

Abstract
CHAMP1 encodes a protein with a function in kinetochore-microtubule attachment and in the regulation of chromosome segregation, both of which are known to be important for neurodevelopment. By trio whole-exome sequencing, we have identified de novo deleterious mutations in CHAMP1 in five unrelated individuals affected by intellectual disability with severe speech impairment, motor developmental delay, muscular hypotonia, and similar dysmorphic features including short philtrum and a tented upper and everted lover lip. In addition to two frameshift and one nonsense mutations, we found an identical nonsense mutation, c.1192C>T (p.Arg398∗), in two affected individuals. All mutations, if resulting in a stable protein, are predicted to lead to the loss of the functionally important zinc-finger domains in the C terminus of the protein, which regulate CHAMP1 localization to chromosomes and the mitotic spindle, thereby providing a mechanistic understanding for their pathogenicity. We thus establish deleterious de novo mutations in CHAMP1 as a cause of intellectual disability.
Main Text
Intellectual disability (ID) is defined as substantial impairment of cognitive and adaptive functions with an onset in childhood and has an estimated prevalence of 1.5%–2.0%.1 The rapid development of new technologies, including next-generation sequencing, has allowed for the elucidation of a large number of previously undiagnosed Mendelian disorders during recent years. This holds especially true for children with seemingly sporadic, non-syndromic or mild dysmorphic forms of intellectual disability and/or global developmental delay (ID/GDD) who could not be analyzed systematically on a genome-wide level before the advent of chromosomal microarrays and whole-exome or genome sequencing. Indeed, recent studies have identified pathogenic de novo mutations in a rapidly growing number of individuals with ID/GDD and thus have established the power of proband-parent trio whole-exome sequencing in particular.2–4
Here, we report five unrelated individuals, three males and two females, with ID/GDD, severe speech impairment, and similar (though subtle) facial dysmorphisms (Table 1; Figure 1). In all five individuals, trio whole-exome sequencing identified causative de novo frameshift or nonsense mutations in CHAMP1 (chromosome alignment-maintaining phosphoprotein 1; also called CAMP, ZNF828, or C13orf8). CHAMP1 (GenBank: NM_032436.2; MIM: 616327) encodes a zinc-finger protein, which is involved in the maintenance of kinetochore-microtubule attachment during mitosis and the regulation of accurate chromosome segregation,5 both known to be crucial for cortical and mental development.6 All biological samples and images were obtained after written informed consent was given by the parents of the affected individuals. The study was performed in accordance with the Declaration of Helsinki protocols and approved by the ethics committees of the respective institutions.
Table 1.
Clinical Characteristics of Individuals with De Novo CHAMP1 Mutations Presented Here
| Individual A:II-1 | Individual B:II-3 | Individual C:II-2 | Individual D:II-2 | Individual E:II-2 | |
|---|---|---|---|---|---|
| Sex | male | male | male | female | female |
| Birth at gestational week | 40 | 39 | 39 | 37 | 40 |
| Birth weight (g/SD) | 3,520/−0.4 | 3,580/+0.2 | 3,100/−0.9 | 3,105/+0.3 | 4,100/+1.5 |
| Birth length (cm/SD) | 52/−0.2 | ND | 49/−1.3 | 50/+1.0 | 53/+0.6 |
| OFC at birth (cm/SD) | 33/−2.0 | ND | 35.6/−2.1 (age 6 weeks) |
29.5/−3.1 | 35/−0.4 (age 4 weeks) |
| Age at last examination (years) | 4 | 3 | 18 | 3 | 8 |
| Weight at last examination (kg/SD) |
16.3/+0.2 | 14.7/0 | 50/−2.7 | 14.7/−0.1 | 45.5/+3.3 |
| Length at last examination (cm/SD) |
111.5/+0.7 | 86/−2.2 | 160/−2.9 | 93.5/−0.9 | 139/+0.6 |
| OFC at last examination (cm/SD) |
48/−2.5 | 47.3/−1.5 | 52.2/−3.1 | 45.5/−2.4 | 52/−0.3 |
| Neurological Signs | |||||
| Muscular hypotonia | + | + | + | + | + |
| Motor development delay | + | + | + | + | + |
| Age of walking without support (months) | 48 | 36 | 30 | 18 | 20 |
| Impaired speech development | + | + | + | + | + |
| Dysarthria, speech defect | no speech | no speech | + | + | + |
| Intellectual disability | severe | severe | severe | moderate | moderate |
| Friendly behavior | + | + | + | + | + |
| Craniofacial Abnormalities | |||||
| Orofacial hypotonia | + | − | + | + | + |
| Long face | + | − | + | − | + |
| Epicanthic folds | − | + | − | + | + |
| Upslanting palpebral fissures | − | + | + | − | + |
| Short philtrum | + | + | + | + | + |
| Open-mouth appearance and salivation | + | − | + | + | + |
| Thin and tented upper lip | + | tented (not thin) |
+ | + | + |
| Everted lower lip | + | + | + | + | + |
| High arched palate | + | + | ND | + | + |
| Pointed chin | + | + | − | − | + |
| Low set ears | − | + | + | − | + |
| Other Findings | |||||
| Strabismus | + | − | + | − | − |
| Hyperopia | + | ND | + | + | + |
| Joint hypermobility | + | − | + | − | − |
| Neonatal feeding difficulties | + | + | + | + | − |
| Umbilical hernia | + | + | − | − | − |
| Decreased pain sensation | + | − | + | + | + |
| Recurrent upper air tract infections | − | + | + | + | − |
| Further Explorations | |||||
| Cerebral MRI | mild brain atrophy and cerebellar cortical dysplasia | slightly delayed myelination | normal (age one year) |
normal (age three months) |
normal (age three years) |
| CHAMP1 Mutation | |||||
| Gene mutation, protein alteration | c.1866_1867delCA, p.Asp622Glufs∗8 |
c.1768C>T, p.Gln590∗ |
c.1192C>T, p.Arg398∗ |
c.635delC, p.Pro212Leufs∗7 |
c.1192C>T, p.Arg398∗ |
Abbreviations are as follows: +, present; −, absent; ND, not done
Figure 1.

Facial Phenotype of Individuals with CHAMP1-Associated Disorder
Facial images of individual A:II-1 at age 4 years (A), individual B:II-3 at the age of 12 months and 6 years (B), individual C:II-2 from the age of 6 till 18 years (C), individual D:II-2 at birth and at age 3 years (D), and individual E:II-2 at the age 4 and 9 years (E). Note the long face, orofacial hypotonia, epicanthic folds, upslanting palpebral fissures, short philtrum, thin and tented upper lip and everted lower lip, pointed chin, and deep-set ears.
The first proband (individual II-1 of family A) is the first child of healthy unrelated parents of European descent. He was born after an uneventful pregnancy at 40 weeks of gestation with normal birth weight and length and a borderline occipitofrontal head circumference (OFC) of 33 cm (−2 SD). Feeding difficulties, vomiting, and weight loss characterized the early postnatal period. Developmental delay, muscular hypotonia, and strabismus first became evident at the age of 3 months. Hyperopia was diagnosed at the age of 9 months. All milestones of motor development were delayed; he was rolling over at 12 months, sitting at 12 months, crawling at 30 months, and walking at 4 years. At the age of four years, he displayed severe developmental delay with microcephaly (OFC −2.2 SD) and truncal and orofacial hypotonia. His gait was slightly ataxic, and he displayed stereotypic movements including frequent hand fluttering and jactitation. His behavior was very friendly and open-minded. In addition to joint hypermobility and an umbilical hernia, mild dysmorphic features were observed, including a flattened occiput, long face, short nose with anteverted nares, short philtrum, thin and tented upper lip, everted lower lip, pointed chin, high arched palate, and dysplastic teeth (Figure 1A). Language development was significantly delayed and limited to three words at the age of 4 years. His parents additionally observed decreased pain sensation. A brain MRI, performed at the age of 4 years, revealed mild generalized atrophy of the brain, a simplified gyral pattern, and mild cerebellar cortical dysplasia resembling a mild form of partial rhombencephalosynapsis with fused superior posterior cerebellar hemispheres but separated inferior anterior cerebellar hemispheres (Figure S1). Conventional chromosome analysis of lymphocytes, interphase fluorescence in situ hybridization (FISH) analysis in buccal swab cells, array comparative genomic hybridization (CGH), SNRPN-locus methylation analysis (MIM: 182279), and direct sequencing of ARX (MIM: 300382) were performed, all of which gave normal results. This prompted us to perform trio exome sequencing with DNA samples of both healthy parents and the proband, as described before.2 In brief, coding DNA fragments were enriched with a SureSelect Human All Exon 50Mb V5 Kit (Agilent), and sequencing was performed on a HiSeq2500 system (Illumina). Reads were aligned to the human genome assembly hg19 (UCSC Genome Browser) with the Burrows-Wheeler Aligner (BWA, v.0.5.87.5), and detection of genetic variation was performed with SAMtools (v.0.1.18), PINDEL (v. 0.2.4t), and ExomeDepth (v.1.0.0). Bioinformatic filtering did not detect rare candidate variants (minor allele frequency [MAF] < 0.01) following an autosomal-recessive or X-linked recessive mode of inheritance. However, we identified a single non-annotated, de novo sequence change with a severe impact on protein structure (Table S1A). This 2 bp deletion at cDNA position 1,866 bp (c.1866_1867delCA) in CHAMP1 is predicted to change the reading frame and to induce a premature stop codon at amino acid 629 (p.Asp622Glufs∗8). Sanger sequencing confirmed the heterozygous c.1866_1867delCA mutation in the proband (Figure S2) and its absence from blood DNA of both parents.
The second proband (individual II-3 of family B) is the third child of non-consanguineous Dutch parents. The boy was born after an unremarkable pregnancy at 39 weeks of gestation. His birth weight was normal. In the neonatal period, he had feeding difficulties and was tube-fed for six weeks. A delayed development and impaired vision was noted in the first month of his life. A brain MRI at the age of 3 months showed slightly delayed myelination. He was affected by severe hypotonia, which improved over time. In addition, he suffered from recurrent upper respiratory tract infections and aspiration pneumonias. At the age of 2 years, an examination revealed several dysmorphic features, including brachycephaly, a low frontal hairline, hypertelorism, epicanthic folds, and a broad nasal bridge. His height was at −2.2 SD, whereas his weight and his OFC were within the normal range. He was able to stand with some support at the age of 2.5 years and walk a few steps without support but with insufficient head balance at the age of 3 years. He showed stereotypic behavior characterized by turning, twisting movements of the arms and hands, sighing, and shaking of his body. Nocturnal frontotemporal epilepsy, probably causative for sleep apnea, was successfully treated with carbamazepine. Even though he has low vision with a suboptimal visual evoked potential and normal electroretinogram, the cause of his vision impairment is still unknown. At the age of 5 years, a re-examination showed a very cheerful boy with stereotypies as described above, no speech and a frontal upsweep of the hair, almond-shaped eyes, a high palate, small teeth, and diasthema, in addition to the previously mentioned dysmorphic features (Figure 1B). He had also developed an umbilical hernia. Over time, basic metabolic studies, sequence analysis of ATRX (MIM: 300032), SNP array, and methylation studies for Prader-Willi syndrome (MIM: 176270) yielded normal results. We therefore performed trio-exome sequencing with DNA samples of the parents and the proband, as described before.3 In brief, exome-sequencing was performed with a SOLiD 5500XL machine (Life Technologies) after enrichment with the Agilent SureSelectXT Human All Exon 50Mb Kit (Agilent). The data were analyzed with LifeScope software (Applied Biosystems, Life Technologies). A de novo analysis was performed with the parents and proband’s DNA, identifying a single non-annotated, de novo sequence change with a severe impact on protein structure (Table S1B). This was single-nucleotide variant (SNV) c.1768C>T in CHAMP1, resulting in a nonsense mutation at amino acid 590 (p.Gln590∗). Sanger sequencing confirmed the heterozygous c.1768C>T mutation in the proband (Figure S3) and its absence from blood DNA of both parents.
The third proband (individual II-2 of family C) is an 18-year-old male, the second of three children born to non-consanguineous Dutch parents. He was born at gestational week 39 after an uneventful pregnancy and with a normal birth weight and height. OFC at age 6 weeks was −2.1 SD. Hypothermia and feeding problems characterized the first days of life and esophageal reflux was suspected. Hypotonia and developmental delay became obvious in the first months. At the age of 1 year, developmental delay and feeding problems associated with weight loss led to an extensive clinical work up, revealing normal results in metabolic analysis, MRI of the brain, determination of bone-age, skull X-ray, and pH-metry. Esophegaeal motility analysis showed diminished swallowing movements, but excluded a structural anomaly. Intermittent exotropia and high hyperopia (+6.25 diopters (dpt), bilaterally) were diagnosed. Clinical examination revealed severe flattening of the occiput and increased spacing of his large and prominent upper incisors. Feeding improved over time. He suffered from repeated upper airway infections and gastritis. Motor milestones were delayed, including rolling over at 9 months, sitting at 13 months, and walking at 30 months. His language development was also significantly delayed. He spoke first words at the age of 3 years. Repeated elevation of pipecolic acid in serum prompted an extensive metabolic workup, including analysis in colony-stimulating factor, fibroblast, and liver tissue. Normal results made a peroxisomal defect unlikely. At the last visit at the age of 18 years, his weight, length, and OFC were below the normal range (weight −2.7 SD; height −2.9 SD; OFC −3.1 SD). Autism had been diagnosed. He spoke in short sentences with slurred speech. He loved music and swimming and was able to operate his computer well. His behavior was friendly. Parents reported a decreased pain sensation. Clinical examination revealed muscular hypotonia, in particular orofacial hypotonia with open-mouth appearance and salivation, a long face, upslanting palpebral fissures, a short philtrum, a thin upper lip and everted lower lip, a pointed chin, and low set ears (Figure 1C). Over the years, karyotyping, array CGH, and FISH and methylation testing on chromosomal region 15q11q13, as well as analyses of ARX, VPS13B (MIM: 607817), and UBE3A (MIM: 601623) had been performed without pathogenic findings. This prompted us to perform diagnostic trio exome sequencing with DNA samples of the proband and both parents. Exomes were enriched with the SureSelect XT Human All Exon V5 Kit (Agilent) and sequenced in rapid run mode on the HiSeq2500 sequencing system (Illumina). Reads were aligned to hg19 with the BWA (BWA-MEM v.0.7.5a) and variants were called with the Genome Analysis Toolkit haplotype caller (v.2.7-2). Detected variants were annotated, filtered, and prioritized with the Bench Lab NGS platform (Cartagenia). De novo mutation analysis, by filtering all detected variants against parental and population variants, identified a single non-annotated, de novo sequence change with a severe impact on protein structure (Table S1C). This was the CHAMP1 SNV c.1192C>T resulting in a nonsense mutation at amino acid 398 (p.Arg398∗). The heterozygous c.1192C>T mutation was validated and proven to be de novo by Sanger sequencing (data not shown).
The fourth proband (individual II-2 of family D) is a dichorionic diamniotic twin of Dutch origin. The mother became pregnant after in vitro fertilization (IVF) by intracytoplasmic sperm injection (ICSI) because of paternal oligoasthenospermia. Pregnancy was complicated by preeclampsia. The girl was born at 37 weeks and 2 days of gestation and delivered by cesarean section. Her birth weight and length were normal, but her OFC was below the third centile. Directly after birth, a mild upslant of the palpebral fissures, a short neck with a nuchal fold, and swollen labia minora were noted. She was slightly hypotonic. Feeding difficulties and esophageal reflux were present during the first weeks of life, and she was tube-fed for 10 days. Aneuploidy for chromosomes X, Y, 13, 18, and 21 was excluded by quantitative fluorescent PCR (qfPCR), and array CGH yielded normal results. Brain MRI first showed reduced gyration and broad sulci, although the diagnosis of simplified gyral pattern could not be established. However, the brain MRI at 3 months of age was reported to be normal. Magnetic resonance spectroscopy showed no abnormalities of choline, creatine, N-acetylaspartate (NAA), inositol, glutamate/glutamine, or lactate. No cardiac or renal abnormalities were noted. Her motor development was delayed. She started walking at the age of 18 months. Bayley Scales of Infant Development (BSID-III) testing at 2 years and 4 months revealed a developmental delay of 12 months; her fine motor skills and speech development were delayed. Her receptive language was better developed than her expressive language. She suffered from recurrent upper respiratory tract infections and a chronic otitis media. In addition, high hyperopia (+7 dpt) and astigmatism were diagnosed. Her sleep was markedly disturbed, and there was no improvement upon treatment with melatonin. A sleep EEG showed no abnormalities, however the parents reported staring spells during which she is not responsive. Her behavior is very open and friendly and she has a decreased pain sensation. She showed hand stereotypies, tactile hypersensitivity, and sexual self-stimulation. Feeding difficulties were still present, especially in swallowing certain solid foods. Clinical examination at age 3 years showed mild dysmorphic features, including full eyebrows, epicanthic folds, a broad nasal bridge, slight lateral eversion of lower eyelids, a short philtrum, orofacial hypotonia with open mouth-appearance, a thin and tented upper lip, a prominent lower lip, a high arched palate, small teeth and diasthema (Figure 1D). Her gait was clumsy and slightly broad-based. We performed trio exome sequencing with DNA samples of the parents and the proband, as described above for individual II-3 of family B, which identified a single non-annotated, de novo sequence change (Table S1D). This 1 bp deletion at cDNA position 635 (c.635delC) in CHAMP1 is predicted to change the reading frame and to induce a premature stop codon at amino acid 218 (p.Pro212Leufs∗7). Sanger sequencing confirmed the heterozygous c.635delC mutation in the proband (data not shown) and its absence from blood DNA of both parents.
An independent deleterious de novo CHAMP1 alteration has previously been identified in a systematic trio exome-sequencing study of 51 individuals with severe non-syndromic ID.2 This CHAMP1 alteration is one of 87 de novo variants in the proband group reported without detailed clinical description in the supplementary material. Due to the lack of further affected individuals, the pathogenicity of this variant remained unclear at that time. Notably, this was the identical nonsense mutation (c. 1192C>T; p.Arg398∗; Figure S4) that we also identified here in individual II-2 from family C. This fifth proband (individual II-2 of family E) is a nine-year-old girl, the second child of healthy non-consanguineous German parents. Her mother developed a Wilms tumor at the age of 24 years; further family history was unremarkable. The proband was born by vacuum extraction after 40 weeks of gestation with normal birth weight and length. OFC at birth is unknown, though it was within normal range at the age of four weeks. Psychomotor developmental delay became obvious in the second year of life. She started crawling at the age of 11 months and walking at the age of 20 months. Denver scale testing at the age of 36 months revealed a delay in all functions, particularly in language development. Indeed, speech development was markedly delayed, and she just spoke ten words at the age of 4 years and 8 months. Her receptive language skills were better in comparison, but still delayed for her age. Basic metabolic studies, EEGs, and a brain MRI at the age of 3 years yielded normal results. Neurological examination at the age of 4 years revealed orofacial hypotonia. The parents described a decreased pain sensation. On examination at age 4 years and 8 months, her body length and OFC were within normal range, but she was overweight (BMI 19.8, +3 SD). She showed mild dysmorphic facial features including upslanting palpebral fissures, epicanthic folds, low-set ears, a prominent nose, a short philtrum, an open-mouth appearance with a thin and tented upper lip, a prominent lower lip, and a high arched palate (Figure 1E). Her gait was clumsy. Her behavior was very friendly and open-minded. On last examination at 8 years and 11 months, her length and OFC remained in the normal range, but obesity had increased (BMI 23.3, +3.3 SD). Her speech was slurred and she spoke in up to three-word sentences and additionally used hand signs. Orofacial hypotonia was still present. The parents also reported truncal hypotonia. Meanwhile, hyperopia (right, +4.50 dpt; left, +3.50 dpt) and bilateral astigmatism were diagnosed. Prior to inclusion in the trio exome-sequencing study, karyotyping, FISH testing on chromosomal region 15q11q13 for Prader-Willi and Angelman (MIM: 105839) syndromes, fragile X analysis (MIM: 300624), MLPA P245 (microdeletion syndromes-1, MRC Holland), and array-CGH analysis had given normal results.
Taken together, all five individuals with a de novo deleterious CHAMP1 mutation are affected by ID and delayed motor development with a particularly severe delay in speech development. Whereas the motor development improved over time, speech impairment remained. All individuals suffered from muscular, in particular truncal, hypotonia. Orofacial hypotonia was observed in four probands. Similar dysmorphic features, including a short philtrum, tented upper lip, and everted lower lip were observed in all individuals. Three individuals presented with upslanting palpebral fissures, low-set ears, and a long face and pointed chin. A friendly behavior was described in all individuals. Decreased pain sensation, feeding difficulties during the neonatal period, hyperopia, and a high arched palate were noted in four individuals. Three individuals displayed stereotypic movements. Three individuals showed microcephaly. Before identifying the causative CHAMP1 mutations, a number of different specific genetic tests had been performed. The most important genetic differential diagnoses were Prader-Willi (PWS) and Angelman syndromes (AS). PWS was suspected in three probands because of feeding difficulties. At toddler age, the severely delayed speech development, the microcephaly, and ataxic gait made Angelman syndrome an important differential diagnosis, which was tested in three probands. ARX analysis was performed because of ID and speech delay in two male probands.
Notably, none of the four CHAMP1 alterations identified in our study were present in dbSNP136, the ExAC database, or the 1000 Genomes data, indicating that they are very rare in the population and unlikely to be disease-unrelated alterations. Additional confirming evidence of the pathogenic nature of CHAMP1 mutations in our probands comes from a very recent large-scale sequencing study (Deciphering Developmental Disorders) in altogether 1,133 individuals with ID and developmental delay. In this study, deleterious de novo CHAMP1 alterations were reported in two independent individuals. The first proband was a boy bearing the SNV c.1002G>A predicted to result in the nonsense variant at amino acid 334 (p.Trp334∗) and who was reported to show ID, obstructive sleep apnea, supernumerary nipples, and plagiocephaly. The second proband was a girl bearing the SNV c.1489C>T, resulting in a nonsense variant at amino acid 497 (p.Arg497∗) and who showed GDD, joint hypermobility, abnormalities of the renal collecting system and the CNS, incoordination, and diastasis recti. The authors classified CHAMP1 as a “novel gene with compelling evidence for a role in developmental disorders.”4
The findings in altogether seven independent individuals thus confirm that mutations in CHAMP1 are a monogenic cause of ID/GDD. In line with this assumption, there is no single loss-of-function CHAMP1 variant deposited in the ExAC database, including more than 110,000 sequenced alleles at this locus, which argues strongly for a deleterious effect of CHAMP1 mutations. Notably, the Residual Variation Intolerance (RVI) score, which quantifies gene intolerance to functional mutations7 of CHAMP1, is −0.75 (13.71th percentile) and thus even lower than the average RVI score for genes involved in developmental disorders (0.56; 19.54th percentile). This suggests that the degree of intolerance to deleterious variants of CHAMP1 is significantly more pronounced than the average degree of intolerance to deleterious variants of genes known to have mutations that cause developmental disorders.
CHAMP1 is located at chromosome 13q34 and contains a single coding exon (as well as two 5′-untranslated exons) encoding for a zinc-finger protein of 812 amino acids. CHAMP1 was shown to interact with the mitotic control protein MAD2L2 and is required for its spindle localization. CHAMP1 localizes to chromosomes and the mitotic spindle and regulates localization of CENPE and CENPF to kinetochores. Furthermore, it regulates kinetochore-microtubule attachment and therefore proper chromosome alignment.5 Mutations affecting genes encoding for proteins that regulate chromosome alignment and/or spindle assembly are a well-established cause of a variety of syndromic and non-syndromic developmental disorders. Examples include POGZ (MIM: 614787),4KIF2A (MIM: 602591), TUBG1 (MIM: 191135),6KIF4A (MIM: 300521) and KIF5C (MIM: 604593),8CENPE (MIM: 117143),9 and BUB1B (MIM: 602860).10 Therefore, CHAMP1 represents an attractive functional candidate gene for a developmental disorder. Interestingly, it has been shown that the localization of CHAMP1 to chromosomes and the mitotic spindle is regulated by its C-terminal region containing zinc-finger domains. The latter were further shown to be required for proper chromosome alignment.5 Notably, all thus-far-identified deleterious de novo mutations, if resulting in a stable protein, are predicted to lead to the loss of this functionally important C-terminal domain (Figure 2).
Figure 2.
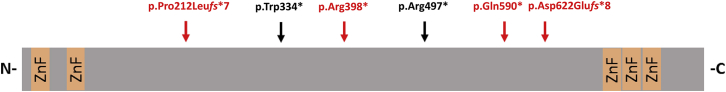
Schematic Protein Structure of CHAMP1
The positions of the de novo mutations identified in this study are marked with vertical arrows and are shown in red; mutations identified in the Deciphering Developmental Disorders study are shown in black. Abbreviations are as follows: ZnF, zinc-finger domain; N, N terminus; C, C terminus.
Notably, several chromosomal deletions involving CHAMP1 have been reported. Microscopically visible deletions of chromosome 13q have been divided into three groups depending on the localization of the deleted regions. Individuals of group 3, i.e., those with deletion breakpoints distal to 13q32, have been reported to be characterized by moderate to severe ID and, in the majority of cases, the absence of major malformations and growth deficiencies.11 More recent reports on individuals with deletions of 13q33–13q34 have expanded this clinical spectrum. For example, one individual was reported to have a cleft palate, microcephaly, and hypospadia in addition to his moderate to severe ID with severe speech delay.12 Another individual with intrauterine growth retardation, generalized hypotonia, severe language delay, and facial dysmorphism including epicanthal folds is reported to be socially outgoing, similar to the SNV-bearing individuals reported here.13 Clinical similarities with the individuals reported here also exist in individuals with submicroscopic deletions including CHAMP1 reported in the DECIPHER database or previous publications. 23 out of 26 individuals bearing a deletion that affects chromosomal bands 13q33 and 13q34 only (maximum size 13.5 Mb, Table S2) are reported to display ID/GDD. One may thus hypothesize that deletion of CHAMP1 may be the main cause for ID/GDD in individuals with deletion CNVs of terminal 13q. Interestingly, several other frequent findings in the five individuals reported here are also present in several individuals with deletions of 13q33–13q34, such as muscular hypotonia (n = 5), a narrow or high arched palate (n = 5), speech delay or an arched upper lip (each n = 3), as well as upslanting palpebral fissures or epicanthic folds (each n = 3). Given the incompleteness of database entries, this should of course not be over-interpreted. However, some of the published probands display three to four of these additional features.14 Thus, CHAMP1 may at least partly be responsible for some of the clinical features besides ID in individuals with group 3 chromosome 13q deletions. Given that the clinical findings of individuals with de novo mutations in CHAMP1 and in probands with larger chromosomal deletions encompassing the complete CHAMP1 gene seem comparable, it is tempting to speculate that this may be an indication for haploinsufficiency as the common etiological mechanism.
In summary, we identified five ID-affected individuals bearing de novo deleterious mutations in CHAMP1. Our data therefore establish deleterious mutations in CHAMP1 as the monogenic cause of a developmental disorder. Moreover, we provide the first detailed clinical description of individuals with CHAMP1 mutations, revealing ID with severely impaired expressive speech and delayed motor development, muscular hypotonia, neonatal feeding difficulties, and hyperopia, as well as similar mild dysmorphic features, as common clinical signs. In addition, we suggest that the genetic analysis of CHAMP1 should be considered in individuals with unclassified, non-syndromic or mild dysmorphic forms of ID/GDD, especially if muscular hypotonia and severe speech delay are present and PWS and AS testing have given normal results. Finally, our data further highlight the importance of the kinetochores and chromosomal alignment for proper human neuronal development and function.
Acknowledgments
We are thankful to the affected individuals and their family members for their participation. This work was supported in part by the German Ministry of Research and Education (grant nos. 01GS08164, 01GS08167, and 01GS08163; German Mental Retardation Network) as part of the National Genome Research Network. T.B.H. was supported by the Federal Ministry of Education and Research through a Juniorverbund in der Systemmedizin “mitOmics” (FKZ 01ZX1405C). We thank Ms. Jennifer Kaiser for excellent technical assistance.
Published: September 3, 2015
Footnotes
Supplemental Data include four figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2015.08.003.
Contributor Information
Hartmut Engels, Email: hartmut.engels@uni-bonn.de.
Davor Lessel, Email: d.lessel@uke.de.
Accession Numbers
The sequence variants p.Asp622Glufs*8, p.Gln590*, p. Arg398*, and p.Pro212Leufs*7 reported in the paper have been deposited in the Leiden Open Variation Database under accession numbers LOVD: 0000076069, 0000076070, 0000076071, and 0000076072, respectively.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
DECIPHER, https://decipher.sanger.ac.uk/
ExAC Browser, http://exac.broadinstitute.org/
OMIM, http://www.omim.org/
Supplemental Data
References
- 1.Leonard H., Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 3.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 4.Deciphering Developmental Disorders S., Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh G., Kanno S., Uchida K.S., Chiba S., Sugino S., Watanabe K., Mizuno K., Yasui A., Hirota T., Tanaka K. CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 2011;30:130–144. doi: 10.1038/emboj.2010.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Pierre B.S., Oger M. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willemsen M.H., Ba W., Wissink-Lindhout W.M., de Brouwer A.P., Haas S.A., Bienek M., Hu H., Vissers L.E., van Bokhoven H., Kalscheuer V. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 2014;51:487–494. doi: 10.1136/jmedgenet-2013-102182. [DOI] [PubMed] [Google Scholar]
- 9.Mirzaa G.M., Vitre B., Carpenter G., Abramowicz I., Gleeson J.G., Paciorkowski A.R., Cleveland D.W., Dobyns W.B., O’Driscoll M. Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum. Genet. 2014;133:1023–1039. doi: 10.1007/s00439-014-1443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanks S., Coleman K., Reid S., Plaja A., Firth H., Fitzpatrick D., Kidd A., Méhes K., Nash R., Robin N. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 11.Brown S., Russo J., Chitayat D., Warburton D. The 13q- syndrome: the molecular definition of a critical deletion region in band 13q32. Am. J. Hum. Genet. 1995;57:859–866. [PMC free article] [PubMed] [Google Scholar]
- 12.Bagherizadeh E., Shafaghati Y., Hadipour F., Behjati F. Phenotypical characterization of 13q deletion syndrome: Report of two cases. Indian J. Hum. Genet. 2014;20:203–205. doi: 10.4103/0971-6866.142912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lance E.I., DuPont B.R., Holden K.R. Expansion of the deletion 13q syndrome phenotype: a case report. J. Child Neurol. 2007;22:1124–1127. doi: 10.1177/0883073807306257. [DOI] [PubMed] [Google Scholar]
- 14.Kirchhoff M., Bisgaard A.M., Stoeva R., Dimitrov B., Gillessen-Kaesbach G., Fryns J.P., Rose H., Grozdanova L., Ivanov I., Keymolen K. Phenotype and 244k array-CGH characterization of chromosome 13q deletions: an update of the phenotypic map of 13q21.1-qter. Am. J. Med. Genet. A. 2009;149A:894–905. doi: 10.1002/ajmg.a.32814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.