provided the diagnostic sample or biopsy is archived, this can be resolved, as in the current case, by comparative genomics.
Acknowledgments
This work was supported by Leukaemia and Lymphoma Research (MG, AMF and CF), the European Hematology Association—EHA Partner Fellowship 2011/01, Instituto Nacional de Cancer—INCA and the Lady Tata Memorial Trust—LTMT International Award for Research in Leukaemia (MBM), The Kay Kendall Leukaemia Fund (FWvD) and by a Wellcome Trust Strategic Award (105104/Z/14/Z) (to MG). We thank Dr Brian Walker for help with custom-library preparation and the members of the ICR Tumour Profiling Unit for providing the targeted sequencing data on the diagnostic material.
Author contributions
MFG designed the study and wrote the manuscript. AMF co-designed and supervised the study, generated and analysed experimental data, and co-wrote the paper. MBM, CF and FWvD generated and analysed copy number variation and next-generation sequencing experimental data. JO, TS, HK and YK performed original patient analyses and provided patient material. All authors critically reviewed and approved the final draft of the manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on the Leukemia website (http://www.nature.com/leu)
Supplementary Material
References
- 1Malogolowkin M, Spreafico F, Dome JS, van Tinteren H, Pritchard-Jones K, van den Heuvel-Eibrink MM et al. Incidence and outcomes of patients with late recurrence of Wilms' tumor. Pediatr Blood Cancer 2013; 60: 1612–1615. [DOI] [PubMed] [Google Scholar]
- 2Tsao H, Cosimi AB, Sober AJ. Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer 1997; 79: 2361–2370. [PubMed] [Google Scholar]
- 3Faries MB, Steen S, Ye X, Sim M, Morton DL. Late recurrence in melanoma: clinical implications of lost dormancy. J Am Coll Surg 2013; 217: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4Mir R, Phillips SL, Schwartz G, Mathur R, Khan A, Kahn LB. Metastatic neuroblastoma after 52 years of dormancy. Cancer 1987; 60: 2510–2514. [DOI] [PubMed] [Google Scholar]
- 5Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer 2010; 10: 871–877. [DOI] [PubMed] [Google Scholar]
- 6Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell 2013; 155: 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014; 14: 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012; 488: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9Frost L, Goodeve A, Wilson G, Peake I, Barker H, Vora A. Clonal stability in late-relapsing childhood lymphoblastic leukaemia. Br J Haematol 1997; 98: 992–994. [DOI] [PubMed] [Google Scholar]
- 10Vora A, Frost L, Goodeve A, Wilson G, Ireland RM, Lilleyman J et al. Late relapsing childhood lymphoblastic leukemia. Blood 1998; 92: 2334–2337. [PubMed] [Google Scholar]
- 11Levasseur M, Maung ZT, Jackson GH, Kernahan J, Proctor SJ, Middleton PG. Relapse of acute lymphoblastic leukaemia 14 years after presentation: use of molecular techniques to confirm true re-emergency. Br J Haematol 1994; 87: 437–438. [DOI] [PubMed] [Google Scholar]
- 12Okamura J, Yamada S, Ishii E, Hara T, Takahira H, Nishimura J et al. A novel leukemia cell line, MR-87, with positive Philadelphia chromosome and negative breakpoint cluster region rearrangement coexpressing myeloid and early B-cell markers. Blood 1988; 72: 1261–1268. [PubMed] [Google Scholar]
- 13van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257–2317. [DOI] [PubMed] [Google Scholar]
- 14Langerak AW, Groenen PJ, Bruggemann M, Beldjord K, Bellan C, Bonello L et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia 2012; 26: 2159–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010; 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19Kodama Y, Okamura J, Fukano R, Nakashima K, Ito N, Nishimura M et al. Re-emerging Philadelphia chromosome-positive acute leukaemia more than 20 years after allogeneic haematopoietic stem cell transplantation. Br J Haematol 2013; 161: 286–289. [DOI] [PubMed] [Google Scholar]
- 20Chen SJ, Chen Z, Font MP, d'Auriol L, Larsen CJ, Berger R. Structural alterations of the BCR and ABL genes in Ph1 positive acute leukemias with rearrangements in the BCR gene first intron: further evidence implicating Alu sequences in the chromosome translocation. Nucleic Acids Res 1989; 17: 7631–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21Bessho F, Takayama N, Fronkova E, Zuna J. Reappearance of acute lymphoblastic leukemia 34 years after initial diagnosis: a case report and study of the origin of the reappeared blasts. Int J Hematol 2013; 97: 525–528. [DOI] [PubMed] [Google Scholar]
- 22Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011; 469: 356–361. [DOI] [PubMed] [Google Scholar]
- 23Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet 2012; 8: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014; 46: 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25Hong D, Gupta R, Ancliffe P, Atzberger A, Brown J, Soneji S et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 2008; 319: 336–339. [DOI] [PubMed] [Google Scholar]
- 26Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M et al. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 2010; 115: 3553–3558. [DOI] [PubMed] [Google Scholar]
- 27Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I et al. Developmental origins and effect of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood 2011; 118: 5559–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Ford AM, Fasching K, Panzer-Grümayer ER, Koenig M, Haas OA, Greaves MF. Origins of ‘late' relapse in childhood acute lymphoblastic leukemia with TEL-AML1 fusion genes. Blood 2001; 98: 558–564. [DOI] [PubMed] [Google Scholar]
- 29van Delft FW, Horsley S, Colman S, Anderson K, Bateman C, Kempski H et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011; 117: 6247–6254. [DOI] [PubMed] [Google Scholar]
- 30Lutz C, Woll PS, Hall G, Castor A, Dreau H, Cazzaniga G et al. Quiescent leukaemic cells account for minimal residual disease in childhood lymphoblastic leukaemia. Leukemia 2013; 27: 1204–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32Zhang X, Zhang Q, Dahlstrom J, Tran AN, Yang B, Gu Z et al. Genomic analysis of the clonal origin and evolution of acute promyelocytic leukemia in a unique patient with a very late (17 years) relapse. Leukemia 2014; 28: 1751–1754. [DOI] [PubMed] [Google Scholar]
- 33Sneddon JB, Werb Z. Location, location, location: the cancer stem cell niche. Cell Stem Cell 2007; 1: 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008; 135: 1118–1129. [DOI] [PubMed] [Google Scholar]
- 35Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 2007; 5: 48–56. [DOI] [PubMed] [Google Scholar]
- 36Pui CH, Pei D, Campana D, Cheng C, Sandlund JT, Bowman WP et al. A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 2014; 28: 2336–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.