

Abstract
Huntington disease (HD) reflects the dominant consequences of a CAG-repeat expansion in HTT. Analysis of common SNP-based haplotypes has revealed that most European HD subjects have distinguishable HTT haplotypes on their normal and disease chromosomes and that ∼50% of the latter share the same major HD haplotype. We reasoned that sequence-level investigation of this founder haplotype could provide significant insights into the history of HD and valuable information for gene-targeting approaches. Consequently, we performed whole-genome sequencing of HD and control subjects from four independent families in whom the major European HD haplotype segregates with the disease. Analysis of the full-sequence-based HTT haplotype indicated that these four families share a common ancestor sufficiently distant to have permitted the accumulation of family-specific variants. Confirmation of new CAG-expansion mutations on this haplotype suggests that unlike most founders of human disease, the common ancestor of HD-affected families with the major haplotype most likely did not have HD. Further, availability of the full sequence data validated the use of SNP imputation to predict the optimal variants for capturing heterozygosity in personalized allele-specific gene-silencing approaches. As few as ten SNPs are capable of revealing heterozygosity in more than 97% of European HD subjects. Extension of allele-specific silencing strategies to the few remaining homozygous individuals is likely to be achievable through additional known SNPs and discovery of private variants by complete sequencing of HTT. These data suggest that the current development of gene-based targeting for HD could be extended to personalized allele-specific approaches in essentially all HD individuals of European ancestry.
Introduction
Huntington disease (HD [MIM: 143100]) is a dominantly inherited neurodegenerative disorder characterized by involuntary movements, motor deficits, cognitive decline, and psychiatric disturbance.1,2 The genetic cause of the disease is expansion to over 35 units of a CAG trinucleotide repeat in the coding sequence of huntingtin (HTT [MIM: 613004]).3 The HTT CAG repeat tract displays both germline and tissue-specific somatic instability,4–7 but the size of the CAG repeat inherited by an individual is the primary determinant of the rate at which the pathogenic process will lead to their clinical diagnosis.3,8–12 There is currently no effective therapy for preventing the onset or delaying the progression of HD, but there is considerable interest in exploring gene-based approaches to suppress production of mutant huntingtin mRNA or protein, given that the dominant disease allele itself might be the most attractive target for effective therapeutic intervention.13 As part of our ongoing effort to characterize HTT, we previously defined the seven most frequent disease-associated HTT haplotypes (here designated hap.01–hap.07) on European HD chromosomes by using 20 common SNPs and one indel (rs149109767 or delta2642: a 3 bp deletion allele at HTT codon 2642; Figure S1). This revealed that a specific haplotype, here referred to as hap.01, accounts for ∼50% of HD chromosomes among individuals of European ancestry and that the region shared by about half of these individuals extends to almost 1 Mb, consistent with a low rate of recombination in the HTT region.14 The same haplotype is less common among normal chromosomes in that it accounts for approximately 9%.14 The eighth most frequent HD-associated haplotype, designated here as hap.08, is present on only ∼2.1% of HD chromosomes but is the most frequent HTT haplotype in the normal European population in that it accounts for ∼26.1% of control chromosomes. With respect to HD chromosomes, there is a relatively strong founder effect, given that hap.01, marked by the delta2642 deletion allele, is present in ∼50% of Europeans with HD. hap.05, the only other frequent disease-associated haplotype bearing the delta2642 deletion allele, comprises an additional ∼5% of disease chromosomes ancestrally related to hap.01, given that they differ at only a single terminal SNP.14 We have now completed a much finer-resolution characterization of this major European HD haplotype by using complete-sequence analysis to (1) fully define it at the nucleotide level, (2) compare it between members of different HD-affected families, (3) compare it to DNA sequences from ancient Europeans, (4) contrast it with the most common HTT haplotype on normal European chromosomes, and (5) provide foundational information for investigating HTT-sequence-dependent allele-specific interventions in HD.
Material and Methods
Whole-Genome Sequencing
DNA samples of 38 individuals from four independent HD-affected families (Figure 1) in whom hap.01 segregates with disease were used for whole-genome sequencing at Complete Genomics, which generated variant-calling files containing sequence variants with confidence scores. Because we aimed to maximize SNP discovery and the inclusion of related samples provided the opportunity to assess sequencing errors, we examined all variants originally reported by Complete Genomics.
Figure 1.

Pedigree Structures of Four Families Used for Haplotype Analysis
Four independent families in whom the hap.01 haplotype segregates with disease were analyzed by whole-genome sequencing followed by haplotype phasing. Of the 38 individuals sequenced, 29 are shown here, permitting the haplotype-phasing analysis of 16 father-mother-child trios (numbered by family and child; filled and open symbols represent subjects with an expanded HTT CAG repeat and normal individuals, respectively). CAG repeats of study subjects are provided under pedigree symbols. Small CAG differences between parent and child pairs (e.g., ±2) are within the margin of error of the CAG genotyping assay, which takes the highest peak among neighbor peaks as the repeat size of the sample.
Haplotyping by Trio Phasing
We performed haplotype phasing by using father-mother-child trio data (summarized in Table 1). We phased the sequence across HTT, plus 10 kb flanking regions at both 5′ and 3′ ends (chr4: 3,066,408–3,255,687; hg19 assembly, UCSC Genome Browser). Trios with a HD parent, normal spouse, and HD or normal child were identified in each family. In some cases, the same parents were members of different trios because full siblings were analyzed. Data pre-processing and trio phasing were performed for each trio independently. In brief, a site (i.e., genetic locus) was eliminated in a given trio if (1) genotypes in all three individuals were unknown, (2) all three individuals were identically heterozygous, or (3) a Mendelian error was detected. Because we aimed to maximize SNP coverage, we included sites with partially missing data even though this makes detection of Mendelian errors difficult. Thus, detecting Mendelian errors was based on checking the genotype data for the following: (1) variant sites without any missing data, (2) sites with no missing data in the child and one or two alleles missing in only one parent, and (3) sites with one allele missing in the child and none missing in the parents. This detection pipeline could still miss some Mendelian errors as a result of missing genotypes, but these errors could be further identified by subsequent merging of multiple phased haplotypes in a given family. After removing sites with Mendelian errors, sites that could not be confidently phased because of missing genotypes, and sites heterozygous in all three members of the trio, we phased the alleles at all remaining sites to produce a detailed haplotype.
Table 1.
Summary of Haplotype Phasing
| Number of Trios | Total Sites | All Missing Sites | Sites of Mendelian Errors | Unphaseable Sites | Inconsistent Sites | Phased Sites | |
|---|---|---|---|---|---|---|---|
| Family 1 | 5 | 189,280 | 1,016 | 21 | 3,282 | 3 | 187,363 (98.98%) |
| Family 2 | 4 | 189,280 | 1,087 | 2 | 3,246 | 2 | 186,505 (98.53%) |
| Family 3 | 3 | 189,280 | 940 | 6 | 789 | 3 | 187,582 (99.10%) |
| Family 4 | 4 | 189,280 | 858 | 39 | 1,691 | 11 | 187,540 (99.08%) |
For a given family, phased haplotypes were generated for 189,280 contiguous sites (chr4: 3,066,408–3,255,687; hg19 assembly) in complete trios from Figure 1. Missing sites were those not called in any trio member. Unphaseable sites were (1) variants heterozygous in all three trio members and (2) variants that could not be phased because of missing genotypes. Inconsistent sites had different phased allele calls within a family.
After data pre-processing, we phased trio genotype data by using the BEAGLE program and further analyzed the HD chromosome haplotypes. Depending on the number of trios in a given family, we obtained multiple HD chromosome haplotypes, and we merged these within each family to discover any inconsistent alleles. Locations and numbers of Mendelian errors and inconsistent alleles are summarized in Table 1 and Figure S4. For each family, we then finalized the HD chromosome haplotype by (1) taking alleles for sites with at least two phased allele calls, (2) assigning “N” to sites that were unphaseable or had a missing genotype or Mendelian errors, and (3) assigning “?” to the inconsistent sites. These procedures yielded phased haplotypes covering at least 98.5% of the bases in the region (summarized in Table 1). We then compared the finalized HD chromosome haplotypes from all four families to identify seven family-specific alleles. For those seven sites, we examined allele frequency in Kaviar315 and performed validation experiments using Sanger sequencing or SNP genotyping. Four of these seven SNPs (rs141511796, rs187059132, rs144933628, rs2798226) were known, and three were not present in dbSNP, the 1000 Genomes Project, or Kaviar3. The latter (rs776711851, rs750632134, and rs765413190) were submitted to NCBI dbSNP for release in build 144.
Comparison to Ancient European Genomes
We compared the consensus hap.01 haplotype sequence to 69 ancient European DNA samples16 for the 20 SNP sites called in the latter within chr4: 3,066,408–3,255,687 (hg19). First, we phased all samples from that study, including ancient and modern DNAs, for those 20 SNPs by using the MACH program in order to obtain haplotypes. Subsequently, we compared the phased haplotypes of 69 ancient DNA samples to hap.01 alleles at those 20 SNP sites.
Identification of Recombination Events
We applied haplotype phasing to the same trio data by using only SNP sites without any missing alleles to identify recombination on the HD chromosome. We analyzed a 1.5 Mb genomic region (chr4: 2,500,000–4,000,000). For a given family, we compared transmitted disease chromosomes (i.e., those with an expanded CAG in HTT) to identify discordant alleles. Discordance between alleles on the disease chromosomes inherited by one sibling and those inherited by the others indicates recombination on the parent’s disease chromosome. When such a discordant site was identified in a child, we compared the parent’s transmitted disease chromosome and non-transmitted normal chromosome, reconstructed from genotypes of the concordant progeny, to the discordant child’s inherited disease chromosome across the flanking region. We examined sites heterozygous in the CAG-expanded parent to localize the region of recombination. By this approach, we identified two recombination events and subsequently compared them to HapMap recombination rates.
Analysis of SNP Heterozygosity
We analyzed SNP heterozygosity by using 1000 Genomes Project imputed genotype data to determine the limit of applicability of allele-specific gene silencing. 2,803 subjects were genotyped on the Illumina HumanOmni2.5-8 v.1 array at the Center for Inherited Disease Research. Study subjects included HD and normal individuals from the Massachusetts General Hospital collection and the PREDICT-HD study.17,18 Genotype data and documents from quality-control analysis are available at dbGaP: phs000371. In brief, we subjected original genotype data to quality-control analysis to generate high-quality genotype data (e.g., sample genotyping call rate > 95%, SNP genotyping call rate > 95%, SNP minor allele frequency > 1%, and SNP Hardy-Weinberg equilibrium p value > 0.000001). Subsequently, we imputed genotypes by using 1000 Genomes Project data as a reference panel with the MACH and MINIMAC programs.19 SNPs with high imputation quality (e.g., R2 value greater than 0.5) were analyzed. Data for 620 SNPs from 2,405 unique HD subjects (i.e., individuals with expanded CAG repeats) were analyzed for heterozygosity in HTT. Starting from all data, we calculated heterozygosity to identify the SNP that had the highest level of heterozygosity. We then excluded heterozygous individuals and re-calculated heterozygosity to identify the SNP that displayed the highest level of heterozygosity in the remaining individuals. We repeated this iteration ten times to evaluate the discriminating power and coverage for allele-specific gene silencing by (1) all SNPs, (2) exon SNPs, and (3) intron SNPs.
Results
Whole-Genome Sequencing of HD-Affected Families
To increase the resolution of hap.01 to the nucleotide level, we sequenced the whole genomes of 38 members (29 subjects with expanded HTT CAG repeats and nine normal relatives) of four HD-affected families in whom hap.01 segregates with disease. The 29 individuals used for haplotype analysis by examination of trios are shown in Figure 1. We compared polymorphisms in the genome sequences to HapMap data for genetic evidence of the ancestry of these families. As shown in the principal-component-analysis plot (Figure S2), samples from all 38 individuals (black circles) co-localize with HapMap CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) samples (red circles) and TSI (Toscani in Italia) samples (green circles), indicating that they are of European ancestry. We assessed familial relationships by using the program PLINK20 to calculate PI_HAT values, which represent the proportion of genome sharing, for all possible pairs of HD subjects. All reported familial relationships were consistent with estimated genome sharing. Average intra-family PLINK PI_HAT values (Figure S3, gray cells) ranged from 0.359 to 0.438, where variation reflects differences in family structure. However, average inter-family PLINK PI_HAT values were quite low (Figure S3, white cells), indicating no evidence of close relationships between any of the families.
A Shared Ancestral HTT Haplotype
We next selected the 189,280 bp DNA sequence spanning HTT (chr4: 3,066,408–3,255,687) for phasing in father-mother-child trios to determine haplotypes at base-pair resolution. Across the four families, it was possible to assess 16 unique trios (Figure 1). We performed data preprocessing and haplotype phasing (see Material and Methods) separately for each trio and then merged phased haplotypes for all trios within a given family to finalize a family-specific HD chromosome haplotype. For any given trio, a small number of bases could not be phased because of missing reads, Mendelian errors, or heterozygosity of all three members. A small number of called variants also appeared inconsistent between members of the same family, as shown in Figure S4. Inconsistent sites were contributed by (1) partially missing genotypes in the trio data for phasing, (2) heterozygous genotypes in both parents and a partially missing genotype in a child, (3) sequencing errors, or (4) possible somatic mutations. Overall, we were able to generate a confidently phased HD haplotype that encompassed at least 98.5% of the bases in the region in each of these four families (Table 1).
The four family-specific haplotypes were nearly identical, arguing strongly for a common ancestral origin. Using only bases that were confidently phased in all four families (185,636 sites, 98% coverage), we reconstructed the shared founder haplotype and compared it with the human reference sequence (hg19). Across HTT, 27 bases (together defining 22 site variants listed in dbSNP) differed between the hap.01 founder and the reference alleles (Figure 2). The 22 site variants include a deletion of four intronic bases, a deletion of three coding bases, and a gain of three bases in hap.01, and 19 single-nucleotide changes. Two of the differences (rs2857790 and the delta2642 indel [rs14910767]) occurred in the coding sequence, three occurred in the UTRs of the mRNA, and the remainder occurred in non-mRNA sequences (introns and non-genic flanking sequences). As in the reference sequence, in hap.01 the proline-encoding trinucleotide repeat downstream of the polymorphic and expanded glutamine-encoding CAG repeat consists of seven CCG units.21
Figure 2.
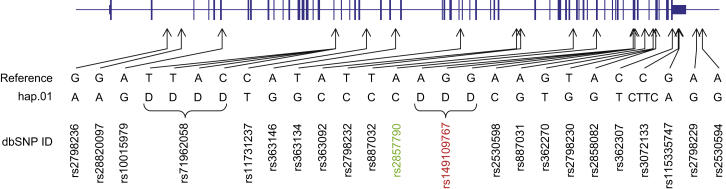
Sequence-Based Haplotype hap.01 Is Compared to the Human Reference Sequence
Site variant alleles shared by the independently established consensus hap.01 disease chromosomes of all four families were compared to the reference genome, identifying 27 differential sites, all of which are reported in dbSNP. The HTT structure is based on GenBank: NM_002111 and is depicted from left (5′) to right (3′) on the line (representing the genomic region); narrow and thickened segments represent non-coding and coding portions of the mRNA, respectively. The locations of variant sites are based on the hg19 genome assembly. “D” indicates a single-nucleotide deletion. The site noted in green is a synonymous coding variant, and a codon loss (three consecutive “D” sites) is shown in red. All other variants are non-coding.
Family-Specific Variants
Although the family-specific haplotypes were identical to the consensus ancestral haplotype at almost all of the 185,636 bases, they diverged from each other at seven variant sites. We confirmed the existence of these seven unexpected variants by additional Sanger sequencing and genotyping (Figure S5). As shown in Figure 3, these sites include three SNPs not present in dbSNP, the 1000 Genomes Project, or Kaviar3 (rs776711851, rs750632134, and rs765413190), two rare (<1% minor allele frequency) known SNPs (rs141511796 and rs187059132), and two common SNPs (rs144933628 and rs2798226) located downstream of the 3′ UTR at the extreme centromeric end of the haplotype. The accumulation (or loss in all but one family) of variants in generations subsequent to the haplotype founder could have occurred by several mechanisms. The previously unidentified SNPs most likely represent point mutations that occurred on the original haplotype, whereas the known SNPs could have resulted from either a recurrent mutation or a very limited segment of gene conversion or double recombination. Whatever the mechanism(s) of divergence, the most parsimonious scenario for the relationships between the four family-specific haplotypes is shown in Figure S6.
Figure 3.

Locations and Control-Population Frequencies of Family-Specific Variants
(A) The final hap.01 sequences revealed family-specific differences at seven sites, whose locations, genomic reference alleles, and disease-chromosome alleles for each family are shown.
(B) Kaviar3 data were used for obtaining allele frequencies for the seven family-specific variants, shown as pie charts. In three cases, alleles for variations not seen previously are shown at 0% frequency in the pie chart.
The accumulation of family-specific variants on an otherwise identical haplotype suggested an ancient origin for hap.01, so we examined recently reported genome-wide SNP data that included 20 SNPs across HTT for 69 ancient Europeans.16 We found that a haplotype in one individual (Motala 2) from the Swedish Mesolithic culture, dated at 5898–5531 cal BC (on the basis of direct radiocarbon dates), was identical at all 20 SNPs with hap.01, consistent with an ancient origin for this haplotype.
New CAG-Expansion Mutations
For most disorders associated with founder haplotypes, the common ancestor possessed the disease-causing mutation. For hap.01 in HD, however, the common ancestor most likely did not carry an expanded CAG repeat, given that several families carrying the delta2642 deletion allele have been previously reported to exhibit de novo expansion of a normal-length CAG allele to an expanded, HD-producing CAG allele.22 Using SNP genotyping, we confirmed in two available families that the “new” mutation to an expanded HTT CAG-repeat length associated with HD occurred on chromosomes containing either the hap.01 or the related hap.05 haplotype (Figure S7), directly documenting that the most common disease-associated haplotype can be traced back in some circumstances to recent non-HD progenitors.
Characterization of Recombination Landscape
Sequence-level haplotypes in multiple transmissions within a family provide a rare opportunity for characterizing the recombination patterns of the mutation-bearing chromosome in HD subjects. We have previously noted the sharing of extended haplotypes of 1 Mb or more in a substantial fraction of HD individuals, so we analyzed a broad region of 1.5 Mb of phased HD chromosome sequence (chr4: 2,500,000–4,000,000, shown in Figure 4) by comparing transmitted HD chromosome haplotypes within a family to identify recombination events. As previously reported in multi-allele-marker linkage studies and revealed by the HapMap recombination rate shown in Figure 4A, the HTT region shows a relatively low recombination rate, arguing against crossovers subsequent to CAG expansion as the source of the bulk of the diversity of HTT haplotypes observed on HD chromosomes.14 Proximal to HTT, recombination rates increase and show a particular hotspot at ∼3,725,000 (expanded in Figure 4B). Of 16 meiotic transmissions of the disease chromosome in the four HD-affected families, two exhibited recombination events: one in family 1 and one in family 4. Both independent events occurred within the same 3 kb segment of DNA, at the hot-spot noted above (Figures 4C and 4D). Although we examined only a limited number of meioses, the sequence-level findings within and centromeric to HTT suggest that the recombination landscape of hap.01 HD chromosomes is comparable to that of normal chromosomes and is not dramatically disrupted by the presence of an expanded CAG repeat.
Figure 4.

Recombination Events on hap.01 HD Chromosomes
(A) RefSeq genes are indicated in blue arrows above the orange traces, which represent the HapMap recombination rates (chr4: 2,500,000–4,000,000 bp region). Recombination rates are very low around HTT, and recombination peaks are located centromerically. The red arrow indicates the highest recombination peak in this region, where two recombination events were detected.
(B) In an expanded view of the highest recombination peak, the reference genomic coordinates with recombination rates are aligned with actual recombination events in (C) and (D). Both recombination events were located within the recombination peak at chr4: 3,725,000 bp.
(C) A recombination event in family 1 is shown. Five transmitted HD chromosomes were compared for chr4: 22,500,000–4,000,000, revealing a recombination event in the maternal transmission in trio 2. The mother’s disease chromosome (red) and normal chromosome (green) are compared to the transmitted disease chromosomes (red and green). Only bases at sites of heterozygosity are shown.
(D) A similar analysis to that in (C) was performed for family 4. One recombination event was detected in the paternal transmission of trio 4.
Potential for Allelic Discrimination in HD
One of the most promising avenues for exploration of HD therapies is sequence-based silencing of the mutant allele, at either the transcriptional or the translational level. Given the potential for negative consequences of loss of normal huntingtin activity, the ideal gene-silencing strategy would be specific to the mutant allele. The capacity to discriminate alleles depends not only on the disease haplotype but also on the normal HTT haplotype with which it is paired in each individual. The most frequent diplotype in European HD individuals naturally combines the most frequent HD haplotype, hap.01, and the most frequent haplotype in the normal population, hap.08, which differ at 19 of 21 sites in the common-marker-based haplotype (Figure S1). The presence of 12 hap.08 control chromosomes in our fully sequenced families (three chromosomes in family 1, three chromosomes in family 3, and six chromosomes in family 4) permitted us to also compare differences at the nucleotide level. We were able to define 97.9% of all bases in hap.08 and discovered that there were 109 differences between hap.01 and hap.08 (Figure S8). Interestingly, there were also 15 consistent differences between the normal hap.08 chromosomes defined in these three families, indicating that, like our hap.01 HD chromosomes, these control chromosomes have accumulated additional variants not found in their shared ancestor. The extensive differences between hap.01 and hap.08 offer numerous potential targets for allele-specific gene silencing. However, this diplotype accounts for only 10.4% of European HD individuals, indicating the need for a personalized approach to extend allele-specific gene-silencing strategies to the bulk of the HD population.
Our data suggest that sequencing of other HTT haplotypes could readily identify sites of genetic variation applicable to allele-specific targeting for other diplotypes. However, to estimate the potential coverage that could already be achieved through known variants, we capitalized on genome-wide SNP array data from HD subjects and 1000 Genomes Project data to impute the SNP variation across HTT in European HD individuals. We validated the approach by comparing SNP variants imputed from the array data for 24 family 1 and 2 individuals who were also fully sequenced. For 405 imputed SNPs in the HTT region, we observed a median genotype concordance of 99%, suggesting that the use of imputed SNPs can provide a relatively accurate assessment of heterozygosity in the HD population.
To identify SNPs providing optimal discrimination of HD and normal chromosomes in European HD subjects, we sequentially selected the SNP that was heterozygous in the maximum proportion of HD subjects, removed those subjects from consideration, and again selected the most heterozygous SNP. We repeated this process ten times (Table S1). The results are shown in Table 2 for HTT as a whole (chr4: 3,076,237–3,245,687; hg19), mRNA-specific SNPs, and intron-specific SNPs, for each of which is provided a list of ten SNPs capable of supporting allele-specific targeting because of their heterozygosity in 97.1%, 93.8%, and 97.0% of HD subjects, respectively. Clearly, variants already known to be polymorphic in the European population can distinguish disease chromosomes from normal chromosomes, allowing targeting of either the mRNA or genomic sequence, in the vast majority of HD heterozygotes. A very small percentage of HD subjects will be homozygous for each of these SNPs, but it is likely that going further down the list of imputed SNPs and/or personalized deep sequencing, as performed here for hap.01 and hap.08, will reveal additional variants that could make almost all HD individuals eligible for an allele-specific gene-silencing approach.
Table 2.
Allele Discrimination by Heterozygous SNPs
| Round |
Exon or Intron SNP |
Exon SNP |
Intron SNP |
||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Percentage | Cumulative Percentagea | ID | Percentage | Cumulative Percentagea | ID | Percentage | Cumulative Percentagea | |
| 1 | rs11731237 | 52.6% | 52.6% | rs362307 | 47.5% | 47.5% | rs11731237 | 52.6% | 52.6% |
| 2 | rs71597207 | 22.2% | 74.9% | rs2530595 | 23.8% | 71.3% | rs71597207 | 22.2% | 74.9% |
| 3 | rs115335747 | 10.9% | 85.8% | rs362331 | 10.9% | 82.2% | rs187059132 | 10.1% | 84.9% |
| 4 | rs3135055 | 5.4% | 91.2% | rs362267 | 3.6% | 85.8% | rs2530596 | 5.7% | 90.6% |
| 5 | rs362312 | 1.9% | 93.1% | rs116419279 | 3.0% | 88.8% | rs362312 | 2.0% | 92.6% |
| 6 | rs191103377 | 1.6% | 94.7% | rs35892913 | 2.0% | 90.8% | rs3135054 | 1.7% | 94.3% |
| 7 | rs3135054 | 0.9% | 95.6% | rs115335747 | 1.6% | 92.3% | rs57666989 | 0.9% | 95.2% |
| 8 | rs2530596 | 0.6% | 96.2% | rs2276881 | 0.6% | 93.0% | rs186403576 | 0.8% | 96.0% |
| 9 | rs186403576 | 0.5% | 96.7% | rs146151652 | 0.5% | 93.4% | rs148125464 | 0.6% | 96.5% |
| 10 | rs187059132 | 0.5% | 97.1% | rs144364475 | 0.4% | 93.8% | rs3135055 | 0.5% | 97.0% |
The cumulative percentage of HD subjects heterozygous for at least one SNP. The most frequent heterozygous SNP remaining was chosen in each round.
Discussion
Our sequence-level analysis of the major HD-associated HTT haplotype supports the previous hypothesis from HD haplotypes that multiple different origins of the CAG-expansion mutation have contributed to the European HD population.23 However, it also provides detailed molecular support for a single ancestral chromosome that contributes to 55% of HD subjects and thus represents a strong founder effect. The fact that de novo CAG expansions are observed from chromosomes with the same ancestral haplotype but with CAG repeats below the HD-producing size range indicates that the common ancestor of today’s hap.01 HD subjects was most likely an individual with a CAG repeat in the normal range, unlike the disease-producing mutations inherent to most other haplotypes associated with human disease. In the present-day population, a group of haplotypes clustered as haplogroup A, which includes hap.01, are associated with chromosomes that bear a higher-than-average proportion of high-normal (27–35 CAGs) HTT CAG repeats,23 and it has been suggested that haplogroup A might be more prone to CAG-repeat instability. However, a direct comparison of HTT CAG-repeat lengths on disease chromosomes showed no significant difference between haplotypes,14 and parent-child transmission data suggested that the intergenerational HTT CAG-repeat instability, at least of expanded alleles, is not restricted to specific haplotypes (unpublished data). The same sequencing approach used here could be applied to defining the origins of the other HD-associated haplotypes that, unlike hap.05, do not show evidence of a clear relationship with hap.01. For example, hap.02 and hap.03 are present at comparable frequencies on both disease and normal chromosomes, and sequence-level analysis could potentially identify variants that distinguish a subset of each that is enriched on disease chromosomes.
Our analysis suggests that hap.01 existed in the European population at least 7,000 years ago and that today’s HD individuals with an expanded CAG repeat on the hap.01 haplotype share a common ancestor who could be quite ancient but apparently had a normal-length CAG repeat. The sequence-level analysis of hap.01 indicates that variants occurring subsequently to this ancestor have contributed to the slight divergence of the original haplotype over an extended time period. In comparing four families in whom hap.01 segregates with disease, we found seven unexpected SNP differences. The three previously unidentified SNPs are most likely due to point mutations occurring at the average genome-wide mutation rate,24 as would be expected for an ancient haplotype. Although the two previously known rare SNPs could theoretically be present because of the same phenomenon, their presence in non-HD individuals could reflect their occurrence in an ancestor prior to CAG expansion or be due to an inherently higher-than-average mutation rate at that site. Alternatively, the alleles could have moved onto hap.01 by some other mechanism (e.g., gene conversion or local double recombination). The common SNPs downstream of the HTT 3′ UTR could theoretically have been moved independently onto hap.01 by simple recombination, although our examination of downstream SNPs in the sequencing data does not provide unequivocal evidence of such events. The possibility that rs2798226 and rs144933628 have been introduced onto hap.01 as point mutations would suggest a high mutation rate for these sites, which is not consistent with their observed disequilibrium patterns in control populations. Thus, like the appearance of the two rarer SNPs, the presence of the unexpected rs2798226 allele on hap.01 in family 1 could have resulted from another mechanism. The unexpected accumulation of these variants on a founder haplotype suggests the intriguing possibility that, in addition to de novo mutation, simple recombination, and structural alteration, other mechanisms such as gene conversion or double recombination contribute significantly to the divergence of human chromosomal haplotypes between individuals. Sequence-level analysis of many more founder haplotypes in human disease could provide a system for exploring the relative importance of different mechanisms in generating sequence variation on an initially fixed background.
There is currently no disease-modifying treatment for HD, but silencing the mutant allele by using any of a variety of technologies is an attractive route for potential intervention because it would target the actual cause of disease pathogenesis. However, for the continuing wellness of an individual with HD, it might be important to preserve wild-type HTT function. Consequently, an allele-specific gene-targeting strategy is likely to be optimal, making it critical to determine the frequency with which an HD individual has distinguishable disease and normal HTT alleles and to define the variants that provide this discrimination in the highest proportion of HD subjects.23,25,26 From our previous SNP-based definition of HTT haplotypes, the seven most frequent HD-associated haplotypes account for 83% of European disease chromosomes and 35% of normal chromosomes.14 The extended haplotypes across HTT are consistent with the very low recombination rate observed in the region. This low recombination rate also predicts that the use of genotype data from a limited number of SNPs should provide accurate imputation of the large number of SNPs known to exist in the European population. Our full-sequence analysis of the HTT region permitted us to directly confirm this assumption, making it possible to use imputed SNPs to assess heterozygosity across the locus. Our data provide estimates of the frequency of heterozygosity in European HD subjects for 1000 Genomes population variants and identify those SNPs with the greatest discriminatory power between HD and normal chromosomes, maximizing the potential for allele-specific targeting approaches aimed at either mRNA or genomic DNA. In the vast majority of cases, the diplotype involves two different haplotypes, and known SNPs can provide the basis for distinguishing between normal and CAG-expanded versions of HTT. In the few remaining cases, personalized sequence analysis is very likely to reveal a discriminating variant. Indeed, in the families studied here, two HD individuals bear two copies of the SNP-defined hap.01 haplotype, but our full-sequence analysis revealed two sites with different alleles on their HD and normal chromosomes. Overall, our sequence-level haplotype analyses led to the encouraging conclusion that virtually all European HD individuals are candidates for allele-specific gene-silencing techniques in a personalized medicine approach and suggest that a similar sequence-based strategy can be readily applied to extend these techniques to other populations.
Acknowledgments
We thank the Huntington-disease-affected families who contributed their time and blood samples to make this work possible. Support was provided by grants from the National Institute of Neurological Disorders and Stroke (“Huntington’s Disease Center Without Walls,” P50NS016367, U01NS082079, R01NS091161), the Center for Systems Biology P50 (GM076547, NIH), the University of Luxembourg Institute for Systems Biology Program, and the Huntington’s Disease Society of America’s Coalition for the Cure. Additional support came from the Taube/Koret Center for Neurodegenerative Diseases (S.F.), the Gladstone Institutes (S.F.), and a grant from the NIH (X01 HG007750 to S.F.).
Published: August 27, 2015
Footnotes
Supplemental Data include eight figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2015.07.017.
Accession Numbers
The accession numbers for the three SNPs reported in this paper are dbSNP: rs776711851, rs750632134, and rs765413190.
Web Resources
The URLs for data presented herein are as follows:
BEAGLE, http://faculty.washington.edu/browning/beagle/beagle.html
Complete Genomics, http://www.completegenomics.com
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Kaviar, http://db.systemsbiology.net/kaviar/cgi-pub/Kaviar.pl
OMIM, http://www.omim.org/
UCSC Genome Browser, https://genome.ucsc.edu/
Supplemental Data
References
- 1.Huntington G. On chorea. Med. Surg. Rep. 1872;26:320–321. [Google Scholar]
- 2.Schoenfeld M., Myers R.H., Cupples L.A., Berkman B., Sax D.S., Clark E. Increased rate of suicide among patients with Huntington’s disease. J. Neurol. Neurosurg. Psychiatry. 1984;47:1283–1287. doi: 10.1136/jnnp.47.12.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy L., Evans E., Chen C.M., Craven L., Detloff P.J., Ennis M., Shelbourne P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy L., Shelbourne P.F. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum. Mol. Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- 6.Lee J.M., Zhang J., Su A.I., Walker J.R., Wiltshire T., Kang K., Dragileva E., Gillis T., Lopez E.T., Boily M.J. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst. Biol. 2010;4:29. doi: 10.1186/1752-0509-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum. Mol. Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 8.Andrew S.E., Goldberg Y.P., Kremer B., Telenius H., Theilmann J., Adam S., Starr E., Squitieri F., Lin B., Kalchman M.A. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 9.Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M., Abbott M. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 10.Lee J.M., Ramos E.M., Lee J.H., Gillis T., Mysore J.S., Hayden M.R., Warby S.C., Morrison P., Nance M., Ross C.A., PREDICT-HD study of the Huntington Study Group (HSG) REGISTRY study of the European Huntington’s Disease Network. HD-MAPS Study Group. COHORT study of the HSG CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–695. doi: 10.1212/WNL.0b013e318249f683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Persichetti F., Srinidhi J., Kanaley L., Ge P., Myers R.H., D’Arrigo K., Barnes G.T., MacDonald M.E., Vonsattel J.P., Gusella J.F. Huntington’s disease CAG trinucleotide repeats in pathologically confirmed post-mortem brains. Neurobiol. Dis. 1994;1:159–166. doi: 10.1006/nbdi.1994.0019. [DOI] [PubMed] [Google Scholar]
- 12.Snell R.G., MacMillan J.C., Cheadle J.P., Fenton I., Lazarou L.P., Davies P., MacDonald M.E., Gusella J.F., Harper P.S., Shaw D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- 13.Aronin N., DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Mov. Disord. 2014;29:1455–1461. doi: 10.1002/mds.26020. [DOI] [PubMed] [Google Scholar]
- 14.Lee J.M., Gillis T., Mysore J.S., Ramos E.M., Myers R.H., Hayden M.R., Morrison P.J., Nance M., Ross C.A., Margolis R.L. Common SNP-based haplotype analysis of the 4p16.3 Huntington disease gene region. Am. J. Hum. Genet. 2012;90:434–444. doi: 10.1016/j.ajhg.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glusman G., Caballero J., Mauldin D.E., Hood L., Roach J.C. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. 2011;27:3216–3217. doi: 10.1093/bioinformatics/btr540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haak W., Lazaridis I., Patterson N., Rohland N., Mallick S., Llamas B., Brandt G., Nordenfelt S., Harney E., Stewardson K. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–211. doi: 10.1038/nature14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen J.S., Hayden M., Stout J.C., Langbehn D.R., Aylward E., Ross C.A., Guttman M., Nance M., Kieburtz K., Oakes D., Predict-HD Investigators of the Huntington Study Group Preparing for preventive clinical trials: the Predict-HD study. Arch. Neurol. 2006;63:883–890. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- 18.Paulsen J.S., Langbehn D.R., Stout J.C., Aylward E., Ross C.A., Nance M., Guttman M., Johnson S., MacDonald M., Beglinger L.J., Predict-HD Investigators and Coordinators of the Huntington Study Group Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almqvist E., Spence N., Nichol K., Andrew S.E., Vesa J., Peltonen L., Anvret M., Goto J., Kanazawa I., Goldberg Y.P. Ancestral differences in the distribution of the delta 2642 glutamic acid polymorphism is associated with varying CAG repeat lengths on normal chromosomes: insights into the genetic evolution of Huntington disease. Hum. Mol. Genet. 1995;4:207–214. doi: 10.1093/hmg/4.2.207. [DOI] [PubMed] [Google Scholar]
- 22.Myers R.H., MacDonald M.E., Koroshetz W.J., Duyao M.P., Ambrose C.M., Taylor S.A., Barnes G., Srinidhi J., Lin C.S., Whaley W.L. De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat. Genet. 1993;5:168–173. doi: 10.1038/ng1093-168. [DOI] [PubMed] [Google Scholar]
- 23.Warby S.C., Montpetit A., Hayden A.R., Carroll J.B., Butland S.L., Visscher H., Collins J.A., Semaka A., Hudson T.J., Hayden M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009;84:351–366. doi: 10.1016/j.ajhg.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conrad D.F., Keebler J.E., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll J.B., Warby S.C., Southwell A.L., Doty C.N., Greenlee S., Skotte N., Hung G., Bennett C.F., Freier S.M., Hayden M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol. Ther. 2011;19:2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfister E.L., Kennington L., Straubhaar J., Wagh S., Liu W., DiFiglia M., Landwehrmeyer B., Vonsattel J.P., Zamore P.D., Aronin N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 2009;19:774–778. doi: 10.1016/j.cub.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.