

Abstract
A 61 year old Caucasian male of European ancestry with significant coronary heart disease (CHD) since age 42 years and marked high density lipoprotein (HDL) deficiency (HDL cholesterol 1 mg/dl) was evaluated. His fasting low density lipoprotein (LDL) cholesterol (C) level was 42 mg/dl, and his triglycerides were 417 mg/dl on therapy with rosuvastatin 40 mg/day, ezetimibe 10 mg/day, fenofibrate 145 mg/day, and extended release niacin 2 g/day. Further analysis of his plasma revealed an apolipoprotein (apo) A-I level of 23.5 mg/dl (about 20% of normal), and the absence of small alpha-4 HDL, medium alpha-3 HDL, and very large alpha-1 HDL, with only very small pre-beta-1 HDL and large alpha-2 HDL being present. ApoA-I gene sequencing revealed a novel heterozygous in-frame insertion mutation with duplication of nucleotides 1535 through 1552 inserted at position 1553, causing a new amino acid glycine at codon 157 and a duplication of amino acids alanine, arginine, alanine, histidine, and leucine at codons 158 – 162. This novel apoA-I mutation results in the formation of apoA-I that appears to have abnormal lipid binding properties, resulting in impaired reverse cholesterol transport, probable enhanced clearance, and premature CHD.
Case Report
A 61 year old Caucasian male of European ancestry presented with marked HDL deficiency. He had a long standing history of coronary artery disease initially diagnosed at age 42 years when he developed angina, and at that time had an angioplasty of his left anterior descending coronary artery. At age 52 years he developed recurrent angina and subsequently underwent coronary artery bypass surgery grafting surgery of his right, circumflex, left anterior descending coronary arteries. He also had a history of hypertension, past cigarette smoking (quit at age 41 years), and peripheral arterial disease. His father died at age 69 years of colon cancer, and his mother was alive at age 88 years, with a history of hypertension and stroke, and reportedly normal lipid levels. His parents were unrelated, so there was no evidence of consanguinity. He had 4 siblings. One sister died at age 49 years of breast cancer. One brother died at age 48 years of a cerebral artery aneurysm. One brother died at age 51 years of a drug overdose. One brother was alive and well at age 52 years, with no history of CHD, with normal lipids, but with an HDL-C value in the 30 mg/dl range. The patient’s wife was in good health. He had two offspring, a daughter age 31 years in good health with normal lipids and an HDL-C of 62 mg/dl, and a son, age 36 years in good health, with unknown lipid levels. The patient declined to have his family members studied.
At the time of his examination in our clinic he was on the following medications: aspirin 81 mg/day, extended release diltiazem 120 mg/day, extended release niacin 2 grams/day, ezetimibe 10 mg/day, fenofibrate 145 mg/day, fish oil 1 g/day, isosorbide 10 mg three times daily, metoprolol 25 mg three times/day, and rosuvastatin 40 mg/day. A physical examination revealed: height 71 inches, weight 231 pounds, blood pressure 140/99 mmHg, and heart rate 66/minute. His eye examination revealed significant circular arcus senilis. However in contrast to the arcus observed with aging or familial hypercholesterolemia, in this patient there was no space between the arcus and the limbus. The arcus was previously observed for some time by his referring physician, but it is unclear precisely at what age it developed. He had normal heart and lung examinations. The pulses in his legs (popliteals, dorsalis pedis, and posterior tibial) were present, but were significantly diminished bilaterally. He had no evidence of hepatosplenomegaly or xanthomas. Blood was drawn after an overnight fast for laboratory analysis.
Laboratory Analysis
Laboratory results on blood obtained after an overnight fast as analyzed by Quest Laboratories (Cambridge, MA) revealed: total cholesterol 112 mg/dl (normal), triglyceride 447 mg/dl (elevated), HDL-C < 5 mg/dl (very low), and LDL-C 77 mg/dl (close to optimal), and lipoprotein (a) 25 nmol/L (normal). His other values were: liver alanine transaminase 24 U/L (normal), aspartate transaminases 39 U/L (slightly increased), alkaline phosphatase 65 U/L (normal), blood urea nitrogen 22 mg/dl (normal), creatinine 0.82 mg/dl (normal), thyroid stimulating hormone 1.53 uIU/ml (normal), C reactive protein 1.5 mg/L (normal), glycosylated hemoglobin 5.4% (normal), and fasting glucose 107 mg/dl, albumin 4.7 g/dl, bilirubin 0.9 mg/dl, and calcium 10.8 mg/dl (elevated). His electrolytes were all normal.
Specialized Laboratory Testing
Because of these abnormal findings a sample of his plasma was analyzed in our laboratory (Lipid Metabolism Laboratory) at Tufts University. In this laboratory all lipid analyses have been standardized by the Lipid Standardization Program of the Centers for Disease Control, Atlanta, GA. His lipid values were: total cholesterol 116 mg/dl (decreased), triglycerides 368 mg/dl (elevated), HDL-C 1.2 mg/dl (markedly decreased), and direct LDL-C 42 mg/dl (decreased), and very low density lipoprotein-C 29.3 mg/dl (elevated), all measured as previously described (1–5). His apoA-I level as measured by immunoassay as previously described was 23.5 mg/dl (markedly decreased, about 20% of normal) (2,3). His insulin as measured by immunoassay as previously described was 19.0 uU/ml (normal range 5 – 15 uU/ml) (4). The percentage of his total cholesterol that was in the unesterified form was normal at approximately 30%.
Two dimensional gel electrophoresis of his plasma followed by immunoblotting with antibody specific for apoA-I as previously described revealed normal levels of apoA-I in very small pre-beta-1 HDL of 7.5 mg/dL and large pre-beta-2 HDL of 1.2 mg/dL, significantly decreased levels of apoA-I in large alpha-2 HDL of 14.8 mg/dL (29% of normal), and undetectable apoA-I in small alpha-4 HDL, medium alpha-3 HDL, and very large alpha-1 HDL (see figures 1 and 2) (2,3). Because of these abnormal findings a sample of his DNA was sent to Dr. Robert Hegele of the Robarts Research Institute, London, Ontario, Canada for sequencing of his ApoA-I gene. This analysis revealed a novel heterozygous A-1 in-frame insertion mutation with a duplication of nucleotides 1535 through 1552 inserted at position 1553, causing a new amino acid glycine at codon 157 and a duplication of amino acids alanine, arginine, alanine, histidine, and leucine at codons 158 – 162 (see figure 4). This novel ApoA-I mutation results in the formation of apoA-I that appears to have abnormal lipid binding properties, resulting in impaired reverse cholesterol transport and premature CHD.
Figure 1.

Two dimensional gel electrophoresis patterns followed by immunoblotting with monospecific apoA-I antibody of a normal subject (left) and of a patient with premature heart disease (center) in the center, and with a schematic of the individual HDL particles on the right. The electrophoresis is carried out in the horizontal dimension to separate pre-beta, alpha, and pre-alpha particles, and in the vertical dimension using a 4–30% gradient gel (particles of 5 to 12 nm in diameter, followed by immunoblotting with specific apoA-I antibody. The pattern from the patient with premature coronary heart disease in the center has decreased large HDL. All of the particles shown contain apoA-I, but only the alpha-3 and alpha-2 HDL particles contain appreciable amounts of apoA-II.
Figure 2.

The two dimensional gel electrophoretic pattern of the proband’s plasma followed by immunoblotting with specific immunopurified apoA-I antibody is shown. Here we document the absence of alpha-4, alpha-3, and alpha-1 HDL particles.
Figure 4.

New Insertion Mutation (ApoA-INashua)
Comment and Discussion
Decreased HDL-C <40 mg/dL has been identified as an independent risk factor for CHD by the National Cholesterol Education Program (6,7). Severe HDL deficiency (< 10 mg/d) can be due to severe liver disease, familial apolipoprotein A-I (apoA-I) deficiency states, apoA-I variants, Tangier disease (defective ATP-binding cassette A1 transporter function), and lecithin:cholesterol acyltransferase (LCAT deficiency) (5). Liver disease was ruled out based on history and normal liver function tests. The patient did have triglyceride elevation, but not enough to account for the severe HDL deficiency. ApoA-I deficiency was ruled out by the presence of apoA-I in plasma at a concentration of 23.5 mg/dl. Tangier disease was ruled out by the finding of HDL particles other than pre-beta-1 HDL (4). LCAT deficiency was ruled by the finding of HDL particles other than pre-beta-1 HDL and alpha-4 HDL, and by lack of significant corneal opacification (5).
ApoA-I is a single polypeptide chain composed of 243 amino acids in the mature sequence. It is composed of four exons located on chromosome 11q23 (see figure 3). It plays an important role as the major protein of HDL and a cofactor for LCAT. Based on the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/all.php) queried on February 15th, 2012, the following types of mutations in the ApoA-I gene have been reported: 29 missense/nonsense, 1 splicing, 4 regulatory, 13 small deletions, 3 small insertions, 2 gross deletions, 1 gross insertion, and 3 complex mutations. With regard to the insertions, the first of these was reported by Nakata et al in a healthy 10 year old Japanese girl whose HDL-C was 27 mg/dl and her apoA-1 levels were 76 mg/dl, both < 1% percentile for her age and gender-specific Japanese population (8). She was found to be heterozygous for a single nucleotide insertion of a C in a region of a seven C run in exon 3 between codons 3 and 5, resulting in a premature stop codon at codon 34. The mutation was designated as apoA-ITsukuba and resulted in a frameshift mutation with the synthesis of an abnormal polypeptide containing only the N terminal 33 amino acids (out of 243) of apoA-I amino acid sequence. Her mother and her maternal aunt had similarly low HDL-C and apoA-I levels, and were also heterozygous for this mutation (see table 1). The second insertion mutation was reported in 2007 by Kiss et al and is an insertion of a C in codon 33 in exon 3, resulting in a premature stop with only the N terminal 32 amino acids of apoA-I, and low HDL-C (9). No other information about the case was provided. The third insertion was reported in 2008 by Pisciotta et al in a 39 year old man with a positive family history of CHD and an HDL-C value of 28 mg/dl and an apoA-I level of 72 mg/dl, with no corneal opacification or xanthomas (10). The patient was noted to have a C insertion at codon 17 in exon 3 resulting in a frameshift and a premature stop at position 26, resulting in only the N terminal 25 amino acids being present in the apoA-I sequence (see table 1).
Figure 3.

Structure of apoA-I
Table 1.
Reported Insertion Mutations in the Human APOAI Gene
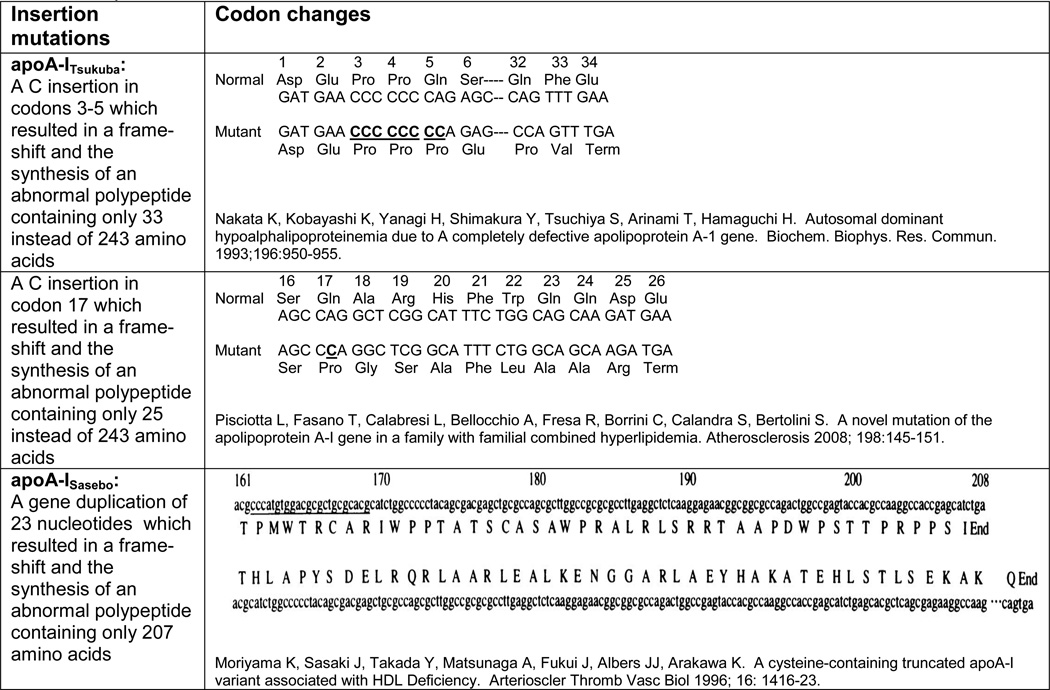 |
A gross insertion mutation was also reported in 1996 by Moriyama et al in a 50-year-old Japanese woman who presented with xanthomas involving the eyelids, elbows, neck, knees, and Achilles tendons, as well as corneal opacities. She had an HDL-C value of 1.2 mg/dl and an apoA-I level of 0.8 mg/dl, and was found to an apoA-I molecular weight of 24,000 instead of the normal 28,000 (11). She was found be homozygous for a mutation with a partial gene reduplication encompassing 23 nucleotides leading to a tandem repeat of bases 333 to 355 from the 5’ end of exon 4, causing a frameshift mutation with a premature stop codon resulting in a truncated apoA-I after residue 207 (see table 1). This mutant apoA-I was designated as apoA-ISasebo (11).
Our case is most similar to the last case, except that our patient was heterozygous for a gross insertion in ApoA-I of a novel heterozygous in-frame insertion mutation (a duplication of nucleotides 1535 through 1552 inserted at position 1553) causing a new amino acid glycine at codon 157 and a duplication of amino acids alanine, arginine, alanine, histidine, and leucine at codons 158 – 162 (see Figure 4). This novel apoA-I mutation results in the formation of apoA-I that appears to have abnormal lipid binding properties, resulting in impaired reverse cholesterol transport, probable enhanced clearance, and premature CHD. We have designated this new apoA-I mutation as apoA-INashua. In homozygous apoA-I deficiency there is clearly premature CHD, but in heterozygotes it is not as clear and premature CHD probably depends on the presence or absence of other risk factors such as cigarette smoking.
Acknowledgements
Dr. Lee was supported as an endocrinology fellow at Tufts Medical Center, Boston, MA. Drs. Schaefer, Polisecki and Asztalos were supported by research grants to the Lipid Metabolism Laboratory at Tufts University from the National Institutes of Health, Bethesda, MD (HL-60935, HL 74753 and PO50HL083813) and the US Department of Agriculture, Washington DC (contract 53-3K06-5-10).
Abbreviations
- (apo) A-I
apolipoprotein
- CHD
coronary heart disease
- HDL
high density lipoproteins
- LDL
low density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ai M, Otokozawa S, Asztalos BF, Ito Y, Nakajima K, White CC, Cupples LA, Wilson PW, Schaefer EJ. Small dense low density lipoprotein cholesterol and coronary heart disease: results from the Framingham Offspring Study. Clin Chem. 2010;56:967–976. doi: 10.1373/clinchem.2009.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asztalos BF, Cupples LA, Demissie S, Horvath KV, Batista MC, Schaefer EJ. High-density lipoprotein subpopulation profile and coronary heart disease prevalence in male participants in the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 2004;24:2181–2187. doi: 10.1161/01.ATV.0000146325.93749.a8. [DOI] [PubMed] [Google Scholar]
- 3.Asztalos BF, Schaefer EJ, Horvath KV, Yamashita S, Miller M, Franceschini G, Calabresi L. Role of LCAT in HDL remodeling: an investigation in LCAT deficiency states. J Lipid Res. 2007;48:592–599. doi: 10.1194/jlr.M600403-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Ai M, Otokozawa S, Schaefer EJ, Asztalos BF, Nakajima K, Shrader P, Kathiresan S, Meigs JB, Williams G, Nathan DM. Glycated albumin and direct low density lipoprotein cholesterol levels in type 2 diabetes mellitus. Clin Chim Acta. 2009;406:71–74. doi: 10.1016/j.cca.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010;21:289–297. doi: 10.1097/MOL.0b013e32833c1ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Expert Panel. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) J Am Med Assoc. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 7.Grundy SM, Cleeman JI, Merz CN, Brewer HB, Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC, Jr, Stone NJ Coordinating Committee of the National Cholesterol Education Program. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol. 2004 Aug 4;44(3):720–732. doi: 10.1016/j.jacc.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Nakata K, Kobayashi K, Yanagi H, Shimakura Y, Tsuchiya S, Arinami T, Hamaguchi H. Autosomal dominant hypoalphalipoproteinemia due to A completely defective apolipoprotein A-1 gene. Biochem. Biophys. Res. Commun. 1993;196:950–955. doi: 10.1006/bbrc.1993.2341. [DOI] [PubMed] [Google Scholar]
- 9.Kiss RS, Kavaslar N, Okuhira K, Freeman MW, Walter S, Milne RW, McPherson R, Marcel YL. Genetic etiology of isolated low HDL syndrome. Arterioscler Thromb Vasc Biol. 2007;27:1139–1145. doi: 10.1161/ATVBAHA.106.137646. [DOI] [PubMed] [Google Scholar]
- 10.Pisciotta L, Fasano T, Calabresi L, Bellocchio A, Fresa R, Borrini C, Calandra S, Bertolini S. A novel mutation of the apolipoprotein A-I gene in a family with familial combined hyperlipidemia. Atherosclerosis. 2008;198:145–151. doi: 10.1016/j.atherosclerosis.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 11.Moriyama K, Sasaki J, Takada Y, Matsunaga A, Fukui J, Albers JJ, Arakawa K. A cysteine-containing truncated apoA-I variant associated with HDL Deficiency. Arterioscler Thromb Vasc Biol. 1996;16:1416–1423. doi: 10.1161/01.atv.16.12.1416. [DOI] [PubMed] [Google Scholar]