

Abstract
Soybean lipoxygenase-1 (SLO-1) is a paradigmatic enzyme system for studying the contribution of hydrogen tunneling to enzymatic proton-coupled electron transfer processes. In this study, the impact of pairs of double mutants on the properties of SLO-1 is presented. Steady-state rates and their deuterium kinetic isotope effects (KIEs) have been measured for the bimolecular reaction of enzyme with free substrate (kcat/Km) and compared to the unimolecular rate constant, kcat. A key kinetic finding is that the competitive KIEs on the second-order rate constant (kcat/Km) are all reduced from Dkcat and, despite large changes in rate and activation parameters, remain essentially unaltered under a variety of conditions. These data implicate a protein reaction coordinate that is orthogonal to the chemical reaction coordinate and controls the concentration of the active enzyme. This study introduces a new means to interrogate the alteration of conformational landscapes that can occur following site-specific mutagenesis.
Enzymes are truly extraordinary catalysts with enormous rate enhancements of up to 1026-fold1 over uncatalyzed reactions and turnover numbers approaching 106 s–1 (e.g., ref (2)). In many instances, the binding and release of substrate(s) and product(s) are slower than the catalyzed bond cleavage events, making measured rate constants an underestimate of the true catalytic power of an enzyme.3 It is generally accepted that this enormous catalytic efficiency is dependent on the specific organization of a large number of protein side chains, cofactors, etc., within the active site that, because of the huge entropic barrier, has no counterpart in comparative uncatalyzed reactions.4 Within this context, the textbook explanation for enzymatic rate accelerations has been largely focused on differences in electrostatic interactions between the transition state and ground state that are intrinsic to folded three-dimensional protein structures.5 As an alternate view, dynamical models of catalysis are gaining ground, implicating conformational sampling and active site motions in the evolution of highly active enzymes.6−9 While single-molecule studies have corroborated a correlation between a changing conformational landscape and altered catalytic competence on the time scale of seconds,10,11 the demonstration of a catalytic role for a more rapidly interconverting landscape, on time scales of milliseconds and faster, can be a daunting task. Toward this goal, the development of new kinetic and spectroscopic tools has become increasingly important for defining the role of protein dynamics in the bond cleavage steps of catalysis.
Soybean lipoxygenase (SLO-1) is one of several paradigmatic systems for studies of the C–H bond activation process via hydrogen tunneling.12−17 The substrate of SLO-1, linoleic acid (LA), undergoes an initial transfer of a net hydrogen atom from C-11 of substrate to the active site ferric hydroxide, generating a pentadienyl radical and ferrous water. In a second step, the substrate-derived intermediate is trapped by addition of molecular oxygen at C-13, leading to the lipid hydroperoxide product and a reoxidized, ferric state for the active site metal ion (Scheme 1). The large size of the primary hydrogen isotope effect (KIE ∼ 80) in WT SLO-1, which increases to an enormous value of 500–700 for a double active site SLO-1 mutant,18 has provided strong evidence of a model in which the barrier to C–H bond activation free energy originates entirely from the heavy atom motions of the protein and substrate.13,15−17
Scheme 1. Mechanism of Oxidation of Linoleic Acid (LA) Catalyzed by SLO-1.

SLO-1 catalyzes regio- and stereospecific conversion of linoleic acid to produce 13(S)-hydroperoxy-9(Z),11(E)-octadecanoic acid [13-(S)-HPOD].
Early mutational studies of SLO-1 were focused on single hydrophobic side chains that are either in the proximity of the reacting C-11 position of substrate (Leu546 and Leu754) or one helix turn away (Ile553)13,15 (Figure 1). Among the six, single-site reduced bulk variants generated, five showed that the size of the KIE remains close to that of the WT enzyme at room temperature (with the one exception being Ile553Gly), while the temperature dependence of the KIE is altered from close to temperature-independent (WT) to highly temperature-dependent.13,15 These experimental trends have been explained within the context of a conformational landscape that can sample very short tunneling-ready donor–acceptor distances (2.8 Å) in the WT enzyme. Replacement of the native, bulky hydrophobic side chains (Figure 1) with smaller side chains simultaneously alters the ability of an enzyme to create such close donor–acceptor distances and enhances a local distance sampling or gating mode that temporarily restores the H donor and acceptor tunneling distance to one approximating that of the native enzyme. Importantly, while the role for a local donor–acceptor distance sampling mode can be uncovered via changes in the temperature dependence of the KIE, kinetic detection of the global conformational landscape remains a major challenge.7
Figure 1.
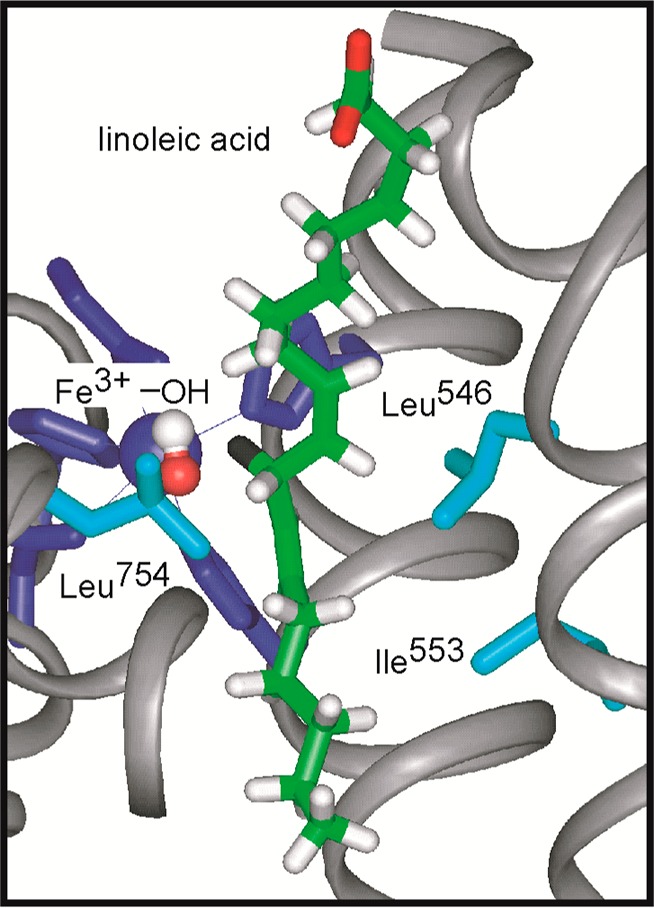
X-ray structure for SLO-1, with LA modeled into the active site. Figure taken from ref (13). Copyright 2002 American Chemical Society.
One question that emerged during our continuing focus on the kinetic implications of the mutational introduction of packing defects within the interior of proteins was the limit to which a protein can be “pushed”, before an active site becomes unable to compensate for structural disruptions, via either global sampling or the increased participation of a donor–acceptor gating mode. Within this context, a series of double mutants of SLO-1 were generated: Leu546Ala/Leu754Ala, Leu546Ala/Ile553Ala, and Ile553Ala/Leu754Ala (cf. Figure 1). Studies of the Leu546Ala/Leu754Ala double mutant have been particularly instructive, revealing an apoprotein structure that is almost identical to that of the WT protein, with the exception of an expanded active site that is partially filled by additional water molecules.18 Perhaps, most significantly, this protein shows one of the largest, if not the largest, primary KIE ever reported for a C–H reaction in the condensed phase, implicating an expanded donor–acceptor distance that is prevented from restoring a suitably short tunneling distance. The kinetic aspects of Leu546Ala/Leu754Ala18 and earlier studies13,19,20 were largely focused on the unimolecular rate constant for conversion of the enzyme substrate complex to product. In this work, we introduce a systematic comparison of second-order (kcat/Km) and first-order (kcat) rate constants and their accompanying isotope effects within the available series of double mutants. The results show the importance of an orthogonal reaction coordinate that controls the concentration of active enzyme via conformational sampling processes. These data and their analysis introduce a new kinetic approach for the detection of impaired protein conformational landscapes resulting from site-specific mutagenesis
Materials and Methods
Mutagenesis, Expression, and Purification of the SLO-1 Double Mutants
Wild-type SLO-1 and double mutants were expressed and purified as described previously.19,20 The SLO-1 Ile553Ala/Leu754Ala plasmid double mutant was prepared starting from the Ile553Ala SLO-1 plasmid with mutation at position 754 using the forward (5′-CTT TCA GTG ATA GAG ATC GCA TCG ACA CAT GCT TCT-3′) and reverse (5′-AGA AGC ATG TGT CGA TGC GAT CTC TAT CAC TGA AAG-3′) primers. The mutant plasmids were isolated, and the double mutation was confirmed by sequencing utilizing three different primers that targeted different regions of the gene: the beginning of the gene, a 500 bp region of the gene containing the mutation site(s), and a region that covers 500 bp up to the end of the gene. The preparation of the remaining double mutants, Leu546Ala/Ile553Ala and Leu546Ala/Leu754Ala, has been previously described.18,20
These double mutants were expressed using the pT7-7 plasmid in Escherichia coli and purified as described before with some minor modifications.19−21 The starting culture was diluted 300-fold and incubated at 37 °C while being shaken until OD600 reached ∼0.7. The temperature was then rapidly lowered to 15 °C and the sample incubated while being shaken for an additional 96 h. The cells were harvested and stored at −80 °C until they were purified.
Cell paste from 3 L (∼18 g) was resuspended in ∼72 mL of lysis buffer [25 mM Tris base (pH 7.5), 0.1 mM EDTA, 1× BugBuster (Novagen), 1250 units of benzonase (Novagen), ∼0.5 mM aminoethylbenzenesulfonyl fluoride (Sigma), and ∼0.2 mg/mL lysozyme (Sigma)]. The lysis reaction was allowed to proceed for 40 min at room temperature (RT) followed by cooling to 4 °C for 20 min. The lysis mixture was centrifuged at 20K rpm for 20 min to pellet the insoluble cellular debris. The soluble supernatant was dialyzed in 20 mM BIS-TRIS (pH 6.0) for 3–4 h, centrifuged, and then loaded onto a column packed with 70 mL of SP Sepharose fast flow (Sigma). The column was then washed with 100 mL of 20 mM BIS-TRIS buffer (pH 6.0) until the absorbance at 280 nm reached zero. The bound proteins were eluted with a 600 mL linear gradient from 0 to 500 mM NaCl [solution A consisting of 20 mM BIS-TRIS (pH 6.0) and 0 mM NaCl; solution B consisting of 20 mM BIS-TRIS (pH 6.0) and 500 mM NaCl]. The fractions containing SLO-1 [identified via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and monitoring of enzyme activity] were pooled and further dialyzed in 20 mM BIS-TRIS (pH 6.0) to remove salt. Protein was then concentrated and further purified using an UNO S6 column (Bio-Rad) with a 210 mL stepwise gradient [A consisting of 0 mM NaCl in 20 mM BIS-TRIS (pH 6.0) and B consisting of 500 mM NaCl in 20 mM BIS-TRIS; gradient steps, 0% B for 10 min, 0 to 18% B over 10 min, 18 to 40% B over the next 20 min, followed by a wash with 100% B and equilibration of the column in buffer A]. The flow rate was 2.5 mL/min, and fraction sizes were 5 mL. The enzyme-containing fractions were pooled (∼120 mM NaCl), buffer exchanged in 100 mM borate (pH 9.0), and concentrated to a minimal volume. SLO-1 double mutants were routinely obtained with a final yield of ∼6 mg/L of cells. The purity of the enzymes was >90% as seen by SDS–PAGE.
Kinetic Measurements
Steady-state kinetics was performed on a Cary50 spectrophotometer in the single-wavelength mode. The reaction progress was monitored by following the generation of the product, 13(S)-hydoperoxy-9(Z),11(E)-octadecanoic acid [13-(S)-HPOD] (λ234 = 23600 M–1 cm–1). All assays were performed in 100 mM borate (pH 9.0) under an ambient atmosphere at a constant temperature regulated by a water-jacketed cuvette holder as described previously with some minor adjustments. Kinetic assays of double mutants with protio-LA were performed at substrate concentrations ranging from 0.70 to 35 μM, and those with dideutero-LA (11,11-2H2-LA) were performed from 5 to 35 μM. Final enzyme concentrations in the analysis of the protio substrate were 0.1–0.3 μM for Ile553Ala/Leu754Ala and 0.6–1 μM for Leu546Ala/Leu754Ala. For reaction of Ile553Ala/Leu754Ala with 11,11-2H2-LA, the enzyme concentration was ∼1.5 μM. It was not possible to assay Leu546Ala/Leu754Ala with the deutero substrate using this assay (see below). The UV kinetic profile shows a lag phase, which is longer for 11,11-2H2-LA at lower substrate concentrations and temperatures (≤30 °C). The lag phase gradually decreases with either an increase in substrate concentration or an increase in assay temperature and becomes much smaller for reactions performed at higher temperatures (≥45 °C) with higher concentrations (>20 μM LA). The lag phase is followed by a linear rate, which, in turn, is followed by a steady decrease in the reaction rate as the substrate concentration is depleted. The initial rates were fitted to the Michaelis–Menten equation to obtain the kinetic parameters kcat and kcat/Km. The errors associated with each kcat measurement were used to weight the exponential Arrhenius fit. The rate constants were corrected for the iron content of the mutant enzyme as determined by ICP (PerkinElmer Optima 3000 DV), using standardized iron solutions.
Solvent Viscosity Studies
The impact of viscosity on WT SLO-1, Leu546Ala/Leu754Ala, and Ile553Ala/Leu754Ala was dertermined at different relative viscosities (ηrel = η/η°, where η° is the viscosity of water at 20 °C) as previously described.22 A buffer with different relative viscosities was prepared by dissolving 0, 8, 14, 21.5, 26, and 30% by weight of glucose in 0.1 M CHES (pH 9.0) with corresponding relative viscosities of 1, 1.25, 1.5, 2.0, 2.5, and 3.0, respectively, at 20 °C. The enzymatic activities of WT SLO-1 and double mutants in 0.1 M CHES (pH 9.0) were found to be similar to those measured in 0.1 M borate (pH 9.0).
Solvent Isotope Effects
Solvent isotope effects for WT SLO-1, Leu546Ala/Leu754Ala, and Ile553Ala/Leu754Ala were obtained by comparing kinetic parameters for protio-linoleic acid at 30 °C in 0.1 M borate (pH 9.0) in D2O (pH meter reading of 8.6) with those in H2O (pH 9.0).
Circular Dichroism
Circular dichroism (CD) measurements for WT SLO-1, Leu546Ala/Ile553Ala, Leu546Ala/Leu754Ala, and Ile553Ala/Leu754Ala were taken on an Aviv 410 spectropolarimeter with a Peltier temperature-controlled cell holder using a 1 cm path length cuvette. A 50 μg/mL sample of either WT SLO-1 or other mutants in 0.1 M borate (pH 9.0) was equilibrated at 25 °C for 5 min before the CD signals from 200 to 300 nm were recorded.
Isolation and Purification of Perdeutero-Linoleic Acid (2H31-LA) from an Algal Fatty Acid Ester Mixture
Perdeutero-LA for the competitive KIE measurements was isolated from the algal fatty acid mixture (Cambridge Isotope Laboratories, methyl esters U-D 97–98%; DLM-2497-0) following Ag-silica column chromatography.21 In short, the algal fatty acid ester mixture (∼0.5 mL) in hexane was loaded onto a Ag-silica column (a mixture of 100 g of silica and a solution of 6.7 g of AgNO3 in 150 mL of methanol, first mixed, then dried completely by evaporation on a rotavap, and packed onto the column as a slurry with hexane). The bound esters were eluted from the Ag-silica column by running 100 mL of hexane, 200 mL of 2% ethyl acetate in hexane, 400 mL of 5% ethyl acetate in hexane, and finally 200 mL of 10% ethyl acetate in hexane. The presence of the perdeutero-LA esters in the 5% ethyl acetate fractions was verified by thin layer chromatography and NaOH hydrolysis followed by ES-MS. The fractions containing perdeutero-LA ester were pooled, evaporated to an oil, and then de-esterified with NaOH in ethanol. After complete hydrolysis, the mixture was acidified by adding ∼100 mL of 0.5 M acetic acid followed by extraction with CH2Cl2. Multiple extractions were performed to ensure complete extractions of perdeutero-LA from the aqueous mixture. The extracted organic layer was dried to an oil, dissolved in methanol, and stored at −80 °C for further purification. This perdeutero-LA was further enzymatically treated with WT SLO-1 to deplete the trace amount of protio-LA contamination (monitored in the spectrophotometer, to ensure 7–10% of substrate depletion) and then acidified, extracted, and dried as before. The protio-LA-depleted perdeutero-LA was dissolved in methanol and purified via RP-HPLC using a Phenomenex semipreparative HPLC column (Luna C18 100A, 250 mm × 10.00 mm, 5 μm) via isocratic elution (87.9% methanol, 12% H2O, and 0.1% acetic acid) at a flow rate of 3 mL/min. The RP-HPLC-purified perdeutero-LA was evaporated to dryness, dissolved in methanol, and stored at −80 °C.
Competitive Kinetic Isotope Effect (KIE) Measurements
The competitive KIEs [D(kcat/Km)] for WT SLO-1, Ile553Ala/Leu546Ala, Leu546Ala/Leu754Ala, and Ile553Ala/Leu754Ala at 10 and 30 °C in 0.1 M borate (pH 9.0) and 0.1 M Tris-HCl (pH 7.0) (ionic strength of 0.2 M) were determined as previously described23 with some minor changes. A known ratio of RP-HPLC-purified protio- and perdeutero-LA (1:4; total concentration of the substrate, 10 μM) was allowed to react with individual SLO-1 variants in the appropriate buffer and at the appropriate temperature. An aliquot of the reaction was monitored at 234 nm and stopped with acetic acid (final acetic acid concentration in the mixture of ∼5%) quenching at <5% of the total substrate consumption. The acidified reaction mixture was extracted with CH2Cl2 (three extractions, 5 mL each), evaporated to dryness, reconstituted in methanol, injected into an analytical C18 column (Phenomenex, Luna, 5 μm, 250 mm × 4.6 mm), and eluted at 1 mL/min with an isocratic mobile phase of 79.4% methanol, 21.5% H2O, and 0.1% acetic acid. The concentrations of the enzymes in the assay were ∼0.2, ∼8, ∼16, and ∼170 nM for WT SLO-1, Ile553Ala/Leu546Ala, Ile553Ala/Leu754Ala, and Leu546Ala/Leu754Ala, respectively. The baseline resolution of protio- and perdeutero-13-HPOD allowed the calculation of the competitive KIE as the ratio of corresponding peak areas, equated to the molar ratio of protio- and perdeutero-13-HPOD. The competitive KIE is calculated as ([P-H]/[P-D])+/([P-H]/[P-D])∞, where ([P-H]/[P-D])+ is the ratio of the integrated peak areas at <5% reaction conversion and ([P-H]/[P-D])∞ is the ratio of peak areas at complete conversion. The ratio of protio to deutero substrate, ([S-H]/[S-D])0, was found to be identical to ([P-H]/[P-D])∞.
Results
Impact of Active Site Defects on Steady-State Kinetics
Initial rate parameters for protio-LA with the double mutants can be obtained using a continuous spectroscopic assay, in which the formation of the dienoic hydroperoxide product is monitored at 234 nm (Materials and Methods). Both kcat and kcat/Km are summarized in Table 1, along with values for WT SLO-1 and the single-site mutants. There is a trend in which the impact of mutation is greatest for positions 754 > 546 > 553. As previously shown, Ile553Ala displays a rate almost identical to that of the WT, although the use of the deuterio substrate is less well tolerated by Ile553Ala and leads to more temperature-dependent KIEs [i.e., a greater impact on Ea(D) than on Ea(H) for Ile553Ala].13 Perhaps not surprisingly, the double mutants with Ile553Ala show a similar trend in kcat and kcat/Km as seen for the single mutants at positions 546 and 754. Additionally, the impact of the single mutants is not additive for Leu546Ala/Leu754Ala, showing a reduction in kcat relative to that of the WT that is ∼1 order of magnitude less than that calculated from the product of the rate reductions for the individual variants. Given the ping-pong nature of these reactions (Scheme 1), kcat/Km(O2) will be unchanged,22 with the result that Km(O2) is reduced in parallel with kcat, ensuring that the concentration of O2 in all of these experiments (237 μM) is sufficient to produce saturation kinetics with regard to O2.13,20,24
Table 1. Kinetic Parameters of SLO-1 and Mutants in 0.1 M Borate (pH 9.0)a.
| enzyme form | kcat (s–1) | kcat/KM (μM–1 s–1) | Ea(H) (kcal/mol) | AH (s–1) | Dkcat |
|---|---|---|---|---|---|
| SLO-1 WTb | 297 (12) | 11 (1) | 2.1 (0.2) | 9 × 103 (2 × 103) | 81 (5) |
| 553Ac | 280 (10) | 12 (1) | 1.9 (0.2) | 7 × 103 (2 × 103) | 93 (4) |
| 546Ab | 4.8 (0.6) | 0.33 (0.1) | 4.1 (0.4) | 4 × 104 (3 × 104) | 93 (9) |
| 546A/553Ad | 2.21 (0.09) | 0.11 (0.02) | 3.8 (0.4) | 1.1 × 103 (5 × 102) | 128 (3) |
| 754Ac | 0.31 (0.02) | 0.07 (0.02) | 4.1 (0.3) | 2 × 102 (2 × 102) | 112 (11) |
| 553A/754A | 0.56 (0.03) | 0.046 (0.020) | 6.9 (0.2) | 5.0 × 104 (2 × 104) | 85 (7) |
| 546A/754Ae | 0.021 (0.001) | 0.0023 (0.0004) | 9.9 (0.2) | 3.3 × 105 (2 × 105) | 729 (26) |
In all of our earlier published kinetic data for single-site mutants of SLO-1, there were either no or modest changes in Ea (from 2 to 4 kcal/mol), together with a reduction in the Arrhenius prefactor, AH, or a value for AH that is within experimental error of that of the WT. In contrast, double mutants Ile553Ala/Leu754Ala and Leu546Ala/Leu754Ala produce significant increases in both Ea (to 6.9 and 9.9 kcal/mol, respectively) and AH relative to those of the single mutants (Table 1 and Figure 2A). Ile553Ala/Leu754Ala is of special interest, as its rate is virtually identical to that of Leu754Ala. As shown in our study of mutated forms of the ht-ADH from Bacillus stearothermophilus, the phenomenon of an unchanged rate together with simultaneous elevation of both Ea and AH is diagnostic of conditions that cause a protein to reversibly occupy new conformational states that are catalytically ineffective.25 This point will be discussed again after the introduction of further probes of the behavior of the double mutants.
Figure 2.

(A) Arrhenius plots of SLO-1 double mutants for kcat with protio-LA. Data points for Ile553Ala/Leu754Ala (◆) from 5 to 50 °C and Leu546Ala/Leu754Ala (○) from 10 to 50 °C in 0.1 M borate (pH 9.0). (B) Impact of substrate deuteration on the kcat for the Ile553Ala/Leu754Ala SLO-1 double mutant. Data points for 11,11-d2-LA (□) from 15 to 45 °C in 0.1 M borate (pH 9.0). For comparison, the data points for protio-LA (◆) from 5 to 50 °C are also shown.
Spectroscopic Monitoring of Isotope Effects on the Unimolecular Rate Constant (kcat)
Using 11,11-2H2-linoleic acid (11,11-2H2-LA or 11,11-d2-LA) as a substrate, KIEs could be pursued with Ile553Ala/Leu754Ala using the same spectroscopic assay that was used for the protio substrate. The kcat isotope effects for Leu546Ala/Ile553Ala and Leu546Ala/Leu754Ala were already reported and are included in Table 1 for comparison. For Ile553Ala/Leu754Ala, the Dkcat at 30 °C remains large (85) and very similar to those of both the WT (81)13 and Ile553Ala/Leu546Ala (128)20 at the same temperature. The temperature dependence of kcat with the deuterated substrate is shown in Figure 2B, with a final value for AH/AD of ∼0.2. This inverse value for the isotope effect on the Arrhenius prefactor ratio is equally represented in the large difference in the enthalpy of activation [Ea(D) – Ea(H) = 3.5 kcal/mol]. Isotope effects on kcat provide a frame of reference for the comparative measurement of KIEs on kcat/Km.
Analysis of Isotope Effects on the Bimolecular Rate Constant (Dkcat/Km) by Competitive Methods
Analogous to earlier efforts to measure Dkcat with the slowest mutant (Leu546Ala/Leu754Ala),18 spectroscopic assays proved to be too insensitive to measure turnover at low substrate concentrations using deuterium-labeled LA, a prerequisite for obtaining D(kcat/Km) values. A discontinuous assay that involves analyzing the reaction of 1:4 mixtures of protio- and perdeutero-LA was pursued. As illustrated in panels A and B of Figure 3, the monitoring of hydroperoxo products from perprotio versus perdeutereo substrates can be achieved with baseline resolution. Time-dependent assays based on the quantification of the peaks in Figure 3B yield the competitive isotope effects of Table 2. As seen previously with native SLO-1,22,26 the recombinant WT shows reduced values for D(kcat/Km) at high pH and low temperature, increasing at pH 7.0 and 30 °C toward the value for Dkcat. The fact that D(kcat/Km) remains below Dkcat under all conditions means that a step other than C–H bond cleavage is contributing to kcat/Km. In the context of an intrinsic KIE for H transfer of ∼80, the data at pH 7.0 indicate that this second step is approximately equal in magnitude to the C–H bond cleavage step. Quite unexpectedly, despite the very large rate reductions observed with the double mutants, their competitive KIEs also remain reduced from Dkcat and almost indistinguishable from those for WT SLO-1 at 30 °C. If the only impact of the mutations at positions 756, 564, and 553 had been on the barrier for the C–H bond abstraction step, a decreased rate for bond cleavage alone should have produced an increase in the observed values for D(kcat/Km). In an effort to explore this anomaly, we turned to solvent isotope and viscogen measurements.
Figure 3.

HPLC traces of separation of product hydroperoxides (HPODs) formed from perprotio-linoleic acid (HLA) and perdeuterio-linoleic acid (2H31LA). (A) HPLC trace of a 1.5 mL aliquot of a reaction mixture that is allowed to react with WT SLO-1 to completion, showing product formation with the composition of starting substrate mixture (1:4, HLA:2H31LA). (B) HPLC trace of a 20 mL aliquot taken 6 min after the addition of Ile553Ala/Leu754Ala to the substrate mixture with the composition described above. The inset shows the gradual increase in the peak area from 4 min (gray trace) to 6 min (black trace) reaction time.
Table 2. Competitive Deuterium Isotope Effects on kcat/Kma.
| enzyme form | pH 9.0, 30 °Cb | pH 9.0, 10 °Cb | pH 7.0, 30 °Cb |
|---|---|---|---|
| WT SLO-1 | 27 (1) | 15 (1) | 50 (4) |
| 546A/553A | 35 (2) | 39 (7) | 44 (3) |
| 553A/754A | 31 (2) | 28 (1) | 49 (4) |
| 546A/754A | 35 (1) | 32 (2) | 40 (2) |
All measurements are new to this study.
Average values for isotope effects are 32, 29, and 46 from left to right columns, respectively.
Solvent Isotope Effects
A comparison of reaction rates for SLO-1 in H2O and D2O at 30 °C and pH 9.0 affords the solvent isotope effects (SIEs) under conditions where a second kinetic step, in addition to C–H bond cleavage step, contributes to kcat/Km. No effort was made to correct for SIEs on the pKa for a protein-bound residue(s) as kcat is pH-independent in this range and kcat/Km has plateaued.22,26 WT SLO-1 shows a small SIE on kcat and a slightly larger value on kcat/Km (Table 3). For comparison, the Ile553Ala/Leu754Ala double mutant shows a slightly larger SIE for kcat and a slightly smaller SIE on kcat/Km, whereas the Leu546Ala/Leu754Ala SLO-1 double mutant has SIEs of unity for both kcat and kcat/Km. In any case, the impact of D2O on rate is small, and in no instance do the differences between the WT and double mutants appear to be very significant within experimental error.
Table 3. Solvent Isotope Effect at 30 °Ca.
| enzyme form | D2Okcat | D2O(kcat/Km) |
|---|---|---|
| WT SLO-1 | 1.20 (0.10) | 1.52 (0.31) |
| 553A/754A | 1.46 (0.11) | 1.27 (0.27) |
| 546A/754A | 1.04 (0.12) | 0.91 (0.28) |
H2O buffer: 0.1 M borate (pH 9.0) in H2O. D2O buffer: 0.1 M borate (pD 9.0) in D2O (with a pH meter reading of 8.6).
Viscogen Effects
Following the protocols of earlier studies with the native SLO-1 from soybeans,22 we employed 0–30% (by weight) glucose as a viscogen and interrogated its impact on recombinant WT SLO-1 and two of its double mutants, Ile553Ala/Leu754Ala and Leu546Ala/Leu754Ala. Assays conducted with protio-LA at 20 °C and pH 9 showed little or no impact of the viscogen on kcat/Km for either WT enzyme or the slowest double mutant, Leu546Ala/Leu754Ala (Figure 4A,C). In the case of Ile553Ala/Leu754Ala (Figure 4B), there is actually a small increase in kcat/Km, opposite to the direction expected if substrate binding were partially rate-determining. As controls, the effect of viscogen on kcat was also examined, indicating a very small increase for the WT and Leu546Ala/Leu754Ala and a decrease of ∼20% over a one-unit increase in viscosity for Ile553Ala/Leu754Ala. Overall, the impact of viscogen is small and fails to reveal a significant contribution of the substrate binding rate to the kcat/Km parameter. Thus, analogous to the results with SIEs, there are no remarkable differences between the WT enzyme and the mutants that can easily rationalize and explain either the large reduction in kcat/Km for the mutants or the lack of variance in D(kcat/Km). These data imply a partially rate determining rearrangement of bound linoleic acid, subsequent to the initial formation of the enzyme–substrate complex, as the origin of the kinetic behaviors seen for kcat/Km.
Figure 4.

Effect of viscogen in (A) WT SLO-1, (B) Ile553Ala/Leu754Ala SLO-1, and (C) Leu546Ala/Leu754Ala SLO-1.
Discussion
The SLO-1-catalyzed oxidation of LA provides an excellent system in which to tease apart the contributions of protein motions to an enzymatic bond cleavage process. The extensive, previously published kinetic data for SLO-112,22,24,26,27 have led to a well-resolved catalytic scheme in which a hydrogen atom is first removed from the C-11 position of the substrate, LA, via an irreversible process,26 followed by the trapping of the substrate-derived radical by molecular O2 (Scheme 1). The presence of a “ping-pong” kinetic mechanism means that kcat/Km for the substrate can be analyzed in a manner independent of any contribution from the O2-dependent half-reaction. The huge size of the primary kinetic hydrogen isotope effect in the SLO-1 reaction, as shown both in the steady state18,22 and under single-turnover conditions,12,18 has established that C–H bond cleavage is fully rate-limiting under conditions of kcat.22 Further, the magnitude of the KIE, together with its temperature dependence, has necessitated a rate analysis that treats the hydrogen transfer step as fully quantum mechanical, with the barriers to reaction arising from the motions of the substrate and protein.13−17
The X-ray structures for SLO-1 have provided an important frame of reference for previous and present mutagenesis studies.15,28,29 Numerous high-resolution structures are available for SLO-1 and for other lipoxygenases.30,31 Neither product32 nor substrate33,34 is seen to give rise to large protein conformational changes, consistent with a highly packed α-helical core that constitutes the catalytic domain of all lipoxygenases. In the case of SLO-1, successful docking of LA into the active site provides a model for the enzyme–LA complex that is compatible with the regio- and stereochemistry of product formation and with the impact of pH on altered distributions of products.19 In the past, single-site mutations in SLO-1 were focused on three hydrophobic side chains, Leu754 and Leu546 that sandwich the reactive position 11 of bound substrate and Ile553 that is one helix turn away from Leu546 (Figure 1). A series of X-ray studies on a series of mutations at position 553 indicated little or no impact on the structure of SLO-1.15 The major kinetic trend that emerged from the studies on the single mutants of SLO-1 was an increase in the temperature dependence of the KIEs that is easily accommodated within a full tunneling model.13,15 It has been concluded that a reduction in the bulk at the targeted amino acid side chains both increases the H donor–acceptor distance and reduces the stiffness within the reactive configurations of the enzyme; the latter allows active distance sampling (or gating) between the hydrogen donor and acceptor atoms.7,15 By contrast, the active site of the WT enzyme produces distances between the H donor and acceptor that are sufficiently short that a tunneling-ready state can be achieved in the absence of significant donor–acceptor distance sampling.7
An initially unexpected feature of WT SLO-1, together with a large number of other native enzyme systems, was the property of weak or temperature-independent KIEs.8 This type of behavior is only rarely seen for reactions in solution35−38 and has stimulated much debate and analysis.39−41 To date, the most robust interpretation of the observations is centered on an ability of proteins to achieve more compact reactant configurations than are present in solution reactions. The large size and inherent flexibility of proteins are essential features in the generation of active site compaction, proposed to arise as a result of the extensive sampling of protein conformational substates under physiological conditions. It is this sampling that allows the transient creation of the short (2.7–2.8 Å) donor–acceptor distances that are a prerequisite for efficient tunneling.7 However, unlike the temperature dependence of the KIE,39 which provides insight into the participation of a temperature- and isotope-dependent distance sampling, it has been more difficult to establish a set of protocols for linking an isotope-independent conformational sampling process directly to chemistry occurring at an enzyme active site.
Comparison of Rates to Activation Parameters
In a previous study of a thermophilic alcohol dehydrogenase (ht-ADH), we presented the dual observations of highly inflated Arrhenius prefactors and greatly elevated values for Ea as an indicator of impaired conformational landscapes.25 A key feature of this insight was the unchanged rate at the break point (30°) that converts the active enzyme to a less than optimal catalyst.
Inspection of the data for SLO-1 in Table 1 indicates several instances in which the introduction of a second mutation has little effect on the rate for the protio substrate, allowing an analogous comparison of the impact of mutation on Ea and AH: (i) WT versus Ile553Ala, (ii) Leu546Ala vs Ile553Ala/Leu546Ala, and (iii) Leu754Ala vs Ile553Ala/Leu754Ala. The fact that Ile553Ala looks almost identical to the WT with regard to all parameters (for reaction of the protio substrate, but not the deuterio substrate) negates any meaningful comparison. The impact of double versus single mutation in the pair of Leu546Ala and Leu546Ala/Ile553Ala is also quite distinct from the data for ht-ADH, with a similar Ea and a reduced value for AH. The remaining pair, however, Leu754Ala and Ile553Ala/ILeu754Ala, shows a very clear trend in which both Ea and AH are elevated substantially, highly analogous to the trends reported for ht-ADH.25 This observation provided the first intimation that the introduction of a second site mutation could skew the distribution of protein substates toward conformers with significantly impaired tunneling properties.
Spectroscopic probes are essential for distinguishing between perturbed protein landscapes with only subtle structural changes among a population of protein substates and the possibility of large protein conformational changes in very different forms of the enzyme. Significantly, X-ray structures for both WT SLO-128 and the slowest double mutant, Leu546Ala/Leu754/Ala,18 indicate almost identical three-dimensional structures for the apoenzyme. As shown in Figure 5, CD analysis of the remaining double mutants yields results that can be superimposed with traces of WT SLO-1 and Leu546Ala/Leu754Ala, effectively ruling out either extensive protein unfolding or the trapping of variants into conformers that differ significantly with regard to the helix and β-sheet content of the WT protein.
Figure 5.

Circular dichroism (CD) analysis of WT and double-mutant SLO-1.
Comparison of Unimolecular to Bimolecular Isotope Effects as a Means of Detecting an Impaired Conformational Sampling Landscape
We now introduce the use of comparative KIEs on kcat versus kcat/Km for the substrate, LA, as a means of demonstrating the presence of reduced activity conformational substates within an enzyme variant. This analysis relies on the fact that the KIE on kcat/Km for WT SLO-1 is smaller than the KIE on kcat, a common observation in enzymology, indicating the contribution of a step(s) other than the chemical step to the second-order rate constant. Previous studies conducted with native SLO-1 included the use of solvent D2O and added viscogen, in an attempt to understand the nature of the additional step(s) in kcat/Km that reduces its KIE below the kcat value of ∼80. Small effects on both D2O(kcat/Km) and relative rates (with or without viscogen) were seen with the native, soybean-derived enzyme, suggesting either a partially rate-limiting binding of LA or a subsequent step that could represent a rearrangement of preassociated LA to a different ground-state configuration.22,26 The fact that all studies reported since these earlier investigations have used recombinant enzyme led to a re-examination of WT SLO-1 in parallel with a study of the double mutants. From the new results summarized in Table 3 and Figure 4A–C regarding the impact of solvent D2O and viscogen on kcat/Km, respectively, we conclude that these are small and very similar to those of the WT and the double mutants. Although we cannot yet give a physical description of the additional step(s) that limits kcat/Km in SLO-1, this must be occurring subsequent to the initial encounter between the enzyme and substrate and most likely involves a D2O-insensitive “diffusion” or rearrangement of the large substrate through the enzyme into a position close to the active site iron center.
We next turn to the trends in D(kcat/Km) values with the double-mutant enzymes (Table 2). Initial efforts to obtain reliable D(kcat/Km) values with these slow variants of SLO-1 via a noncompetitive spectroscopic approach were greatly hampered by a poor signal-to-noise ratio with the protio substrate, exacerbated by a further large reduction in rate with the deutero substrate. For this reason, we turned to a competitive method (Materials and Methods) that provides both high sensitivity and reproducibility. What is immediately obvious is that the D(kcat/Km) values for the double mutants are essentially identical to those of the WT enzyme at pH 7.0 and 9.0 and 30 °C, despite large reductions in the rate for kcat/Km. The kinetic expression for D(kcat/Km) in SLO-1 contains the isotope effect on the C–H bond cleavage step, kchem, moderated by a ratio of rate constants that expresses the relative importance of the chemical step versus the release of bound substrate back into solution, koff
| 1 |
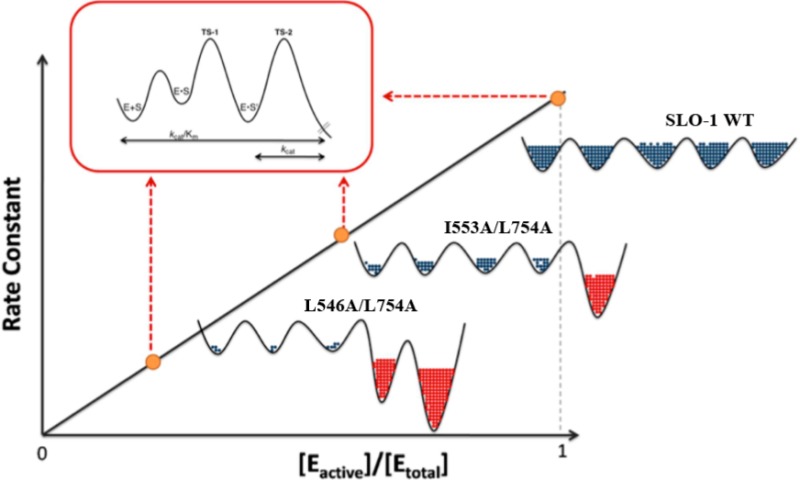
If the impact of each of the double mutants had been solely on the reorganization barrier that controls H tunneling (kchem = kcat), the barrier for this chemical step would have been expected to rise in preference to the barrier controlling the additional rate-determining step in kcat/Km (designated koff), decreasing the magnitude of kchem/koff and leading to values of D(kcat/Km) that become closer to Dkcat. We note that the insensitivity of D(kcat/Km) to mutation could have been due to a coordinated and equivalent decrease in both kchem and koff after each mutation, such that the ratio of these rate constants was unchanged. This seems unlikely across the board, given that the pairs of mutations span quite different positions in the active site. A more compelling interpretation is that the impact of these mutations is on the concentration of the active enzyme and not on the catalytic properties of the fraction of the enzyme that can support catalysis, allowing the ratio of rate constants within the kinetically competent pathway to remain invariant upon mutation (Scheme 2). As controls in this regard, enzyme levels of mutant proteins were determined prior to the assay and shown to be stable; further, no evidence of a time-dependent loss of protein activity was evident under any of the conditions for the measurement of noncompetitive rates or KIEs. As a first approximation, the change in “protein concentration” upon mutation can be imagined as arising from a shift in the conformational landscape that fine-tunes the positioning of the substrate and active Fe(III)-OH. Importantly, such a shift in the conformational ensemble would need to take place on a time scale rapid enough to allow full expression of the kchem on kcat as required by the very large magnitude of the KIEs; as a corollary, such a rapid, non-rate-determining conformational interconversion cannot be the source of the reduction in size of D(kcat/Km) in relation to Dkcat, though it is the likely source of the reduced magnitude of the second-order rate constant itself. That a similar perturbation of the conformational landscape affects kcat and kcat/Km is supported by a similar impact of mutation (within 2–3-fold) on both first- and second-order rate constants. The small, consistent differential likely reflects an underestimate of kcat/Km for the WT enzyme that was used as a single point of reference in comparison to rate constants for the variants.
Scheme 2. Decrease of Enzyme Activity, Together with Changes in Ea and AH, Contrasts with the Lack of Change in D(kcat/Km).

This is attributed to different coordinates representing the chemical reaction coordinate for SLO-1 (in the red box) that is orthogonal to the protein conformational landscape coordinate. The latter causes a decrease in kinetic parameters (together with elevated values for Ea and AH) that comes from a reversible population of enzyme substates that are either inactive or very poorly active (represented by red cross hatching to the right).
The schematic given in Scheme 2 illustrates a conformational landscape that takes place orthogonal to the chemical reaction coordinate. As shown, WT SLO-1 is optimized to allow access to a smooth conformational landscape that ensures efficient sampling of the entire protein in its search for the most catalytically relevant protein substrates. The introduction of packing defects into the interior of SLO-1, especially those introduced via the reduction in size of two centrally located hydrophobic side chains, alters this property of the conformational landscape, leading to inactive or poorly active conformers that effectively reduce the concentration of the active enzyme. Further, a rapid equilibration between inactive and active enzyme states can be expected to reduce both kcat and kcat/Km by the same factor (1 + K1):
| 2 |
where K1 = Einactive/Eactive. Only when protein enters into the region of conformational landscape that is catalytically competent can catalysis proceed. Once the catalytic substates are achieved, the chemical nature of the steps that make up kcat and kcat/Km is retained; hence, the ability to observe values for D(kcat/Km) that are independent of the impact of mutation on rate is possible.
An Outlier: Leu546Ala/Leu754Ala
The one dramatic outlier in this study is Leu546Ala/Leu754Ala, which shows a primary KIE on kcat of ∼729 (Table 1) versus a D(kcat/Km) that is in line with the values of the other variants (Table 2). We considered the possibility that the huge primary KIE for Leu546Ala/Leu754Ala on kcat but not kcat/Km might be a result of “nonproductive” binding of the deuterated substrate.42 However, binding isotope effects of this type and magnitude are unknown in enzymes, indicating the need for an alternate explanation. One possibility is a conformational landscape for the free enzyme [affecting D(kcat/Km)] that is distinct from the landscape for the enzyme–substrate complex (affecting Dkcat). While this is an attractive possibility, consistent with the proposal of conformational selection proposed by Kern and co-workers,43,44 we note that the decreases in kcat and kcat/Km for all of the double mutants, including Leu546Ala/Leu754Ala, are within 2–3-fold of each other. At this juncture, we offer a more traditional explanation for the behavior of Leu546Ala/Leu754Ala, in which the mutational impairment of the enzyme has a greater impact on koff for substrate than kchem, altering the ratio of kchem/koff in a manner distinct from that seen in the WT and other mutants. Given the very elevated values for both Ea(H) and AH with Leu546Ala/Leu754Ala (Table 1), it seems likely that defects in the protein conformational landscape for the double mutant are severe enough to selectively impair the egress of substrate from the active site.
Conclusions
We introduce a new approach for demonstrating the impact of site-specific mutagenesis on protein conformational landscapes. This method involves a comparative analysis of initial rate parameters together with deuterium KIEs on the second-order (kcat/Km) and first-order (kcat) kinetic constants of an enzyme reaction. SLO-1 offers an excellent system in which to showcase this diagnostic, because of its large primary KIEs on kcat that indicate a single rate-determining C–H bond cleavage under substrate saturation conditions. The key observations are (i) large and similar decreases in both kcat/Km and kcat upon mutation and (ii) invariant and reduced KIEs on kcat/Km relative to kcat. A model that introduces a rapidly equilibrating protein coordinate that is orthogonal to the chemical coordinate will explain these data with SLO-1, where the reduction in rate comes from a decrease in the concentration of the active enzyme according to the expression [Et]/(1 + K1), where K1 is the equilibrium constant for the production of protein conformational substates that are inactive or have greatly reduced enzyme activity. At the same time, the mutation leaves the relative rate constants for substrate release and protium transfer largely unaltered. The ability to parse the impact of a mutation on the global conformational landscape versus a more local, H donor–acceptor distance sampling mode9 provides a potent context for further biophysical probes of SLO-1. The application of this method to other enzyme systems will be possible in those instances in which measured KIEs on kcat/Km are seen to be reduced in relation to kcat.
Acknowledgments
Glossary
Abbreviations
- SLO-1
soybean lipoxygenase-1
- BIS-TRIS
2-bis(2-hydroxyethyl) amino-tris(hydroxymethyl)methane
- HPOD
13(S)-hydroperoxy-9(Z),11(E)-octadecanoic acid
- CHES
N-cyclohexyl-2-aminoethanesulfonic acid
- KIE
kinetic isotope effect
- CD
circular dichroism
- WT
wild-type
- ES-MS
electrospray mass spectrometry
- RP-HPLC
reversed phase high-performance liquid chromatography.
Supporting Information Available
Tables of rate constants versus temperature for the Ile553Ala/Leu754Ala double mutant, numerical values for the impact of glucose as a viscogen on WT SLO-1, Ile553Ala/Leu754Ala, and Leu546Ala/Leu754Ala SLO-1, and a calibration curve for the HPOD concentration determined via HPLC. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00374.
This work was supported by a grant from the National Institutes of Health (GM025765) to J.P.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Wolfenden R. (2006) Degrees of difficulty of water-consuming reactions in the absence of enzymes. Chem. Rev. 106, 3379–3396 10.1021/cr050311y. [DOI] [PubMed] [Google Scholar]
- Silverman D. N.; Lindskog S. (1988) The catalytic mechanism of carbonic anhydrase: implications of a rate-limiting protolysis of water. Acc. Chem. Res. 21, 30–36 10.1021/ar00145a005. [DOI] [Google Scholar]
- Cleland W. W. (1975) What limits the rate of an enzyme-catalyzed reaction. Acc. Chem. Res. 8, 145–151 10.1021/ar50089a001. [DOI] [Google Scholar]
- Page M. I.; Jencks W. P. (1971) Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. U. S. A. 68, 1678–1683 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. (1948) Chemical achievement and hope for the future. Am. Sci. 36, 51–58. [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes G. G.; Hammes-Schiffer S. (2008) Free-energy landscape of enzyme catalysis. Biochemistry 47, 3317–3321 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. (2009) A 21st century revisionist’s view at a turning point in enzymology. Nat. Chem. Biol. 5, 543–550 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. (2006) Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 106, 3095–3118 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. (2013) Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 82, 471–496 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. P.; Xun L.; Xie X. S. (1998) Single-molecule enzymatic dynamics. Science 282, 1877–1882 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- Lu H. P. (2012) Biochemistry. Enzymes in coherent motion. Science 335, 300–301 10.1126/science.1217170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T.; Glickman M. H.; Sun S.; Klinman J. P. (1996) Experimental evidence for extensive tunneling of hydrogen in the lipoxygenase reaction:implications for enzyme catalysis. J. Am. Chem. Soc. 118, 10319–10320 10.1021/ja961827p. [DOI] [Google Scholar]
- Knapp M. J.; Rickert K.; Klinman J. P. (2002) Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 124, 3865–3874 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- Meyer M. P.; Klinman J. P. (2005) Modeling temperature dependent kinetic isotope effects for hydrogen transfer in a series of soybean lipoxygenase mutants: The effect of anharmonicity upon transfer distance. Chem. Phys. 319, 283–296 10.1016/j.chemphys.2005.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M. P.; Tomchick D. R.; Klinman J. P. (2008) Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl. Acad. Sci. U. S. A. 105, 1146–1151 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. (2010) Impact of distal mutation on hydrogen transfer interface and substrate conformation in soybean lipoxygenase. J. Phys. Chem. B 114, 6653–6660 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. (2007) Proton-coupled electron transfer in soybean lipoxygenase: dynamical behavior and temperature dependence of kinetic isotope effects. J. Am. Chem. Soc. 129, 187–196 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- Hu S.; Sharma S. C.; Scouras A. D.; Soudackov A. V.; Carr C. A.; Hammes-Schiffer S.; Alber T.; Klinman J. P. (2014) Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C-H activation. J. Am. Chem. Soc. 136, 8157–8160 10.1021/ja502726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert K. W.; Klinman J. P. (1999) Nature of hydrogen transfer in soybean lipoxygenase 1: separation of primary and secondary isotope effects. Biochemistry 38, 12218–12228 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- Sharma S. C.; Klinman J. P. (2008) Experimental evidence for hydrogen tunneling when the isotopic arrhenius prefactor (A(H)/A(D)) is unity. J. Am. Chem. Soc. 130, 17632–17633 10.1021/ja806354w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman T. R.; Zhou J.; Solomon E. I. (1998) Spectroscopic and functional characterization of a ligand coordination mutant of soybean lipoxygenase-1: First coordination sphere analogue of human 15-lipoxygenase. J. Am. Chem. Soc. 120, 12564–12572 10.1021/ja982844c. [DOI] [Google Scholar]
- Glickman M. H.; Klinman J. P. (1995) Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 34, 14077–14092 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- Lewis E. R.; Johansen E.; Holman T. R. (1999) Large competitive kinetic isotope effects in human 15-lipoxygenase catalysis measured by a novel HPLC method. J. Am. Chem. Soc. 121, 1395–1396 10.1021/ja983083g. [DOI] [Google Scholar]
- Knapp M. J.; Klinman J. P. (2003) Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry 42, 11466–11475 10.1021/bi0300884. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Dong M.; Bahnson B. J.; Klinman J. P. (2011) Impaired protein conformational landscapes as revealed in anomalous Arrhenius prefactors. Proc. Natl. Acad. Sci. U. S. A. 108, 10520–10525 10.1073/pnas.1104989108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M. H.; Klinman J. P. (1996) Lipoxygenase reaction mechanism: demonstration that hydrogen abstraction from substrate precedes dioxygen binding during catalytic turnover. Biochemistry 35, 12882–12892 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- Knapp M. J.; Seebeck F. P.; Klinman J. P. (2001) Steric control of oxygenation regiochemistry in soybean lipoxygenase-1. J. Am. Chem. Soc. 123, 2931–2932 10.1021/ja003855k. [DOI] [PubMed] [Google Scholar]
- Minor W.; Steczko J.; Stec B.; Otwinowski Z.; Bolin J. T.; Walter R.; Axelrod B. (1996) Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution. Biochemistry 35, 10687–10701 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- Boyington J. C.; Gaffney B. J.; Amzel L. M. (1990) Crystallization and preliminary x-ray analysis of soybean lipoxygenase-1, a non-heme iron-containing dioxygenase. J. Biol. Chem. 265, 12771–12773. [PubMed] [Google Scholar]
- Skrzypczak-Jankun E.; Amzel L. M.; Kroa B. A.; Funk M. O. (1997) Structure of soybean lipoxygenase L3 and a comparison with its L1 isoenzyme. Proteins: Struct., Funct., Genet. 29, 15–31. [DOI] [PubMed] [Google Scholar]
- Gillmor S. A.; Villasenor A.; Fletterick R.; Sigal E.; Browner M. F. (1997) The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol. 4, 1003–1009 10.1038/nsb1297-1003. [DOI] [PubMed] [Google Scholar]
- Skrzypczak-Jankun E.; Bross R. A.; Carroll R. T.; Dunham W. R.; Funk M. O. (2001) Three-Dimensional Structure of a Purple Lipoxygenase. J. Am. Chem. Soc. 123, 10814–10820 10.1021/ja011759t. [DOI] [PubMed] [Google Scholar]
- Neau D. B.; Bender G.; Boeglin W. E.; Bartlett S. G.; Brash A. R.; Newcomer M. E. (2014) Crystal Structure of a Lipoxygenase in Complex with Substrate: The Arachidonic Acid-Binding Site of 8R-Lipoxygenase. J. Biol. Chem. 289, 31905–31913 10.1074/jbc.M114.599662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe M. J.; Neau D. B.; Mitchell C. E.; Bartlett S. G.; Newcomer M. E. (2014) The Structure of Human 15-Lipoxygenase-2 with a Substrate Mimic. J. Biol. Chem. 289, 8562–8569 10.1074/jbc.M113.543777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart H.; Brechbiel M. W.; Acheson R. M.; Ward D. C. (1982) Observations on the geometry of hydrogen transfer in [1,5] sigmatropic rearrangements. J. Am. Chem. Soc. 104, 4671–4672 10.1021/ja00381a026. [DOI] [Google Scholar]
- Kwart H. (1982) Temperature dependence of the primary kinetic hydrogen isotope effect as a mechanistic criterion. Acc. Chem. Res. 15, 401–408 10.1021/ar00084a004. [DOI] [Google Scholar]
- Roth W. R.; Konig J. (1966) [Hydrogen displacements. IV. Kinetic isotope effect of the 1.5 hydrogen displacement in cis-pentadiene-(1.3)]. Justus. Liebigs. Ann. Chem. 699, 24–32 10.1002/jlac.19666990103. [DOI] [PubMed] [Google Scholar]
- Braun J.; Schwesinger R.; Williams P. G.; Morimoto H.; Wemmer D. E.; Limbach H. H. (1996) Kinetic H/D/T isotope and solid state effects on the tautomerism of the conjugate porphyrin monoanion. J. Am. Chem. Soc. 118, 11101–11110 10.1021/ja961313q. [DOI] [Google Scholar]
- Klinman J. P.; Kohen A. (2013) Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 82, 471–496 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacki D. R.; Harvey J. N.; Mulholland A. J. (2012) Taking Ockham’s razor to enzyme dynamics and catalysis. Nat. Chem. 4, 169–176 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. (2012) Good vibrations in enzyme-catalysed reactions. Nat. Chem. 4, 161–168 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- Fersht A. (1998) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, 1st ed., W. H. Freeman and Co., New York. [Google Scholar]
- Eisenmesser E. Z.; Millet O.; Labeikovsky W.; Korzhnev D. M.; Wolf-Watz M.; Bosco D. A.; Skalicky J. J.; Kay L. E.; Kern D. (2005) Intrinsic dynamics of an enzyme underlies catalysis. Nature 438, 117–121 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Fraser J. S.; Clarkson M. W.; Degnan S. C.; Erion R.; Kern D.; Alber T. (2009) Hidden alternative structures of proline isomerase essential for catalysis. Nature 462, 669–673 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.