

Abstract
Fibrosis is one of the most prevalent features of age-related diseases like obesity, diabetes, non-alcoholic fatty liver disease, chronic kidney disease, or cardiomyopathy and affects millions of people in all countries. Although the understanding about the pathophysiology of fibrosis has improved a lot during the recent years, a number of mechanisms still remain unknown. Although TGF-β1 signaling, loss of metabolic homeostasis and chronic low-grade inflammation appear to play important roles in the pathogenesis of fibrosis, recent evidence indicates that oxidative stress and the antioxidant system may also be crucial for fibrosis development and persistence. These findings point to a concept of a redox-fibrosis where the cellular oxidant and antioxidant system could be potential therapeutic targets. The current review aims to summarize the existing links between TGF-β1 signaling, generation and action of reactive oxygen species, expression of antioxidative enzymes, and functional consequences including epigenetic redox-mediated responses during fibrosis.
Keywords: ROS, Antioxidants, Antioxidative enzymes, Matrix, Diet, Fibrosis
Graphical abstract
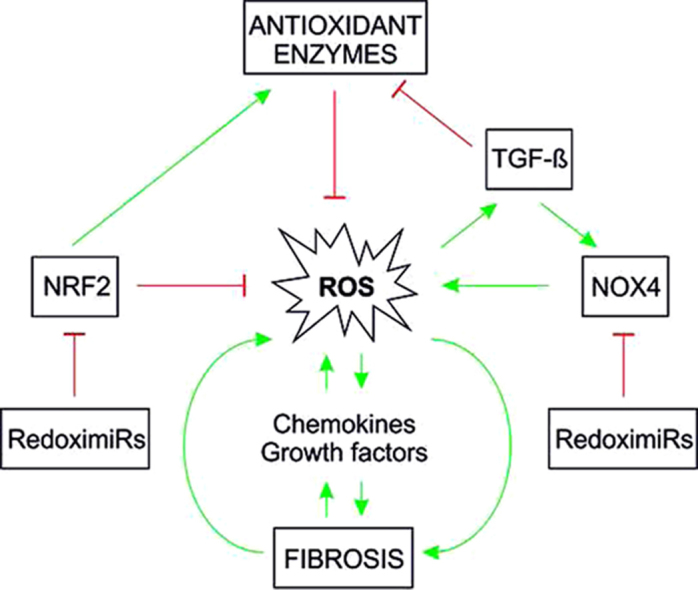
Highlights
-
•
Oxidative stress can be both a cause or a consequence of fibrotic events.
-
•
TGF-β1 is a key cytokine which contributes to production and reduction of ROS.
-
•
Feedback: ROS are able to activate TGF-β1.
-
•
ROS are powerful post-transcriptional and epigenetic regulators in fibrosis.
-
•
Are antioxidants a treatment option during fibrosis?
1. Introduction
Fibrosis is a major health problem and considered to be one of the most prevalent features of age-related diseases affecting millions of people in all countries. It can be considered to be a non-physiological scarring process in response to chronic diseases where an excessive extracellular matrix (ECM) deposition leads to irreversible tissue damage and failure or disturbance of proper organ function. Fibrosis has been found to affect all major organs and tissues including lungs, kidney, liver, heart, and skin. Moreover, the stroma of solid tumors can be considered to be partially fibrotic [1–4]. The pathophysiology of fibrosis has generally been studied in the context of the particular organ or tissue affected. However, recent research on the underlying molecular mechanisms provided evidence that common aspects for initiation and progression of fibrosis appear to exist in each of the affected organs/tissues. In particular, loss of metabolic homeostasis and chronic low-grade inflammation appear to play new emerging roles in the pathogenesis of fibrosis. Recent evidence indicated that oxidative stress, a condition where the balance between formation of reactive oxygen species (ROS) and their degradation by the antioxidant system is shifted in favor of a pro-oxidant state, appears to link metabolic homeostasis and inflammation thereby underpinning the concept of common fibrosis pathways that could be potential therapeutic targets. The current review aims to summarize the role of ROS and especially the anti-oxidative enzymes in fibrosis.
2. Fibrosis: common features and aspects
Fibrogenesis is a dynamic process and was proposed to occur in all organs in four common phases: i) initiation, due to injury of the organ/tissue; ii) inflammation and activation of effector cells, iii) enhanced synthesis of ECM; and iv) deposition of ECM with progression to end-organ failure [4]. The common aspects of a fibrotic pathogenesis can also be “visualized” because the same macroscopic and microscopic features are shared between the fibrotic organs. Macroscopically fibrotic organs usually display an uneven surface, are non-elastic, hard, and pale; signs resulting from accumulated ECM, contracted fibroblasts and reduced vasculature. Microscopically clear signs of an injured parenchyma, excess appearances of fibroblasts and fibrillar ECM, lack of capillaries (i.e. microvasculature), and a mononuclear infiltrate can be visualized [3]. Although there are obvious common elements in the fibrotic developments between organs, it appears that there are also some differences, which account for some organ specific aspects which become visible during the disease.
3. Fibroblasts: key players during fibrosis
The name fibrosis is based on the very well and long-term known observation that fibroblasts are the major source of the ECM; in fibrosis fibroblasts are called “activated fibroblasts” or “myofibroblasts” due to their increased synthetic capacity. Although the name “activated fibroblasts” may also imply a higher proliferative capacity, they are in fact less proliferative but more metabolically active in particular in ECM production [5]. Moreover, fibroblasts are not homogenous; they were found to be heterogenous within single organs and also between organs [6]. The heterogeneity may result, at least in part, from different cells and modes used for fibroblast recruitment/activation. While the most common view considers that activated fibroblasts derive via proliferation from tissue resident fibroblasts, various studies have shown that bone marrow-derived fibrocytes [7], endothelial cells that have undergone endothelial-to-mesenchymal transition [8], vascular smooth muscle cells and pericytes shed off from vessels [9,10], and epithelial cells after epithelial-to-mesenchymal transition [11] are also able to contribute to ECM deposition and fibrosis (Fig. 1). This makes the molecular analysis of fibroblasts particularly challenging, and though α-smooth muscle actin, vimentin, fibroblast-specific protein 1, or desmin are used to identify fibroblasts, it may explain why a specific common marker for all fibroblasts has not been found yet (for review see [3] and [12]). Moreover, processes like aging also affect the fibroblast protein expression profiles [13] which further complicates the molecular analyses of fibroblasts.
Fig. 1.

Fibroblasts as major extracellular matrix (ECM) producers. Fibroblasts can be derived from different cells like vascular smooth muscle cells, pericytes, fibrocytes, endo-and epithelial cells or resident fibroblasts. Depending on their origin these cells undergo differentiation, poliferation, epithelial-to-mesenchymal transition (EMT) or endothlial-to-mesenchymal transition (EndoMT). The so emerged myofibroblasts show an increased synthesis of matrix proteins contributing to excessive ECM deposition and fibrosis.
4. Fibrosis, tissue injury and inflammation
Commonly the regenerative capacity of parenchymal cells is able to cope with the loss of parenchyma occurring during single tissue injury events. However, this regenerative ability is lost upon repeated injury with the consequence of deregulated wound healing and a chronic activation of the immune response with appearing inflammation. Although fibrosis and its associated inflammation can be caused by infectious agents (e.g. viral infection of the liver or bacterial infections of lungs and kidneys), in most cases no underlying infection could be determined. This indicates that the inflammation is due to other not yet fully understood mechanisms, likely involving cell death responses [14,15]. Interestingly, anti-inflammatory therapies are rather ineffective in the treatment of fibrosis suggesting a complex interplay between the fibroproliferative process and the inflammatory response where pro- and retrograde cellular crosstalk of parenchymal cells, fibroblasts, and immune cells needs to be differently considered (for review see [16–18]).
5. ROS and oxidative stress in fibrosis
In addition to activation of fibroblasts and immune cells, fibrosis-inducing events also cause release of profibrotic metabolites (e.g., ROS), and secretion of chemokines and growth factors. Increased evidence indicates that production of ROS and oxidative stress are interlinked with production and activation of various growth factors and cytokines [19–21]; thereby feed-back as well as feed-forward cycles appear to exist (Fig. 2).
Fig. 2.

Feed-forward and feed-back events where ROS can contribute to fibrosis Infection, injury, toxic chemicals, drugs as well as radiation (UV, ionizing) may lead to formation of ROS. As a consequence ROS might directly contribute to fibrosis or indirectly via enhanced inflammation. Fibrosis itself may feedback to ROS formation or foster generation of cytokines and growth factors which also can contribute to generation of ROS. Under normal circumstances (non-fibrotic response) the transient induction of inflammation by ROS is followed by tissue regeneration.
Indeed, increased levels of 4-hydroxy-2′-nonenal (4-HNE), a marker of lipid peroxidation [22], have been detected in biopsy samples from liver fibrosis patients. In addition, 8-isoprostane, another marker of oxidative stress, was found to be present in exhalants and bronchoalveolar lavage fluid from patients with lung fibrosis [23,24]. In particular ROS were supposed to play a key role in the development of silicosis and asbestosis since nitro-tyrosine adducts and indicators for enhanced oxidative DNA damage like 8-hydroxy-2′-deoxyguanosine (8-OHdG) were found in those patients [25,26]. Moreover, oxidative stress was shown to be present during development of renal fibrosis [27,28] as well as to play a role in several aspects of fibrotic cardiac repair/remodeling after infarction [29–31].
5.1. Sources of ROS in fibrosis
ROS can be produced either non-enzymatically or enzymatically. The major non-enzymatic triggers for ROS formation in vivo are ionizing and UV radiation, toxic chemicals and drugs, which all are well known inducers of fibrosis in different organs. While the majority of intracellular ROS is generated as by-products of respiratory chain function, various enzymes such as xanthine oxidoreductase (XOR), several peroxisomal oxidases, enzymes of the cytochrome P450 family, cyclooxygenases, lipoxygenases, and NADPH oxidases (NOX) were found to contribute to ROS production (for review see [32].
From the enzymatic ROS producers NADPH oxidases appear to be most important during the pathological progression of fibrosis [33,34] (Fig. 3). Production of O2¯• by the NOX complex in phagocytes is a well-known phenomenon of the oxidative burst involved in bacteriocidal activity [35,36]. The heterodimeric transmembrane parts of the classical NOX complexes from phagocytes consist of the NOX2 protein and NOX subunit p22phox. Up to now, further NOX proteins have been identified and currently the NOX family members are designated NOX1–5 and DUOX1/2 [33,34]. While the expression of NOX2 appears to be confined to polymorphonuclear cells, macrophages and endothelial cells, NOX1 seems to be present in the plasma membrane of various cell types [33,37,38]. In general, NOX activity is supposed to be tightly controlled by regulatory subunits. Among these are the classical NOX2 regulators p47phox and p67phox, their homologs NOXO1 and NOXA1, the DUOX1/2 regulators DUOXA1 and 2, and the GTPase Rac.
Fig. 3.

Generation of ROS and action of antioxidant enzymes The ROS superoxide (O2•−) is the major precursor for the production of other ROS (light gray); it is generated due to the action of various enzymes. Antioxidant enzymes (dark gray) like superoxide dismutase (SOD) are able to convert O2•− to H2O2 which can be neutralized through the action of glutathione peroxidases (GPX), peroxiredoxins (Prx) or catalase (CTL). NOX, NADPH oxidase; XOR, xanthine oxidoreductase; COX, cyclooxygenase; NOS, nitric oxide synthase; NO●, nitric oxide; ONOO●−, peroxynitrite; NO2●, nitrogen dioxide; ●OH, hydroxyl radical; OH−, hydroxide anion; Trx, thioredoxin; TrxR, thioredoxin reductase; GR, glutathione reductase; GSH, glutathione; GSSG, oxidized glutathione.
NOX-derived ROS were found to be associated with fibrosis in several organs such as lung [39], heart [40] kidney [41], pancreas [42], and liver [43–45]. From the NOX proteins NOX4 is unique in that its activity depends on NOX4 expression levels [20,46] and except p22phox does not require further regulatory proteins. NOX4 was found to be associated with endothelial cell dysfunction linked to conditions such as hypoxia known to cause further up-regulation of NOX4 expression [47]. In respect to these findings and in relation to the activity of the other NOX proteins, NOX4 appears to be the most direct link between oxidative stress, ROS production and fibrosis.
6. TGF-β a key cytokine in the fibrotic process: links to ROS
In addition to enhanced ROS levels, various chemokines and growth factors are also crucial for the onset and progression of fibrosis. Among the chemokines and growth factors secreted, the cytokine transforming growth factor-β (TGF-β) appears to be a key mediator of the fibrotic process, which plays a role in fibrogenesis in almost, if not all, organs [48–50]. Originally TGF-β was found to be a potent inducer of anchorage-independent growth of human embryonic kidney fibroblasts [51]. In the meantime, three TGF-β isoforms have been identified, which are called TGF-β1, TGF-β2 and TGF-β3. In most cases and until the three isoforms were discovered, TGF-β refers to TGF-β1. In addition to fibrosis, TGF-β1 plays also a role in various other diseases like Marfan syndrome, Parkinson's disease, various cancers and diabetes (for review see [52–54]). Importantly TGF-β1 is overexpressed in all fibrotic tissues, and it induces collagen production in cultured fibroblasts irrespective of their origin [55–58]. Increased evidence indicates that production of ROS and oxidative stress are interlinked with TGF-β1 production and activation and thus are key to the fibrotic process [21,59].
6.1. Production of ROS: involvement of TGF-β1
TGF-β1 was shown to promote ROS formation mainly via induction of NOX4 expression and hence activity in various types of cells [40,60–62]. The ROS produced in response to TGFβ1 are able to activate various signaling pathways, thereby influencing gene transcription networks downstream from TGF-β1 [21,41,63,64]. An important role for NOX4 was found in patients with idiopathic pulmonary fibrosis where NOX4 was selectively up-regulated in the lungs and where it was associated with endothelial cell dysfunction and hypoxia which can cause a further up-regulation of NOX4 expression [65,66]. In line, TGF-β1–dependent pro-fibrotic responses and ECM deposition in the lungs were inhibited by siRNA-mediated NOX4 knockdown [39]. In line, mice deficient in NOX4 were found to be protected from bleomycin-induced acute lung injury and the onset of fibrosis [67]; mice deficient for NOX2 were less protected from pulmonary fibrosis [68]. Moreover, therapeutic treatment with a low-molecular-weight NOX4 antagonist attenuated the progression of a fibrotic response in mice with bleomycin-induced pulmonary fibrosis [69]. A role of NOX4 was also established for liver fibrosis where it was found to be increased in patients with hepatitis C virus (HCV)-associated liver fibrosis, and in patients with non-alcoholic steatohepatitis (NASH) [70]. Further, the hepatic stellate cells (HSC) which are key in the fibrotic process of the liver responded to TGF-β1 with an induction of NOX4-dependent ROS formation [71], The TGF-β-induced fibrotic process was inhibited either by down-regulation of NOX4 or in Nox4−/−mice [72]. In addition to these data underlining the role of NOX4, other studies indicated also the importance of NOX1 and NOX2 in the fibrogenic process [45,73–75].
The so far dominant role of NOX4 in fibrosis appears to be also connected with aging since NOX4 was found to promote myofibroblast differentiation and wound healing in young mice whereas it promoted persistent fibrosis in aged mice [39,64,76]. These effects in aged animals could be due to a loss of the antioxidant response driven by the transcription factor NRF2 (nuclear factor-erythroid 2 related factor 2) (see below). It was proposed that the increase in the ratio between NOX4/NRF2 promotes fibrosis and development of senescent and apoptosis-resistant myofibroblasts in those aged mice [76]. Thus, it appears that TGF-β1 mediated ROS production, mainly via NOX4, is crucially involved in the pathogenesis of the fibrotic response which is more severe and persistent in elder subjects.
6.2. Reduction of ROS: involvement of TGF-β1
In addition to induction of expression of ROS producing enzymes, TGF-β1 may also suppress expression of several antioxidant enzymes (Fig. 4). Under normal physiological conditions, formation and removal of ROS are balanced to avoid oxidative stress. In principle, an antioxidant can be any substance that delays/prevents formation or promotes removal of ROS or oxidative damage of a target molecule [32]. The antioxidant defense system consists of a number of molecules among them glutathione (GSH) and several enzymes, like superoxide dismutases (SODs), glutathione peroxidases (GPXs), and catalase (CTL) represent major players [32].
Fig. 4.

Involvement of ROS in TGF-ß mediated fibrosis TGF-ß is released by different cell types like platelets, parenchymal cells, and inflammatory cells (e.g. lymphocytes, macrophages). After the conversion from the latent to the active form, TGF-ß binds to its receptor and induces expression of various genes, among them NOX4, via signaling through SMAD2/3 and/or PI3K. NOX4 expression leads to ROS production. Enhanced ROS may activate proliferation, migration and differentiation of fibroblasts as well as epithelial-to-mesenchymal transition (EMT), apoptosis of epithelial cells and/or excessive extracellular matrix deposition (ECM). Further, TGF-ß contributes to ROS enhancement by decreasing the expression of antioxidant enzymes like glutaredoxin (Grx), catalase (CTL), glutathione peroxidase (GPX), glutathione S transferase (GST), superoxide dismutase (SOD) and the heavy subunit of gamma-glutamylcysteine synthetase (gamma-GCSh).
GSH is a three-residue peptide (γ-l-glutamyl-l-cysteinyl glycine) which in the reduced state can donate an electron upon which two molecules of GSH form the oxidized glutathione disulfide (GSSG). Glutathione reductase (GR) is able to reconvert GSSG back to GSH. The ubiquitous presence of GSH in a high concentration (1–10 mM) and this recycling system allow that it acts as a major cellular antioxidant [77]. It can directly react with O2•¯ and some other ROS but more important are its indirect ROS-scavenging functions [78–80]. These are achieved due to the involvement of GSH in revitalizing other antioxidants; e.g. it can reduce α–tocopherol radicals and semihydroascorbate radicals [80]. Moreover, GPXs couple oxidation of GSH with detoxification of hydrogen peroxidase. In addition, GSH is cofactor and substrate for glutaredoxins and glutathione S transferase (GST) which conjugates reactive electrophilic compounds, including environmental toxins, and products of oxidative stress, with GSH to detoxify them [81].
Importantly, GSH levels were found to be decreased in patients suffering from various fibrotic diseases like chronic liver disease and cirrhosis, viral hepatitis, chronic obstructive lung diseases and asbestosis, conditions already reported to show increased ROS levels [82–84]. Interestingly, alcohol consumption, a major reason for chronic liver disease, did not only cause an 80–90% depletion of GSH in liver but also in lungs [85]. Mechanistically the decrease in GSH levels in liver cirrhosis could be explained by reduced production. Thereby TGF-β1 appeared to be crucial since it affects the function of gamma-glutamylcysteine synthetase (gamma-GCS), the rate-limiting enzyme of GSH synthesis [86,87]. This enzyme consists of the catalytical heavy (gamma-GCSh) subunit and the regulatory light (gamma-GCSl) subunit. TGF-β1 decreased gamma-GCSh expression in human lung alveolar epithelial cells [88] and in the fibrotic areas of usual interstitial pneumonia [89]. In addition, the decrease in gammaGCSh and GSH levels along with increased protein and lipid peroxidation due to TGF-β1 was also found in a mouse model of lung fibrosis [88]. Thus, the depletion of GSH in response to TGF-β1 during fibrosis may contribute to the appearance of oxidative stress as seen during fibrosis.
The function of GSH is closely associated with glutaredoxins (Grx) which are thiol-disulfide oxidoreductases. Interestingly TGF-β1 decreased Grx1 expression, again highlighting that the GSH driven antioxidant system is of importance in fibrotic lung diseases [90,91].
Superoxide dismutases are another enzyme family with antioxidant function. Three SODs catalysing all the dismutation of O2•¯ to H2O2 and O2, but which are localized in the cytosol (SOD1), mitochondria (SOD2), and extracellular (SOD3) can be distinguished. Moreover, the peroxisomal enzyme catalase is best known to convert H2O2 to oxygen and water [32,92].
In addition, peroxiredoxins (Prx) have been shown to react with H2O2 at an exceptionally high rate [93]. The action of Prx is not restricted to H2O2 since Prx were also shown to reduce and detoxify peroxynitrite and a wide range of organic hydroperoxides. Like SODs, Prx members also display a different cellular location. Prx1, 2 and 4 are mainly cytoplasmic but also found in nuclei; Prx1 is also present in mitochondria and peroxisomes while Prx4 is found in lysosomes [94–96]. Mitochondria contain also Prx3, and Prx5. In addition to mitochondria Prx5 is found in cytoplasm, nuclei and peroxisomes [97]. It is believed that Prx2 because of its high reaction rate and abundance traps almost all H2O2 in vivo [79]. Oxidized Prx are reduced by thioredoxins (Trx) which can be oxidized by the action of various stimuli [98]. Oxidized Trx (as well as other oxidized cellular proteins) can be reversibly reduced by thioredoxin reductase (TrxR) in a NADPH-dependent manner [98]. Again, Trx1 as well as TrxR1 can be found in the cytoplasm and nucleus whereas Trx2 and TrxR2 are localized to mitochondria [94,95].
Glutathione peroxidases from which GPX1 is the ubiquitous memeber reducin cytosolic, mitochondrial and in some cells peroxisomal peroxides while GPX2 is an epithelial-specific form with highest expression in intestine [99]. A secreted GPX3 form expressed in lung and kidney can be important in the protection against external peroxides [99]. The fourth member GPX4 has three isoforms derived from a single gene: cytosolic (c-GPX4), mitochondrial (m-GPX4), and nuclear (n-GPX4) [100,101]. Unlike other glutathione peroxidases, GPX4 has a broad range of substrates; in addition to H2O2, these include derivatives from cholesterol and cholesteroyl esters and thymine hydroperoxide [100].
Overall, the conversion of H2O2 avoids formation of hydroxyl radicals (•OH) via an iron catalyzed Fenton reaction. In line, free iron is sequestered by iron-binding proteins like ferritin and transferrin to reduce intracellular ROS production. Dietary micronutrients such as vitamin C, vitamin E, and minerals like selenium as a component of peroxidases GPX1-4 and TrxRs are contributing to the antioxidant activity [32].
Although not much is known about the particular role of the antioxidant enzyme expression during fibrosis, some cell culture experiments have indicated a crucial role of TGF-β1 as a negative regulator. It was shown that TGF-β1 inhibited the mRNA expression as well as the activities of GPX1 and CTL in a hamster pancreatic beta-cell line (HIT). Subsequently, the reduced expression of these enzymes was associated with an increase in intracellular ROS and oxidized proteins [102]. Furthermore, TGF-β1 also suppressed the expression of SOD1, SOD2, CTL and GST in cultured rat hepatocytes and in airway smooth muscle cells. Again, the reduced expression of these antioxidant enzymes was associated with an increase in cellular ROS levels [102–104].
Together, all the above mentioned findings suggest that the fibrotic action of TGF-β1 is coupled to an augmented production of ROS due to induction of ROS generating systems and to suppression of antioxidant systems.
7. NRF2 signaling in fibrosis
As mentioned above the transcription factor NRF2 (nuclear factor-erythroid 2 related factor 2) and its partner Keap1 (Kelch-like ECH-associated protein 1) are considered to be the major transcriptional regulators in the defense against ROS [105,106]. Thereby the sulfhydryl groups within Keap1 appear to be sensitive to oxidant changes [105,107]. In the absence of ROS Keap1 is bound to NRF2 and promotes its proteasomal degradation. Upon an increase in ROS, the cysteine residues in Keap1, with Cys151 being the most critical, become oxidized and Keap1 can no longer bind to NRF2. In addition, ROS induce a dephosphorylation of Keap1 at Tyr141 which promotes Keap1 degradation [108]. As a result, NRF2 is stabilized and transported to the nucleus; its nuclear presence is promoted due to ROS-dependent oxidation of Cys183 which inhibits binding to the nuclear export protein CRM1. Once in the nucleus NRF2 binds to a small Maf protein to activate genes of the antioxidant response [105,107].
NRF2 signaling seems to be important for the pathogenesis of fibrosis since several studies have demonstrated that NRF2−/− mice are more susceptible to chemically induced oxidative stress than wild-type mice; in particular in liver [109,110]. In the liver NRF2 protects mice from carbon tetrachloride induced hepatic fibrosis [111] and enhanced iron accumulation, steatosis, inflammation and fibrosis when fed a methionine-and choline-deficient diet [112–115]. Apparently, the NRF2 protective mechanisms become less efficient with aging, which is expected to contribute to disease progression [49]. Moreover, the NRF2 system appeared also to be involved in mediating the defense against high fat diets or chronic alcohol abuse. Clinical studies and experimental models indicate that alcohol abuse increases the risk for liver fibrosis and cirrhosis as well as for acute lung injury. With respect to the latter it was shown that chronic alcohol abuse in mice primed the lung for bleomycin-induced fibrosis [116]. The alcohol ingestion further caused glutathione depletion and an increased TGFβ-1 expression. In addition, the expression of the NRF2-dependent antioxidant enzymes glutathione S-transferase theta 2, and glutamate-cysteine ligase catalytic subunit were found to be reduced. The effects of alcohol appeared to be mediated via TGFβ 1 expression since blocking TGFβ 1 signaling attenuated the alcohol-induced suppression of NRF2 [117].
Although the above given examples point to the proper NRF2, primarily regulated due to the sensitivity of Keap1, it became evident that upstream or parallel regulatory circuits exist. For example recent findings indicated that the selenoprotein thioredoxin reductase 1 (TrxR1) is a potent NRF2 regulator. Deficiency of selenium and concomitant loss of TrxR1 reactivity is combined with induction of NOX activity and oxidative stress [118].
Moreover, critical feed-forward responses also appear to exist and one includes the NRF2-dependent expression of the transcription factor Kruppel-like factor 9 (Klf9) when the levels of intracellular ROS are above a critical threshold. The induction of Klf9 results in further increases in ROS and subsequent cell death. The importance of this mechanism for the pathogenesis of fibrosis was underlined by recent findings demonstrating that Klf9 independently increased ROS in-vitro and in-vivo in mice where it promotes bleomycin-induced pulmonary fibrosis. The effects of Klf9 are achieved via its DNA binding function; it binds to promoters and alters expression antioxidant genes like thioredoxin reductase 2 [119].
Interestingly, a number of natural products are NRF2 activating substances; among them the plants, fruits and vegetables components quercetin, genistein, curcumin and sulforaphane; therapeutics such as oltipraz, auranofin and acetaminophen; environmental agents like paraquat, and metals as well as endogenous substances like hydrogen peroxide, NO, or 4-hydrohynonenal [120]. This somehow may imply that an activation of the NRF2 pathway may be of therapeutical benefit. However, Keap1 knockout mice die shortly after birth. This finding and the rescue of this lethal phenotype in Keap1/ NRF2 double knockout mice [121] suggests that excessive NRF2 activity may be detrimental for normal life and may render cellular behavior as indicated by a study showing that constitutive activation of the NRF2 pathway is beneficial for tumor survival [122]. Altogether, the NRF2 system seems to be critically involved in the redox-dependent regulation of fibrosis, in particular due to its inactivation in response to TGFβ1. However, due to the multilayered control steps which are currently unraveled it becomes clear that the present knowledge about NRF2 in fibrosis is a piece of an interesting puzzle.
7.1. Activation of TGF-β: involvement of ROS
As the name implies, TGF-β is not only an inducer of ROS forming enzymes and suppressor of ROS degrading enzymes. However, to become active TGF-β needs to be free since it normally exists in a latent complex where it is bound to two other polypeptides, latent TGF-β binding protein (LTBP) and latency-associated peptide (LAP). Although some aspects of the TGF-β activation process are quite well known, the mechanisms are far from being understood. Serum proteases such as plasmin and a number of matrix metalloproteases as well as thrombospondin-1 can induce the release of TGF-β from the latent complex with LTBP and LAP [123,124] In addition, integrins, pH, and ROS have been shown to be able to activate TGF-β [125–127].
In particular the latter mechanism appears to be important since enhanced levels of ROS are not only associated with fibrosis but also with a number of other diseases which display high TGF-β1 levels like cancer and diabetes. This implies that ROS would have the potential to activate TGF-β1 Indeed, recent studies from a cell-free system in which ROS were generated in solution by ionizing radiation or metal ion-catalyzed ascorbate reactions indicate that recombinant latent TGF-β1 can be activated directly by ROS [127]. Thereby, the metal-catalyzed ascorbate oxidation mediated TGF-β1 activation appeared to be very efficient, and dependent on ascorbate concentrations and presence of transition metal ions. Moreover, the ROS dependent activation of TGF-β1 appeared to be isoform specific and to be restricted to TGF-β1 particularly only oxidizing the LAP-beta1 protein at methionine 253 [127]. Together, ROS are not only a result of TGF-β action but appear also to be crucial for the activation of TGF-β1 indicating that they are causally involved in the fibrogenic process.
8. ROS, hypoxia, and fibrogenesis
Changes in ROS levels are not only coupled to the action of various chemokines, hormones, and growth factors but also associated with hypoxia which is a major sign of fibrosis due to loss of endothelial cells, rarefication of capillaries and subsequent malperfusion. The loss of endothelial cells may be due to endothelial-mesenchymal transition (EndMT), a process where endothelial cells undergo a transformation and acquire a mesenchymal (fibroblast-like) phenotype. EndMT is dormant in the adult organs, injuries, or inflammatory events which contribute to fibrosis may reactivate this embryonic process [128–130]. TGF-β and hypoxia are drivers of EndMT. Although hypoxia through activation of hypoxia-inducible transcription factor-1α (HIF-1α) [131,132] drives expression of endothelial growth factors to induce angiogenesis with the aim to improve the capacity to regenerate (or form new) vessels, the link to EndMT suggests that higher fibroblast formation through hypoxia-driven EndMT will occur [57]. While injection of VEGF was beneficial in some experimental models of organ fibrosis [133–135], a recent study demonstrated that hypoxia is a stimulus for human coronary endothelial cells to undergo EndMT via a HIF1-α driven mechanism. Thereby, HIF-1α induced expression of the transcription factor Snail which is a master regulator of EndMT [136]. Together, it is likely that hypoxia, depending on the regenerative capacity of the injured tissue, may contribute to vessel regeneration via HIF-driven VEGF or to progression of fibrosis via Snail-driven EndMT.
8.1. ROS as post-transcriptional and epigenetic regulators in fibrosis
Although our understanding in the pathogenesis of fibrosis has improved tremendously, a huge number of aspects remain still to be resolved. One such aspect is the often occurring inter-individual severity and progression of fibrosis, and the responsiveness to therapeutic regiments in different patients with similar underlying diseases. The basis for the differences may lay in varying post-transcriptional gene regulation events, genetic variability, and epigenetic mechanisms [137].
Recently, microRNAs (miRNAs) which mediate post-transcriptional regulation by promoting mRNA degradation and translational repression have been shown to be regulators of pro- and anti-fibrotic processes [138] (Fig. 5). TGF-β1 was also found to be a powerful driver of miRNA expression. The link between ROS, fibrosis and miRNA expression became especially evident in the regulation of NOX4 expression. As mentioned earlier, TGF-β regulates the expression of NOX4, the major catalytic subunit of the ROS generating NADPH oxidase NOX4. NOX4–dependent generation of hydrogen peroxide was required for TGF-β–induced myofibroblast differentiation, ECM production and contractility [39]. The so called “redoximiRs” miR-146a, and miR-25 [139] downregulate the expression of NOX4 and thus ROS generation whereas miR-135b and miR-708 can be upregulated by H2O2. Moreover, the expression of the major antioxidant transcription factor NRF2 can be abrogated by miR-153, miR-27a, miR-142-5p, and miR144. Further, the activity of NRF2 is influenced via the action of miR-200a which targets Keap1, that represses NRF2, and via miR-34a that targets sirtuin1 (Sirt1), deacetylates NRF2 and promotes its nuclear export (for review see [139,140]). In addition, miR-433 was shown to downregulate expression of the catalytic and regulatory subunits of the major glutathione synthesizing enzyme gamma-GCS [141]. Another miRNA, miR-27a/b, was recently shown to suppress the mitochondrial protein prohibitin 1, whose deficiency results in downregulation of NRF2 [142] and appearance of NASH-like symptoms and liver fibrosis. Furthermore, miR-30e which directly targets the mitochondrial protein UCP-2 was found to be downregulated in fibrotic kidneys [143]. Thus, miRNAs appear to represent another link where redox signaling and regulatory mechanisms of fibrosis converge.
Fig. 5.

Interconnection of ROS with miRNA expression and epigenetic modifications MiRNAs are able to regulate ROS via post-transcriptional degradation of NOX4 and Nrf2 mRNA. Further, ROS are associated with DNA hypermethylation causing decreased expression of different genes like O-6-methylguanine-DNA methyltransferase (MGMT), mutL homolog 1 (MLH1), thymocyte differentiation antigen-1 (Thy-1), Krüppel-like factor 4 (Klf4), methyl CpG binding protein 2 (MeCP2), RAS protein activator like 1 (RASAL1), peroxisome proliferator-activated receptor (PPAR-γ) and patched1 (PTCH1).
Epigenetic changes, defined as heritable traits that are not linked to changes in DNA sequence, but where DNA methylation, and post-translational modifications of histones and other chromatin-associated proteins regulate transcription [144], are another basis for observed differences in fibrosis. Epigenetic changes like DNA methylation can also be influenced by ROS [145]. For example, ROS-mediated DNA base modifications like 8-oxodG formation adjacent to a cytosine may prevent methylation of the latter [146]. The resulting hypomethylation may contribute to induction and suppression of gene expression thus enabling the cells to achieve novel characteristics which may contribute to tumorigenesis. ROS are also associated with hypermethylation [145] that can lead to increased overall mutation rates by downregulating expression of repair genes like O-6-methylguanine-DNA methyltransferase and MLH1 (mutL homolog 1) [147]. Although a direct link to ROS has not been shown, hypermethylation of phosphatase and tension homolog (PTEN) with consequent gene repression favoring ERK and AKT signaling, cell proliferation and migration appears to promote liver fibrosis [148]. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts was found to be associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype [149] and Tet3-mediated silencing of genes due to hydroxymethylation contributed to bone morphogenic protein 7-induced reversal of kidney fibrosis [150]. In addition, hypermethylation and repression of Krüppel-like factor 4 through Dnmt1 [151], and hypermethylation of (methyl CpG binding protein 2 (MeCP2) [152], RAS Protein Activator Like 1 (RASAL1) [153], peroxisome proliferator-activated receptor (PPAR-γ) [154], and patched1 (PTCH1) [152] appear also to promote fibrosis.
While epigenetic modifications in general are acquired in response to environmental stimuli, recent evidence links epigenetic modifications in three regions of the SOD2 gene with loss of SOD2 activity and a feed-forward mechanism promoting further epigenetic aberrancies [155–157]. Thereby, increased O2•− promotes generation of further ROS with the result that a number of metabolites critical for maintaining the cell homeostasis such as NAD(+), S-adenosyl methionine, and 2-oxoglutarate are altered. Since these metabolites are critical cofactors for enzymes like sirtuins, histone methyltransferases, and histone demethylases as well as DNA demethylases, respectively, fluctuations in these metabolites may thus have direct effects on the epigenetic landscape with the consequence of changes in gene expression [157]. Although the direct link of an epigenetic silencing of SOD2 in fibrosis remains to be determined, it appears to be obvious that loss of SOD2 activity promotes further epigenetic aberrancies which may be detrimental for a progression of cells from a fibrotic into a cancer phenotype.
9. Fibrosis and beyond: antioxidants as treatment options?
Fibrosis frequently develops as an own entity on the basis of most common pro-fibrotic co-morbidities like obesity, diabetes, non-alcoholic fatty liver disease) chronic kidney disease, idiopathic pulmonary fibrosis, or cardiomyopathy and is a key determinant for progression into diseases like liver cirrhosis, end-stage renal disease, lung fibrosis or even cancer. These diseases are to a large extend associated with a “Western life style” where dietary caloric intake has increased and energy expenditure has been reduced. Moreover, a lot of changes in the food composition with polyunsaturated fatty acids, excess proteins and cereals have been observed. In addition, chronic alcoholism and tobacco smoking increased severely keeping pace with ever worsening environmental toxicity. Thus, our established antioxidant capability is confronted with more difficulties than it is supposed to encounter, underpinning, at least in part, increased occurrence of the above mentioned diseases and fibrosis.
In light of this, it could be imagined that supplementation of diets with antioxidants would be a therapeutic option. This could, in addition to therapies specific to the underlying disease, and inhibition of TGF-β signaling, be helpful in terms of fibrosis. However, the expectations which were already put into the role of antioxidants and their health beneficial effects could not be met. With respect to lung fibrosis and respiratory diseases like asthma or chronic obstructive pulmonary disease, recent evidence suggested the therapeutic use of resveratrol, a natural antioxidative polyphenol present in grapes and various berries. However, though the in vitro and animal data would support resveratrol use, large clinical trials need to be performed to estimate its real benefit [158]. Moreover, recent meta-analyses of clinical studies analyzing the effects of β-carotene, vitamin A, and vitamin E on mortality indicate that nutrient supplementation with the “antioxidants” is paradoxically rather health threatening than beneficial since a high intake of these substances either as a single compound or in different combinations, had no beneficial effects [159]. In particular, vitamin A was even found to be associated with increased mortality [160–162]. Moreover, large scale studies with respect to cancer and nutritional supplementation with particular antioxidants also showed that the outcome is quite heterogeneous. The risk for cancer might be prevented or delayed by using a selective diet preferentially from an early stage of life; thereby a balanced energy intake and expenditure as well as individual genetic predispositions have to be considered. In line, analyses of antioxidant treatment in multicenter patient trials of idiopathic lung fibrosis showed also no or only modest beneficial effects [163–165]. Although ROS seem to have an important role in fibrosis, these studies make it difficult to interpolate and to make any further prediction/recommendation in terms of antioxidant therapy. However, the low evidence from the large scale studies available so far, indicates the demand to further increase our knowledge about the interconnection between nutrition, lifestyle and the dynamics of ROS/antioxidant action as well as to improve and to standardize techniques measuring reliable biomarkers in terms of fibrosis and underlying diseases.
Acknowledgments
The authors are grateful to all researchers who contributed to the field and apologize to all those whose work could not be cited due to space limitations. Work of the authors is supported by Grants from the Biocenter Oulu, the Academy of Finland (251314; 2012–2017), and Sigrid Juselius Foundation. KR, AK, TK were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU‐ROS).
References
- 1.Dvorak H.F., Form D.M., Manseau E.J., Smith B.D. J. Natl. Cancer Inst. 1984;73:1195–1205. [PubMed] [Google Scholar]
- 2.Dvorak H.F. N. Engl. J. Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 3.Kalluri R., Zeisberg M. Nat. Rev. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 4.Rockey D.C., Bell P.D., Hill J.A. N. Engl. J. Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 5.Hecker L., Jagirdar R., Jin T., Thannickal V.J. Exp. Cell Res. 2011;317:1914–1921. doi: 10.1016/j.yexcr.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisberg M., Yang C., Martino M., Duncan M.B., Rieder F., Tanjore H. J. Biol. Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 7.Bucala R., Spiegel L.A., Chesney J., Hogan M., Cerami A. Mol. Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 8.Strutz F., Okada H., Lo C.W., Danoff T., Carone R.L., Tomaszewski J.E. J.Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S.L., Kisseleva T., Brenner D.A., Duffield J.S. Am. J. Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ronnov-Jessen L., Petersen O.W., Koteliansky V.E., Bissell M.J. J. Clin. Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E. Nat.Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 12.Sugimoto H., Mundel T.M., Kieran M.W., Kalluri R. Cancer. Biol. Ther. 2006;5:1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 13.Bayreuther K., Rodemann H.P., Hommel R., Dittmann K., Albiez M., Francz P.I. Proc. Natl. Acad. Sci. USA. 1988;85:5112–5116. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anders H.J., Muruve D.A. J. Am. Soc. Nephrol. 2011;22:1007–1018. doi: 10.1681/ASN.2010080798. [DOI] [PubMed] [Google Scholar]
- 15.Sanjuan M.A., Dillon C.P., Tait S.W.G., Moshiach S., Dorsey F., Connell S. Nature. 2007;450 doi: 10.1038/nature06421. 1253<last_page>1257. [DOI] [PubMed] [Google Scholar]
- 16.Murray L.A., Rubinowitz A., Herzog E.L. Curr. Opin. Rheumatol. 2012;24:656–662. doi: 10.1097/BOR.0b013e3283588de4. [DOI] [PubMed] [Google Scholar]
- 17.Wynn T.A. J. Exp. Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wynn T.A., Ramalingam T.R. Nat. Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babalola O., Mamalis A., Lev-Tov H., Jagdeo J. Arch. Dermatol. Res. 2014;306:313–330. doi: 10.1007/s00403-013-1416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paik Y.H., Kim J., Aoyama T., De Minicis S., Bataller R., Brenner D.A. Antioxid. Redox Signal. 2014;20:2854–2872. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes J.L., Gorin Y. Kidney Int. 2011;79:944–956. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seki S., Kitada T., Sakaguchi H. Hepatol. Res. 2005;33:132–134. doi: 10.1016/j.hepres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Dekhuijzen P.N., Aben K.K., Dekker I., Aarts L.P., Wielders P.L., van Herwaarden C.L. Am. J. Respir. Crit. Care Med. 1996;154:813–816. doi: 10.1164/ajrccm.154.3.8810624. [DOI] [PubMed] [Google Scholar]
- 24.Pratico D., Basili S., Vieri M., Cordova C., Violi F., Fitzgerald G.A. Am. J. Respir. Crit. Care Med. 1998;158:1709–1714. doi: 10.1164/ajrccm.158.6.9709066. [DOI] [PubMed] [Google Scholar]
- 25.Pilger A., Germadnik D., Schaffer A., Theiler A., Pils P., Sluka F. Int. Arch. Occup. Environ. Health. 2000;73:305–310. doi: 10.1007/s004200000117. [DOI] [PubMed] [Google Scholar]
- 26.Pelclova D., Fenclova Z., Kacer P., Kuzma M., Navratil T., Lebedova J. Ind. Health. 2008;46:484–489. doi: 10.2486/indhealth.46.484. [DOI] [PubMed] [Google Scholar]
- 27.Ha H., Lee H.B. J. Am. Soc. Nephrol. 2003;14:S246–S249. doi: 10.1097/01.asn.0000077411.98742.54. [DOI] [PubMed] [Google Scholar]
- 28.Djamali A., Vidyasagar A., Adulla M., Hullett D., Reese S. Am. J. Transplant. 2009;9:74–82. doi: 10.1111/j.1600-6143.2008.02463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murdoch C.E., Zhang M., Cave A.C., Shah A.M. Cardiovasc. Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Sirker A., Zhang M., Murdoch C., Shah A.M. Am. J. Nephrol. 2007;27:649–660. doi: 10.1159/000109148. [DOI] [PubMed] [Google Scholar]
- 31.Aragno M., Mastrocola R., Alloatti G., Vercellinatto I., Bardini P., Geuna S. Endocrinology. 2008;149:380–388. doi: 10.1210/en.2007-0877. [DOI] [PubMed] [Google Scholar]
- 32.Samoylenko A., Hossain J.A., Mennerich D., Kellokumpu S., Hiltunen J.K., Kietzmann T. Antioxid. Redox Signal. 2013;19:2157–2196. doi: 10.1089/ars.2012.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown D.I., Griendling K.K. Free Radic. Biol. Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neill S., Brault J., Stasia M.J., Knaus U.G. Redox Biol. 2015;6:135–156. doi: 10.1016/j.redox.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nauseef W.M. Biochim. Biophys. Acta. 1840;2014:757–767. doi: 10.1016/j.bbagen.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deffert C., Cachat J., Krause K.H. Cell. Microbiol. 2014;16:1168–1178. doi: 10.1111/cmi.12322. [DOI] [PubMed] [Google Scholar]
- 37.Aguirre J., Lambeth J.D. Free Radic. Biol. Med. 2010;49:1342–1353. doi: 10.1016/j.freeradbiomed.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badid N., Ahmed F.Z., Merzouk H., Belbraouet S., Mokhtari N., Merzouk S.A. Pathol. Oncol. Res. 2010;16:159–167. doi: 10.1007/s12253-009-9199-0. [DOI] [PubMed] [Google Scholar]
- 39.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C. Nat. Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S. Circ. Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 41.Sedeek M., Nasrallah R., Touyz R.M., Hebert R.L. J. Am. Soc. Nephrol. 2013;24:1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masamune A., Watanabe T., Kikuta K., Satoh K., Shimosegawa T. AJP: Gastrointest. Liver Physiol. 2007;294:G99–G108. doi: 10.1152/ajpgi.00272.2007. [DOI] [PubMed] [Google Scholar]
- 43.De Minicis S., Brenner D.A. Arch. Biochem. Biophys. 2007;462:266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui W., Matsuno K., Iwata K., Ibi M., Matsumoto M., Zhang J. Hepatology. 2011;54:949–958. doi: 10.1002/hep.24465. [DOI] [PubMed] [Google Scholar]
- 45.Paik Y.H., Iwaisako K., Seki E., Inokuchi S., Schnabl B., Osterreicher C.H. Hepatology. 2011;53:1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petry A., Djordjevic T., Weitnauer M., Kietzmann T., Hess J., Gorlach A. Antioxid. Redox Signal. 2006;8:1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 47.Bernard K., Hecker L., Luckhardt T.R., Cheng G., Thannickal V.J. Antioxid. Redox Signal. 2014;20:2838–2853. doi: 10.1089/ars.2013.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akram K.M., Samad S., Spiteri M., Forsyth N.R. Adv. Biochem. Eng. Biotechnol. 2013;130:105–129. doi: 10.1007/10_2012_140. [DOI] [PubMed] [Google Scholar]
- 49.Aravinthan A., Scarpini C., Tachtatzis P., Verma S., Penrhyn-Lowe S., Harvey R. J. Hepatol. 2013;58:549–556. doi: 10.1016/j.jhep.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 50.Rudolph K.L., Chang S., Millard M., Schreiber-Agus N., DePinho R.A. Science. 2000;287:1253–1258. doi: 10.1126/science.287.5456.1253. [DOI] [PubMed] [Google Scholar]
- 51.Roberts A.B., Anzano M.A., Lamb L.C., Smith J.M., Frolik C.A., Marquardt H. Nature. 1982;295:417–419. doi: 10.1038/295417a0. [DOI] [PubMed] [Google Scholar]
- 52.Zheng W. Methods Mol. Biol. 2009;472:265–277. doi: 10.1007/978-1-60327-492-0_11. [DOI] [PubMed] [Google Scholar]
- 53.Leask A., Abraham D.J. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 54.Leask A., Denton C.P., Abraham D.J. J. Invest. Dermatol. 2004;122:1–6. doi: 10.1046/j.0022-202X.2003.22133.x. [DOI] [PubMed] [Google Scholar]
- 55.Abraham D.J., Shiwen X., Black C.M., Sa S., Xu Y., Leask A. J. Biol. Chem. 2000;275:15220–15225. doi: 10.1074/jbc.275.20.15220. [DOI] [PubMed] [Google Scholar]
- 56.Hogaboam C.M., Blease K., Mehrad B., Steinhauser M.L., Standiford T.J., Kunkel S.L. Am. J. Pathol. 2000;156:723–732. doi: 10.1016/S0002-9440(10)64775-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim Y., Ratziu V., Choi S.G., Lalazar A., Theiss G., Dang Q. J. Biol. Chem. 1998;273:33750–33758. doi: 10.1074/jbc.273.50.33750. [DOI] [PubMed] [Google Scholar]
- 58.Romanelli R.G., Caligiuri A., Carloni V., DeFranco R., Montalto P., Ceni E. Br. J. Pharmacol. 1997;122:1047–1054. doi: 10.1038/sj.bjp.0701484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radwan M.I., Pasha H.F., Mohamed R.H., Hussien H.I., El-Khshab M.N. Cytokine. 2012;60:271–276. doi: 10.1016/j.cyto.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Sturrock A., Cahill B., Norman K., Huecksteadt T.P., Hill K., Sanders K. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 61.Carmona-Cuenca I., Roncero C., Sancho P., Caja L., Fausto N., Fernandez M. J. Hepatol. 2008;49:965–976. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 62.Boudreau H.E., Casterline B.W., Rada B., Korzeniowska A., Leto T.L. Free Radic. Biol. Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crestani B., Besnard V., Boczkowski J. Int. J. Biochem. Cell Biol. 2011;43:1086–1089. doi: 10.1016/j.biocel.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 64.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T. Sci.Transl.Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diebold I., Flugel D., Becht S., Belaiba R.S., Bonello S., Hess J. Antioxid. Redox Signal. 2010;13:425–436. doi: 10.1089/ars.2009.3014. [DOI] [PubMed] [Google Scholar]
- 66.Diebold I., Petry A., Hess J., Gorlach A. Mol. Biol. Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carnesecchi S., Deffert C., Donati Y., Basset O., Hinz B., Preynat-Seauve O. Antioxid. Redox Signal. 2011;15:607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manoury B., Nenan S., Leclerc O., Guenon I., Boichot E., Planquois J.M. Respir. Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jarman E.R., Khambata V.S., Cope C., Jones P., Roger J., Ye L.Y. Am. J. Respir. Cell Mol. Biol. 2014;50:158–169. doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 70.Bettaieb A., Jiang J.X., Sasaki Y., Chao T.I., Kiss Z., Chen X. Gastroenterology. 2015;149:468–480. doi: 10.1053/j.gastro.2015.04.009. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Proell V., Carmona-Cuenca I., Murillo M.M., Huber H., Fabregat I., Mikulits W. Comp. Hepatol. 2007;6:1. doi: 10.1186/1476-5926-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sancho P., Mainez J., Crosas-Molist E., Roncero C., Fernandez-Rodriguez C.M., Pinedo F. PLoS One. 2012;7:e45285. doi: 10.1371/journal.pone.0045285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aram G., Potter J.J., Liu X., Wang L., Torbenson M.S., Mezey E. Hepatology. 2009;49:911–919. doi: 10.1002/hep.22708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Imura Y., Stassen J.M., Kurokawa T., Iwasa S., Lijnen H.R., Collen D. Blood. 1992;79:2322–2329. [PubMed] [Google Scholar]
- 75.Jiang J.X., Venugopal S., Serizawa N., Chen X., Scott F., Li Y. Gastroenterology. 2010;139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thannickal V.J. Int. J. Biochem. Cell Biol. 2010;42:1398–1400. doi: 10.1016/j.biocel.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foyer C.H., Noctor G. Plant Physiol. 2011;155:2–18. doi: 10.1104/pp.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winterbourn C.C., Metodiewa D. Arch. Biochem. Biophys. 1994;314:284–290. doi: 10.1006/abbi.1994.1444. [DOI] [PubMed] [Google Scholar]
- 79.Winterbourn C.C. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 80.Blokhina O., Fagerstedt K.V. Plant Physiol. Biochem. 2010;48:359–373. doi: 10.1016/j.plaphy.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 81.Fernandes A.P., Holmgren A. Antioxid. Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 82.Choi J., Corder N.L., Koduru B., Wang Y. Free Radic. Biol. Med. 2014;72:267–284. doi: 10.1016/j.freeradbiomed.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geybels M.S., Hutter C.M., Kwon E.M., Ostrander E.A., Fu R., Feng Z. Prostate. 2013;73:734–742. doi: 10.1002/pros.22617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao B., Bataller R. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bianchi G., Brizi M., Rossi B., Ronchi M., Grossi G., Marchesini G. Metabolism. 2000;49:1434–1439. doi: 10.1053/meta.2000.16554. [DOI] [PubMed] [Google Scholar]
- 86.Arsalane K., Dubois C.M., Muanza T., Begin R., Boudreau F., Asselin C. Am. J. Respir. Cell Mol. Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- 87.Ramani K., Tomasi M.L., Yang H., Ko K., Lu S.C. J. Biol. Chem. 2012;287:36341–36355. doi: 10.1074/jbc.M112.370775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu R.M., Vayalil P.K., Ballinger C., Dickinson D.A., Huang W.T., Wang S. Free Radic. Biol. Med. 2012;53:554–563. doi: 10.1016/j.freeradbiomed.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tiitto L.H., Peltoniemi M.J., Kaarteenaho-Wiik R.L., Soini Y.M., Paakko P.K., Sormunen R.T. Hum. Pathol. 2004;35:832–839. doi: 10.1016/j.humpath.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 90.Peltoniemi M.J., Rytila P.H., Harju T.H., Soini Y.M., Salmenkivi K.M., Ruddock L.W. Respir. Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harju T.H., Peltoniemi M.J., Rytila P.H., Soini Y., Salmenkivi K.M., Board P.G. Respir. Res. 2007;8:48. doi: 10.1186/1465-9921-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McCord J.M., Fridovich I. Antioxid. Redox Signal. 2014;20:1548–1549. doi: 10.1089/ars.2013.5547. [DOI] [PubMed] [Google Scholar]
- 93.Wood Z.A., Poole L.B., Karplus P.A. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 94.Oberley T.D., Verwiebe E., Zhong W., Kang S.W., Rhee S.G. Free Radic. Biol. Med. 2001;30:412–424. doi: 10.1016/s0891-5849(00)00486-x. [DOI] [PubMed] [Google Scholar]
- 95.Go Y.M., Jones D.P. Antioxid. Redox Signal. 2010;13:489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Immenschuh S., Baumgart-Vogt E., Tan M., Iwahara S., Ramadori G., Fahimi H.D. J. Histochem. Cytochem. 2003;51:1621–1631. doi: 10.1177/002215540305101206. [DOI] [PubMed] [Google Scholar]
- 97.Seo M.S., Kang S.W., Kim K., Baines I.C., Lee T.H., Rhee S.G. J. Biol. Chem. 2000;275:20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- 98.Holmgren A., Johansson C., Berndt C., Lonn M.E., Hudemann C., Lillig C.H. Biochem. Soc. Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 99.Lubos E., Loscalzo J., Handy D.E. Antioxid. Redox Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Imai H., Nakagawa Y. Free Radic. Biol. Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 101.Nomura K., Imai H., Koumura T., Kobayashi T., Nakagawa Y. Biochem. J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Islam K.N., Kayanoki Y., Kaneto H., Suzuki K., Asahi M., Fujii J. Free Radic. Biol. Med. 1997;22:1007–1017. doi: 10.1016/s0891-5849(96)00493-5. [DOI] [PubMed] [Google Scholar]
- 103.Kayanoki Y., Fujii J., Suzuki K., Kawata S., Matsuzawa Y., Taniguchi N. J. Biol. Chem. 1994;269:15488–15492. [PubMed] [Google Scholar]
- 104.Michaeloudes C., Sukkar M.B., Khorasani N.M., Bhavsar P.K., Chung K.F. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011;300:L295–L304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hayes J.D., Dinkova-Kostova A.T. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 106.Levonen A.L., Hill B.G., Kansanen E., Zhang J., Darley-Usmar V.M. Free Radic. Biol. Med. 2014;71:196–207. doi: 10.1016/j.freeradbiomed.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kensler T.W., Wakabayashi N., Biswal S. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 108.Jain A.K., Mahajan S., Jaiswal A.K. J. Biol. Chem. 2008;283:17712–17720. doi: 10.1074/jbc.M709854200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 109.Aleksunes L.M., Manautou J.E. Toxicol. Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 110.Liu J., Wu K.C., Lu Y.F., Ekuase E., Klaassen C.D. Oxid. Med. Cell. Longev. 2013:305861. doi: 10.1155/2013/305861. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu W., Hellerbrand C., Kohler U.A., Bugnon P., Kan Y.W., Werner S. Lab. Invest. 2008;88:1068–1078. doi: 10.1038/labinvest.2008.75. [DOI] [PubMed] [Google Scholar]
- 112.Okada K., Warabi E., Sugimoto H., Horie M., Tokushige K., Ueda T. J. Gastroenterol. 2012;47:924–935. doi: 10.1007/s00535-012-0552-9. [DOI] [PubMed] [Google Scholar]
- 113.Chowdhry S., Nazmy M.H., Meakin P.J., Dinkova-Kostova A.T., Walsh S.V., Tsujita T. Free Radic. Biol. Med. 2010;48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 114.Sugimoto H., Okada K., Shoda J., Warabi E., Ishige K., Ueda T. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;298:G283–G294. doi: 10.1152/ajpgi.00296.2009. [DOI] [PubMed] [Google Scholar]
- 115.Zhang Y.K., Yeager R.L., Tanaka Y., Klaassen C.D. Toxicol. Appl. Pharmacol. 2010;245:326–334. doi: 10.1016/j.taap.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sueblinvong V., Kerchberger V.E., Saghafi R., Mills S.T., Fan X., Guidot D.M. Alcohol. Clin. Exp. Res. 2014;38:336–343. doi: 10.1111/acer.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sueblinvong V., Tseng V., Smith T., Saghafi R., Mills S.T., Neujahr D.C. Alcohol. Clin. Exp. Res. 2014;38:2731–2742. doi: 10.1111/acer.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cebula M., Schmidt E.E., Arner E.S. Antioxid. Redox Signal. 2015 doi: 10.1089/ars.2015.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zucker S.N., Fink E.E., Bagati A., Mannava S., Bianchi-Smiraglia A., Bogner P. Mol. Cell. 2014;53:916–928. doi: 10.1016/j.molcel.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ma Q., He X. Pharmacol. Rev. 2012;64:1055–1081. doi: 10.1124/pr.110.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S. Nat. Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 122.Jiang T., Chen N., Zhao F., Wang X.J., Kong B., Zheng W. Cancer Res. 2010;70:5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Robertson I.B., Horiguchi M., Zilberberg L., Dabovic B., Hadjiolova K., Rifkin D.B. Matrix Biol. 2015 doi: 10.1016/j.matbio.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Munger J.S., Harpel J.G., Giancotti F.G., Rifkin D.B. Mol. Biol. Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lyons R.M., Keski-Oja J., Moses H.L. J. Cell Biol. 1988;106:1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jobling M.F., Mott J.D., Finnegan M.T., Jurukovski V., Erickson A.C., Walian P.J. Vol. 166. 2006. pp. 839–848. (Radiat. Res.). [DOI] [PubMed] [Google Scholar]
- 128.Abraham D.J., Varga J. Trends Immunol. 2005;26:587–595. doi: 10.1016/j.it.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 129.Asada N., Takase M., Nakamura J., Oguchi A., Asada M., Suzuki N. J. Clin. Invest. 2011;121:3981–3990. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dimmeler S., Zeiher A.M. J. Am. Coll. Cardiol. 2004;44:464–466. doi: 10.1016/j.jacc.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 131.Distler O., Distler J.H., Scheid A., Acker T., Hirth A., Rethage J. Circ.Res. 2004;95:109–116. doi: 10.1161/01.RES.0000134644.89917.96. [DOI] [PubMed] [Google Scholar]
- 132.Moon J.O., Welch T.P., Gonzalez F.J., Copple B.L. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G582–G592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Corpechot C., Barbu V., Wendum D., Kinnman N., Rey C., Poupon R. Hepatology. 2002;35:1010–1021. doi: 10.1053/jhep.2002.32524. [DOI] [PubMed] [Google Scholar]
- 134.Ioannou M., Pyrpasopoulou A., Simos G., Paraskeva E., Nikolaidou C., Venizelos I. Mod. Rheumatol. 2013;23:1245–1248. doi: 10.1007/s10165-012-0787-6. [DOI] [PubMed] [Google Scholar]
- 135.Yoon Y.S., Uchida S., Masuo O., Cejna M., Park J.S., Gwon H.C. Circulation. 2005;111:2073–2085. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]
- 136.Xu X., Tan X., Tampe B., Sanchez E., Zeisberg M., Zeisberg E.M. J. Biol. Chem. 2015 doi: 10.1074/jbc.M115.636944. jbc.M115.636944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wynn T.A. Nat. Med. 2010;16:523–525. doi: 10.1038/nm0510-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pottier N., Cauffiez C., Perrais M., Barbry P., Mari B. Trends Pharmacol.Sci. 2014;35:119–126. doi: 10.1016/j.tips.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 139.Cheng X., Ku C.H., Siow R.C. Free Radic. Biol. Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 140.Narasimhan M., Patel D., Vedpathak D., Rathinam M., Henderson G., Mahimainathan L. PLoS One. 2012;7:e51111. doi: 10.1371/journal.pone.0051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Espinosa-Diez Cristina, Fierro-Fernández Marta, Sánchez-Gómez Francisco, Rodríguez-Pascual Fernando, Alique Matilde, Ruiz-Ortega Marta, Beraza Naiara, Martínez-Chantar Maria L., Fernández-Hernando Carlos, and Lamas Santiago. Antioxidants & Redox Signaling, ahead of print. 10.1089/ars.2014.6025 [DOI] [PMC free article] [PubMed]
- 142.H.Yang, T.W.Li, Y.Zhou, H.Peng, T.Liu, E.Zandi, et al., Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice.-PubMed-NCBI 2015. [DOI] [PMC free article] [PubMed]
- 143.Jiang L., Qiu W., Zhou Y., Wen P., Fang L., Cao H. Kidney Int. 2013;84:285–296. doi: 10.1038/ki.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Helin K., Dhanak D. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 145.Franco R., Schoneveld O., Georgakilas A.G., Panayiotidis M.I. Cancer Lett. 2008;266:6–11. doi: 10.1016/j.canlet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 146.Turk P.W., Laayoun A., Smith S.S., Weitzman S.A. Carcinogenesis. 1995;16:1253–1255. doi: 10.1093/carcin/16.5.1253. [DOI] [PubMed] [Google Scholar]
- 147.Ziech D., Franco R., Pappa A., Panayiotidis M.I. Mutat. Res. 2011;711:167–173. doi: 10.1016/j.mrfmmm.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 148.Bian E.B., Huang C., Ma T.T., Tao H., Zhang H., Cheng C. Toxicol. Appl. Pharmacol. 2012;264:13–22. doi: 10.1016/j.taap.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 149.Robinson C.M., Neary R., Levendale A., Watson C.J., Baugh J.A. Respir. Res. 2012;13 doi: 10.1186/1465-9921-13-74. 74-9921-13-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tampe B., Tampe D., Muller C.A., Sugimoto H., LeBleu V., Xu X. J. Am. Soc. Nephrol. 2014;25:905–912. doi: 10.1681/ASN.2013070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xiao X., Tang W., Yuan Q., Peng L., Yu P. Int. J. Mol. Med. 2015;35:1596–1602. doi: 10.3892/ijmm.2015.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang J.J., Tao H., Huang C., Shi K.H., Ma T.T., Bian E.B. Cell. Signal. 2013;25:1202–1211. doi: 10.1016/j.cellsig.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 153.Tao H., Huang C., Yang J.J., Ma T.T., Bian E.B., Zhang L. Toxicology. 2011;290:327–333. doi: 10.1016/j.tox.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 154.Zhao Q., Fan Y.C., Zhao J., Gao S., Zhao Z.H., Wang K. J. Viral Hepat. 2013;20:430–437. doi: 10.1111/jvh.12048. [DOI] [PubMed] [Google Scholar]
- 155.Hitchler M.J., Wikainapakul K., Yu L., Powers K., Attatippaholkun W., Domann F.E. Epigenetics. 2006;1:163–171. doi: 10.4161/epi.1.4.3401. [DOI] [PubMed] [Google Scholar]
- 156.Teoh-Fitzgerald M.L., Fitzgerald M.P., Jensen T.J., Futscher B.W., Domann F.E. Mol. Cancer. Res. 2012;10:40–51. doi: 10.1158/1541-7786.MCR-11-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cyr A.R., Hitchler M.J., Domann F.E. Antioxid. Redox Signal. 2013;18:1946–1955. doi: 10.1089/ars.2012.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Conte E., Fagone E., Fruciano M., Gili E., Iemmolo M., Vancheri C. Histol. Histopathol. 2015;30:523–529. doi: 10.14670/HH-30.523. [DOI] [PubMed] [Google Scholar]
- 159.Vrolijk M.F., Opperhuizen A., EHJM Jansen R.W., Godschalk F.J., Van Schooten A. Bast. Redox Biol. 2015;4:272–278. doi: 10.1016/j.redox.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee S.H. Science. 2001;292:2083–2086. doi: 10.1126/science.1059501. [DOI] [PubMed] [Google Scholar]
- Bjelakovic G., Nikolova D., Gluud L.L., Simonetti R.G., Gluud C. JAMA. 2007;297:842. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 162.Bjelakovic G., Nikolova D., Gluud C. Curr. Opin. Clin. Nutr. Metab. Care. 2013:1. doi: 10.1097/MCO.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 163.Martinez MJ, de Andrade JA, Anstrom KJ, King TE, Jr, Raghu G, Idiopathic Pulmonary Fibrosis Clinical Research Network N. Engl. J. Med. 2014;370:2093–2101. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Demedts M., Behr J., Buhl R., Costabel U., Dekhuijzen R., Jansen H.M. N. Engl. J. Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 165.Day B.J. Antioxid. Redox Signal. 2008;10:355–370. doi: 10.1089/ars.2007.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]