

Abstract
Typing of healthcare-associated methicillin-resistant Staphylococcus aureus (MRSA) from Australia in the 1970s revealed a novel clone, ST2249-MRSA-III (CC45), present from 1973 to 1979. This clone was present before the Australian epidemic caused by the recombinant clone, ST239-MRSA-III. This study aimed to characterize the genome of ST2249-MRSA-III to establish its relationship to other MRSA clones. DNA microarray analysis was conducted and a draft genome sequence of ST2249 was obtained. The recombinant structure of the ST2249 genome was revealed by comparisons to publicly available ST239 and ST45 genomes. Microarray analysis of genomic DNA of 13 ST2249 isolates showed gross similarities with the ST239 chromosome in a segment around the origin of replication and with ST45 for the remainder of the chromosome. Recombination breakpoints were precisely determined by the changing pattern of nucleotide polymorphisms in the genome sequence of ST2249 isolate SK1585 compared with ST239 and ST45. One breakpoint was identified to the right of oriC, between sites 1014 and 1065 of the gene D484_00045. Another was identified to the left of oriC, between sites 1185 and 1248 of D484_01632. These results indicate that ST2249 inherited approximately 35.3% of its chromosome from an ST239-like parent and 64.7% from an ST45-like parent. ST2249-MRSA-III resulted from a major recombination between parents that resemble ST239 and ST45. Although only limited Australian archival material is available, the oldest extant isolate of ST2249 predates the oldest Australian isolate of ST239 by 3 years. It is therefore plausible that these two recombinant clones were introduced into Australia separately.
Keywords: Australia, genome sequence, methicillin-resistant Staphylococcus aureus, recombination, ST2249, Staphylococcus aureus
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) first appeared in Australia in 1965 in hospitals in Sydney [1] and Melbourne [2]. The responsible clone, ST250-MRSA-I by multilocus sequence typing (MLST) and SCCmec typing, caused epidemics in numerous countries in the first global wave of MRSA [3,4]. Contemporary phage typing and phenotypic data suggest that ST250-MRSA-I was replaced in Australia in the early 1970s by another clone with chromosomal determinants for penicillinase and resistance to tetracycline, kanamycin, neomycin, erythromycin and mercury [5–7]. MLST and SCCmec typing of isolates collected in two Melbourne hospitals in the 1970s have shown that a second multiresistant clone, ST2249-MRSA-III, a previously unrecorded member of clonal complex (CC) 45, was also present in Melbourne hospitals at least from 1973 to 1979 [2–7].
When MLST loci are ordered by chromosomal position, ST2249 and ST45 have identical alleles at five contiguous loci (aroE-14, glpF-8, gmk-6, pta-10 and tpiA-3), whereas ST2249 and ST239 have identical alleles at two contiguous loci (arcC-2 and yqiL-3) that span the origin of replication (oriC) [4]. In addition, ST2249 carries a type III SCCmec element, located downstream of oriC and between arcC and yqiL, of the same type as that carried by ST239. This arrangement suggests the possibility that ST2249 arose as the result of a major chromosomal recombination involving parents that resemble ST45 and ST239. Of note, ST239 is itself the result of a major chromosomal recombination involving ST8-like and ST30-like parents [8]. Such recombinations between parents of distinct genetic backgrounds that result in new multilocus sequence types are unusual in S. aureus with only three reported to date [8,9]. The aim of this study is to test the hypothesis that ST2249-MRSA-III is a hybrid resulting from chromosomal recombination involving ST45- and ST239-like parent strains.
Methods
Isolates
Thirteen isolates belonging to ST2249-MRSA-III were available from culture collections from two inner Melbourne teaching hospitals from the 1970s (Table 1). Nine isolates from a children’s teaching hospital had been studied previously [2,4,5]. Four additional isolates were obtained from a collection from an adult teaching hospital.
TABLE 1.
Origin and dru type of ST2249 isolates
| Isolate | Year of isolation | City | Hospital | dru type |
|---|---|---|---|---|
| SK1585 | 1973 | Melbourne | Children’s teaching | dt11j |
| SK1814 | 1974 | Melbourne | Children’s teaching | dt11j |
| SK1821 | 1974 | Melbourne | Children’s teaching | dt11j |
| SK1696 | 1975 | Melbourne | Children’s teaching | dt11j |
| AH1413 | 1976 | Melbourne | Adult teaching | dt9aj |
| AH1414 | 1976 | Melbourne | Adult teaching | dt9aj |
| AH1415 | 1976 | Melbourne | Adult teaching | dt11j |
| AH1431 | 1976 | Melbourne | Adult teaching | dt9aj |
| SK1582 | 1976 | Melbourne | Children’s teaching | dt9aj |
| SK1717 | 1977 | Melbourne | Children’s teaching | dt9aj |
| SK1774 | 1977 | Melbourne | Children’s teaching | dt11j |
| SK1734 | 1978 | Melbourne | Children’s teaching | dt9aj |
| SK1783 | 1979 | Melbourne | Children’s teaching | dt9aj |
DNA microarray analysis
Arrays and reagents were obtained from Alere Technologies (Jena, Germany). The principle of the assay, related procedures and a list of targets has been described previously [10]. Target genes included species markers, markers for accessory gene regulator (agr) alleles and capsule types, virulence factors, resistance genes, staphylococcal superantigen-like/exotoxin-like genes (set/ssl genes) and genes encoding adhesion proteins and immune evasion factors. Positive, negative and ambiguous results for individual markers including those requiring discrimination of allelic variants were interpreted as described previously [11]. Our initial strategy was to examine microarray results for available ST2249 isolates for evidence of likely recombination and for diversity within the lineage.
dru PCR and coagulase typing
Sequencing of SCCmec direct repeat units (dru) of ten isolates of ST2249 from 1973 to 1979 [4] was performed using the forward (GTTAGCATATTACCTCTCCTTGC) and reverse (GCCGATTGTGCTTGATGAG) primers described by Goering et al. [12] and the gel-based method described by Tohda et al. [13]. Coagulase restriction fragment length polymorphism was determined as previously described [14].
Genome sequencing and data analysis
Purified genomic DNA from ST2249 isolate SK1585, isolated in 1973 in Melbourne, was sheared to ~3 kb using a Covaris S220 focused ultrasonicator (Covaris, Woburn, MA, USA). A Mate Pair library suitable for sequencing on the IonTorrent PGM was prepared according to the manufacturer’s instructions, and a single 318 chip of data was generated using an Ion Torrent PGM (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Reads were split into pairs with SFFextract 2.0.13, and file headers were modified with in-house perl scripts before de novo assembly with Newbler v2.6 (Roche, Branford, CT, USA). Contigs were annotated with Prokka v1.4 (Prokka: Prokaryotic Genome Annotation System—http://vicbioinformatics.com/), and scaffolds were ordered against S. aureus JKD6008 [15] using Mauve Contig Mover Tool [16]. The resulting set of ordered, annotated scaffolds was deposited in GenBank (AYLT00000000), and all raw data were organized under NCBI bio-project PRJNA178070.
Identification of major recombination breakpoints
The recombinant structure of the ST2249 chromosome was characterized with two separate analyses. The publicly available genome sequences of S. aureus strains JKD6008 and BK21252 were used as examples of ST2249’s putative parents, ST239 and ST45, respectively (accession numbers NC_017341 and NZ_AHJV00000000.1, respectively). Based on Mauve analysis, these two strains were closer in overall genome content to ST2249 than other strains of similar sequence types with publicly available genome sequences.
The first analysis was based on the method of Brochet et al. [17] Briefly, the contigs of the draft genome sequence of ST2249 were ordered using the genome coordinates of strain JKD6008. The ST2249 genome sequence was then subdivided into 500 bp, non-overlapping windows and subsequently BLASTed against local databases of ST239 and ST45 genome sequences. Only windows that produced a BLAST hit with 100% coverage and no gaps were considered. E-values of 10−2, 10−4 and 10−6 were considered but these identified the same breakpoints, so 10−4 was selected for further analysis. Windows with multiple hits to a parent and windows absent from either of the two parents were discarded to filter out paralogs and accessory regions, respectively. The number of nucleotide polymorphisms within eligible windows, between ST2249 and each of its two parents, was plotted according to the ST2249 genome coordinates.
For the second analysis, the ST2249 genome sequence was aligned with those of its two putative parents using the progressive Mauve algorithm of Mauve v2.3.1 [18], with default parameters. Locally collinear blocks were included in the subsequent analysis provided that they contained sequence from all three strains. Locally collinear blocks were ordered based on the genome coordinates of strain JKD6008 and then concatenated. All gapped positions were removed. A sliding window analysis was performed using DnaSP v5 [19] to determine the number of nucleotide polymorphisms between ST2249 and each of its two parents, using 500 bp, non-overlapping windows, and plotting these polymorphisms according to the ST2249 genome coordinates.
Once the approximate breakpoint coordinates were determined, precise coordinates were identified through visual inspection of the sequences. The gene sequences that contained the two major recombination breakpoints were then aligned using the ClustalW algorithm, implemented in MegAlign v7.1 (Lasergene, DNAStar, Madison, WI, USA).
Results and discussion
Microarray analysis of genomic DNA of 13 ST2249 isolates showed general similarities with CC45/agr IV for most genomic markers (Table 2, Fig. 1, and see Supporting information Table S1). “CC45/agr IV” refers to a lineage within CC45 that differs from the better known, more common and more widespread “CC45/agr I” lineage to which, among others, MRSA strains Berlin Epidemic Strain, USA 600, WA-MRSA 4 and WA-MRSA 106 belong. Differences between the two CC45 lineages include not only the agr group affiliation but also alleles of fnbA/B, sdrD, vwb and lmrP as well as the presence of sasG. CC45/agr IV MRSA became common in Australia (WA-MRSA-23 and -84) and Hong Kong [11] but there are no data on the distribution of CC45/agr IV in the 1970s when ST2249 emerged nor on CC45/agr IV-methicillin-susceptible S. aureus (MSSA) in general.
TABLE 2.

Summary of the major differences between ST2249-MRSA-III, ST239-MRSA-III, CC8, CC30, CC45 (agrI) and CC45 (agrIV) by microarray analysis
FIG. 1.

Approximate localization of array targets in ST2249-MRSA-III isolate SK1585 matching with targets found in CC8, CC30 and CC45/agr IV (outer circle) in comparison with ST239 chromosome (inner circle).
However, several genes in ST2249 yielded microarray signals that were not in accordance with CC45/agr IV alleles but rather with CC8 alleles (sasG and fnbA/B, sdrC/D, ssl/set-locus) whereas others matched hybridization patterns from CC30 (clfB, lmrP, capsule locus) (Table 2). The MLST gene arcC had a CC30 and ST239 sequence, whereas yqiL was identical to the sequence from CC8 and ST239. The spa type was identical to that of ST239 (t037). In addition, coagulase restriction fragment length polymorphism PCR revealed the same type as AUS-2 and AUS-3 strains, both Australian variants of ST239-MRSA-III [20]. Isolates of ST2249 collected over 7 years belonged to two closely related dru types, 11j (six isolates from 1973 to 1977) or 9aj (seven isolates from 1976 to 1979) (Table 1). By array hybridization, the SCCmec element was identified as type III, and the presence of ccrC and the mercury resistance operon resembled AUS-3.
Assuming a constant order of genes within any S. aureus chromosome, these observations were in accordance with a larger fragment of CC8 origin having been inserted into a CC45/agr IV chromosome and a smaller fragment of CC30 origin around oriC having been inserted into that larger CC8 fragment. Because of the gene content similarities identified through microarray analysis, as well as similarities identified by MLST, spa, coagulase, dru and SCCmec typing, it was assumed that the entire insert into the CC45/agr IV chromosome originated from an ST239-MRSA-III strain that itself originated from a previously characterized recombination involving CC8 and CC30.
To test this hypothesis and to more precisely characterize the genome of ST2249, one isolate, SK1585, was sequenced. Ion Torrent PGM sequencing generated 1699425 read pairs (average length 90 bp). De novo assembly with Newbler resulted in ~150 contigs that were further organized into 15 scaffolds with an N50 = 456 855 bp, and an Nmax = 1 154 088 bp.
Recombination breakpoints were visually detected by examining the changing pattern of nucleotide polymorphism in the ST2249 genome sequence, in comparison with the ST239 and ST45 genomes (Fig. 2). Across the origin of replication, the ST2249 chromosome is much more similar to the ST239 chromosome (ST2249 versus ST239: 0.0000872 nucleotide polymorphisms/site) than to the ST45 chromosome (ST2249 versus ST45: 0.0175 nucleotide polymorphisms/site). However, across the remainder of the chromosome, ST2249 is much more similar to the ST45 chromosome (ST2249 versus ST45: 0.00012 nucleotide polymorphisms/site) than to the ST239 chromosome (ST2249 versus ST239: 0.014865 nucleotide polymorphisms/site) (Fig. 2). The above analysis was based on BLASTN comparisons of genome sequences. Essentially the same overall pattern of similarity between ST2249 and the parent-like genomes of ST239 and ST45 was found using a Mauve alignment of genome sequences (see Supporting information, Fig. S1).
FIG. 2.
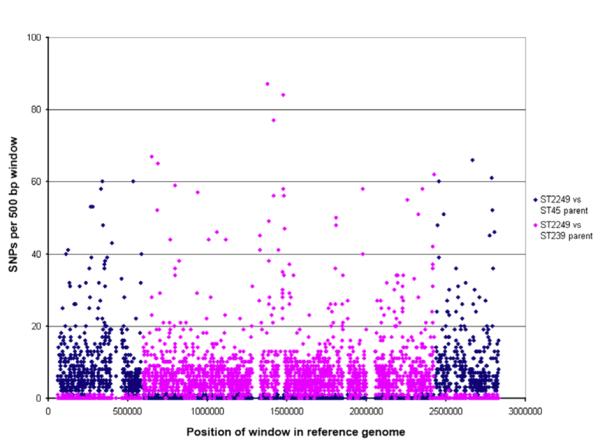
Comparison of single nucleotide polymorphisms (SNP) per site of ST2249-MRSA-III (SK1585) with putative parents ST45 and ST239, respectively.
The genomic comparisons identified recombination break-points to the right and left of the origin of replication, which were investigated in more detail. The right recombination breakpoint occurs between sites 1014 and 1065 of the ST2249 gene D484_00045 (Fig. 3). BLASTP searches indicate that this gene encodes a putative poly(glycerol-phosphate) α-glucosyltransferase. The left recombination breakpoint occurs between sites 1185 and 1248 of the ST2249 gene D484_01632 (Fig. 3). BLASTP searches indicate that this gene encodes nasD, which is a nitrate reductase. Both recombination breakpoints occurred within the coding sequences of the indicated genes, and the recombinations did not introduce frameshift mutations into these genes. Neither of these genes are the breakpoints of the previously identified ST239, ST34 and ST42 hybrids of S. aureus [8,9].
FIG. 3.

Right recombination breakpoint between sites 1014 and 1065 of the ST2249 gene D484_00045 and left recombination breakpoint between sites 1185 and 1248 of the ST2249 gene D484_01632.
These two recombination breakpoints indicate that ST2249 inherited approximately 981.5 kb (35.3%) of its chromosome from an ST239-like parent, i.e. a CC8/CC30 hybrid strain itself, and approximately 1798 kb (65.7%) from a CC45/agr IV-like parent. These sizes are estimated from the draft genome sequence of the ST2249 strain, SK1585. Minor uncertainties in the exact size of the parental contributions arise from potential sequencing/assembly errors, the undefined location of one scaffold that comprises ribosomal DNA operon that can be found at multiple locations across S. aureus genomes, and the possibility that ST2249 subsequently acquired or lost genes (described below). We have shown that ST2249-MRSA-III most probably resulted from a major recombination between parents that resemble ST239-MRSA-III and CC45/agr IV, although a recombination involving ST239-MSSA and CC45/agr IV followed by an independent acquisition of an SCCmec III/mer element might have been possible. However, this possibility seems unlikely as ST239-MSSA is very rare and ST2249-MSSA has never been described.
ST2249 isolates have some unique gene content variations. The gene bbp (bone binding protein) that is normally located near the right recombination breakpoint was absent from the draft genome sequence of strain SK1585 and it was not detected by array hybridization in that strain. However, bbp was present in 12 of 13 ST2249 isolates, suggesting that it has been lost in some isolates and is not related to the major recombination event. MAUVE misaligned a phiSa3-like phage with phiNM3 and phiSa2-like phage, which is visible as a spike of ST45 SNPs in the middle of the genome in Fig. S1 (see Supporting information).
Although only limited Australian archival material is available, the oldest extant isolate of ST2249 predates the oldest Australian isolate of ST239 by 3 years [4]. Even older isolates of ST239 are known from Europe [21]. It is therefore plausible that these two recombinant clones were introduced into Australia separately, or that ST2249 emerged in Australia before the importation and spread of ST239 there was noted. While comprehensive data are lacking, the parent CC45/agr IV lineage appears to be more common in Asia than in either Europe or North America, where it is virtually unknown (with CC45/agr I being dominant), so suggesting a possible origin for ST2249.
Another unanswered question is the mechanism by which such large portions of the chromosome are transferred. It is remarkable that transfer involves both core and accessory regions of the chromosome as a block of contiguous DNA, and that the recombination breakpoints can fall within genes without introducing frameshifts: these characteristics are consistent with homologous recombination. A high-frequency of recombination (Hfr-like) conjugative process remains the most likely mechanism of transfer, because of the large sizes of the transferred DNA. Recent work on patterns of recombination in the S. aureus chromosome has presented evidence of elevated recombination rates around the origin of replication and an association of localized recombination hot-spots with integration sites of certain mobile genetic elements [22]. Whether or not megabase-scale and kilobase-scale recombination events are mediated through the same mechanisms of transfer is unknown. Furthermore, it is possible that more hybrid strains of S. aureus remain to be discovered. To date, hybrid strains have been identified based on alleles from MLST, spa and other strain-typing schemes that are at odds with the rest of the strain’s typing profile. Contemporary assays such as array hybridization and full genome sequencing might identify more hybrid strains, by interrogating at higher resolution across the entire genome.
Hence the frequency and causation of such events remains at issue. As mentioned above, only three strains have been proven to originate from genomic replacements, giving the impression that megabase-scale recombinations are very rare events in the evolution of S. aureus. It is therefore all the more intriguing that ST2249 was derived from two major and temporally sequential chromosomal recombinations (CC8 and CC30 to ST239; and ST239 and CC45/agrIV to ST2249). If these events are indeed rare and result from uncommon environmental conditions, it is remarkable that they occurred twice in the formation of ST2249.
Supplementary Material
Acknowledgements
We thank Neville Firth and Ron Skurray for provision of historical isolates. DAR was supported in part by National Institutes of Health grant GM080602.
Footnotes
Transparency declaration
SM and RE are employed by Alere Technologies GmbH.
Appendix A. Supplementary data
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.cmi.2014.12.018.
References
- [1].Rountree PM, Beard MA. Hospital strains of Staphylococcus aureus, with particular reference to methicillin-resistant strains. Med J Aust. 1968;2:1163–8. doi: 10.5694/j.1326-5377.1968.tb83502.x. [DOI] [PubMed] [Google Scholar]
- [2].Gillespie MT, May JW, Skurray RA. Antibiotic resistance in Staphylococcus aureus isolated in an Australian hospital between 1946 and 1981. J Med Microbiol. 1985;19:137–47. doi: 10.1099/00222615-19-2-137. [DOI] [PubMed] [Google Scholar]
- [3].Enright MC, Robinson DA, Randle R, Feil EJ, Grundmann G, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA) Proc Natl Acad Sci USA. 2002;99:7687–92. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lancashire JF, Jones A, Bergh H, Huygens F, Nimmo GR. Typing early Australian Healthcare-Associated MRSA: confirmation of major clones and emergence of ST1-MRSA-IV and novel ST2249-MRSA-III. Pathology. 2013;45:492–4. doi: 10.1097/PAT.0b013e3283632667. [DOI] [PubMed] [Google Scholar]
- [5].Gillespie MT, May JW, Skurray RA. Antibiotic susceptibilities and plasmid profiles of nosocomial methicillin-resistant Staphylococcus aureus: a retrospective study. J Med Microbiol. 1984;17:295–310. doi: 10.1099/00222615-17-3-295. [DOI] [PubMed] [Google Scholar]
- [6].Rountree PM. History of staphylococcal infection in Australia. Med J Aust. 1978;2:543–6. doi: 10.5694/j.1326-5377.1978.tb131715.x. [DOI] [PubMed] [Google Scholar]
- [7].Vickery AM. Strains of methicillin-resistant Staphylococcus aureus isolated in Australian hospitals from 1986 to 1990. Australian Group for Antimicrobial Resistance. J Hosp Infect. 1993;24:139–51. doi: 10.1016/0195-6701(93)90076-c. [DOI] [PubMed] [Google Scholar]
- [8].Robinson DA, Enright MC. Evolution of Staphylococcus aureus by large chromosomal replacements. J Bacteriol. 2004;186:1060–4. doi: 10.1128/JB.186.4.1060-1064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thomas JC, Godfrey PA, Feldgarden M, Robinson DA. Draft genome sequences of Staphylococcus aureus sequence type 34 (ST34) and ST42 hybrids. J Bacteriol. 2012;194:2740–1. doi: 10.1128/JB.00248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Monecke S, Jatzwauk L, Weber S, Slickers P, Ehricht R. DNA microarray-based genotyping of methicillin-resistant Staphylococcus aureus strains from Eastern Saxony. Clin Microbiol Infect. 2008;14:534–45. doi: 10.1111/j.1469-0691.2008.01986.x. [DOI] [PubMed] [Google Scholar]
- [11].Monecke S, Coombs G, Shore AC, Coleman DC, Akpaka P, Borg M, et al. A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One. 2011;6:e17936. doi: 10.1371/journal.pone.0017936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goering RV, Morrison D, Al-Doori Z, Edwards GF, Gemmell CG. Usefulness of mec-associated direct repeat unit (dru) typing in the epidemiological analysis of highly clonal methicillin-resistant Staphylococcus aureus in Scotland. Clin Microbiol Infect. 2008;14:964–9. doi: 10.1111/j.1469-0691.2008.02073.x. [DOI] [PubMed] [Google Scholar]
- [13].Tohda S, Maruyama M, Nara N. Molecular typing of methicillinresistant Staphylococcus aureus by polymerase chain reaction: distribution of the typed strains in hospitals. Intern Med. 1997;36:694–9. doi: 10.2169/internalmedicine.36.694. [DOI] [PubMed] [Google Scholar]
- [14].Goh S-H, Byrne SB, Zhang JL, Chow AW. Molecular typing of Staphylococcus aureus on the basis of coagulase gene polymorphisms. J Clin Microbiol. 1992;30:1642–5. doi: 10.1128/jcm.30.7.1642-1645.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Howden BP, Seemann T, Harrison PF, McEvoy CR, Stanton JA, Rand CJ, et al. Complete genome sequence of Staphylococcus aureus strain JKD6008, an ST239 clone of methicillin-resistant Staphylococcus aureus with intermediate-level vancomycin resistance. J Bacteriol. 2010;192:5848–9. doi: 10.1128/JB.00951-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics. 2009;25:2071–3. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brochet M, Rusniok C, Couve E, Dramsi S, Poyart C, Trieu-Cuot P, et al. Shaping a bacterial genome by large chromosomal replacements, the evolutionary history of Streptococcus agalactiae. Proc Natl Acad Sci U S A. 2008;105:15961–6. doi: 10.1073/pnas.0803654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- [20].Coombs GW, Nimmo GR, Bell JM, Huygens F, O’Brien FG, Malkowski MJ, et al. Community methicillin-resistant Staphylococcus aureus in Australia: genetic diversity in strains causing outpatient infections. J Clin Microbiol. 2004;42:4735–43. doi: 10.1128/JCM.42.10.4735-4743.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smyth DS, McDougal LK, Gran FW, Manoharan A, Enright MC, Song JH, et al. Population structure of a hybrid clonal group of methicillin-resistant Staphylococcus aureus, ST239-MRSA-III. PLoS One. 2010;5:e8582. doi: 10.1371/journal.pone.0008582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Everitt RG, Didelot X, Batty EM, Miller RR, Knox K, Young BC, et al. Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat Commun. 2014;5:3956. doi: 10.1038/ncomms4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.