

Abstract
There is a consensus that oxidative stress promotes neurodegeneration and may be linked to plaque formation. α-Synuclein is the main component of neurodegenerative plaques. We have found that α-synuclein binds strongly to the enzyme phospholipase Cβ1 (PLCβ1) in vitro and in cells affecting both its G protein activation and its degradation. Because PLCβ1 binds to α-synuclein in cells, we tested whether decreasing its level would promote α-synuclein aggregation and whether overproducing PLCβ1 would inhibit aggregation. By imaging fluorescent α-synuclein in living HEK293, PC12, and SK-H-SH cells, we find that α-synuclein aggregation is directly related to the level of PLCβ1. Importantly, we found that oxidative stress does not affect the cellular levels of α-synuclein but results in the down-regulation of PLCβ1 thereby promoting α-synuclein aggregation. A peptide that mimics part of the α-synuclein binding site to PLCβ prevents aggregation. Our studies indicate that PLCβ1 can reduce cell damage under oxidative stress and offers a potential site that might be exploited to prevent α-synuclein aggregation.
α-Synuclein is a highly conserved neuronal protein that constitutes the major component in neurodegerative plaques called Lewy bodies that are found in diseases such as Parkinson’s and Alzheimer’s (see for background refs 1–4). α-Synuclein is classified as a natively unfolded protein, but it can be purified as a helical tetramer under nondenaturing conditions.5,6 Under some conditions, α-synuclein forms β-stranded fibrils that resemble those found in neurodegenerative plaques. Studies of point mutants of α-synuclein suggest that neuropathicity results from α-synuclein oligomerization that precedes fibrillation.7 While α-synuclein mutants have been directly linked to familial Parkinson’s disease,8 the mechanisms underlying sporadic forms of Lewy body diseases are unclear.
Many environmental factors are thought to contribute to neurodegeneration due to synuclein pathology and of these, oxidative stress has been clearly established.9 The effect of oxidative stress on tissue and cells is profound. Oxidation results in modification of protein side chains, and in α-synuclein, oxidation promotes nitrosylation and tyrosine crosslinking.10,11 Typically, oxidized proteins with modified side chains are degraded in proteosomes. However, this is not the case for α-synuclein12 which is down-regulated by chaperone-assisted autophagy.13
Despite its importance in disease, the cellular function of α-synuclein is not clear. The high degree of sequence conservation suggests a specific function.3 Many studies point to a role in vesicle transport, endocytosis, and neural plasticity (e.g., ref 14). Being ‘natively unfolded’, α-synuclein has been found to bind to many cellular proteins. We have found that α-synuclein binds to strongly to the enzyme PLCβ both in solution and in cells.15,16 PLCβ is activated by Gαq which mediates signals from ligands such as angiotensin, acetylcholine, dopamine, and bradykinin.17,18 Activation of PLCβ triggers a series of events that lead to an increase in intracellular calcium and mitogenic and proliferative changes in the cell.
α-Synuclein binds to the C-terminal region of PLCβ which also contains binding site for Gαq and for the endonuclease TRAX, which plays a key role in RNA interference.19 Additionally, α-synuclein protects PLCβ from degradation by the calcium-activated protease calpain, which promotes feedback regulation of the enzyme.16 The impact of α-synuclein on these PLCβ functions in cells is not clear. It is also not clear how α-synuclein is distributed between PLCβ1 and its other protein partners.
In this study, we have monitored the aggregation of α-synuclein in cells using fluorescence methods. We find that overexpression of α-synuclein results in the formation of oligomers which can be reversed by overexpression of PLCβ. Conversely, down-regulation of PLCβ promotes α-synuclein aggregation. Oxidative stress results in a pronounced reduction in the level of cellular PLCβ while the level of α-synuclein appears unchanged. This loss of PLCβ1 promotes α-synuclein aggregation. However, we find that α-synuclein aggregation can be prevented using a peptide that mimics the α-synuclein-PLCβ1 interaction site. Our studies point to a model in which oxidation promotes α-synuclein toxicity through the reduction of native α-synuclein partners.
METHODS
Cell Culture and Transfection
HEK293 TAP-PLCβ1 (abbreviated as HEK293β1) cells were a generous gift from Dr. Loren Runnels (UMDNJ Piscataway, NJ) and have been described.19 SK–N–SH cells and PC12 cells were obtained from ATCC (HTB-11, CRL-1721). Both HEKβ1 and SK–N–SH cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO 11965) supplemented with 10% fetal bovine serum, 50 units/mL penicillin, and 50 µg/mL streptomycin sulfate. PC12 cells were cultured in DMEM medium supplemented with 10% horse serum, 5% fetal bovine serum, 50 units/mL penicillin, and 50 µg/mL streptomycin sulfate. All cells were incubated at 37 °C with 5% CO2. Tetracycline was added to a final concentration 1 µg/mL to induce PLCβ1 expression in HEKβ1 cells. Nerve growth factor (NGF, Sigma, St. Louis, MO) was added to a final concentration of 100 ng/mL to induce PC12 cells differentiation.
Human α-synuclein was introduced into pmCherry-C1 (Clonetech, Inc.) and pAcGFP1 using BglII and KpnI sites. Plasmids were introduced into cells grown at 80–90% confluency by electroporation for PC12 cells and calcium phosphate precipitation for HEKβ1 cells. The protein expression of endogenous PLCβ1 in SK–N–SH cells was knocked down using small interference RNA to human PLCβ1 (Dharmacon Inc.) according to the manufacturer’s instructions along with the negative control purchased from the manufacturer. The cells were incubated for 72 h post knock down at 37 °C in 5% CO2. Western blot analysis showed this procedure results in ~80% gene silencing.
Protein Purification
His6-PLCβ1 was expressed in Sf9 cells using a baculovirus system with minor modifications.20 α-Synuclein was expressed in Escherichia coli and purified on 15Q column as previously described.4 Expression and purity were assessed by Western blot analysis using commercial antibodies, purchased from Invitrogen (α-synuclein), and Santa Cruz Biochemicals (PLCβ1).
For FRET studies in solution, PLCβ was labeled with the Cys-reactive fluorophore CPM (Invitrogen) and α-synuclein was labeled on the N-terminus with Dabcyl (Invitrogen) using the manufacturer’s protocol. Typically, the probe:protein labeling ratio ranged between ~0.7–0.85:1 as estimated by measuring the absorption of CPM or Dabcyl of the labeled protein and comparing these values to the protein concentration as measured by BCA analysis. PLCβ contains many Cys residues that are accessible to CPM and we have not identified the primary labeling site. Additionally, even though N-terminal labeling was carried out at pH 7.2 to target the primary amine of the N-terminus, there is a very small possibility that a Lys side chain may be reactive which may affect the observed affinity. We also note that the small percentage of unlabeled protein will decrease the dynamic range of the FRET measurements but would not affect the observed affinities.
Peptide Studies
A peptide was synthesized by American Peptide Company (www.americanpeptide.com) whose sequence was identical to 733–746 of PLCβ1 and 739–752 of PLCβ2 (Cys-Lys-Lys-Val-Val-Leu-Pro-Thr-Leu-Ala-Ser-Leu-Arg) which was identified as the main AS binding site by previous mass spectrometry studies.
Förster Resonance Energy Transfer (FRET) Measurements
Twenty-four hours after transfection, cells from 60-mm dishes were plated onto glass bottom culture dishes (MatTek). Images of fluorescent cells were collected 48–72 h after transfection on an Olympus Fluoview1000 confocal microscope equipped with a 40 × 1.4 numerical aperture oil immersion objective. Analysis of the FRET images was performed using standard routine incorporated into software provided for the Olympus microscope that calculates FRET in terms of sensitized emission. For a more complete discussion, see refs 21,22. In vitro FRET studies were carried out on an ISS PC1 spectrofluorometer as previously described.23 Briefly, the integrated emission intensity of CPM-PLCβ was monitored from 380 to 480 nM (λ(exc) = 360), as either Dabcyl-α-synuclein or dialysis buffer was incrementally added.
Microinjection Studies
Cells were grown in MatTek dishes for 48 h to 50–60% confluence. Prior to measurements, the medium was changed to phenol-free Leibovitz L-15 medium. 2 µM Alexa488 labeled α-synuclein or 50 µM peptide in 100 mM KCl, 5 mM K2HPO4, pH 7.4 with 5 µM of DAPI (Invitrogen) to identify injected cells. The control solution did not contain α-synuclein protein or peptide.
Microinjections were performed on an Axiovert 200 M from Zeiss using InjectMan NI2 with a FemtoJet pump from Eppendorf. Samples were microinjected for 0.4 s into cytoplasm with the injection pressure Pi = 30 hPa and the compensation pressure (Pc) at 15 hPa. We examined the microinjected cells under the phase microscope (Zeiss Axiovert 200 M with a 40× phase 2 objective) to select viable cells. Cells were then transferred to the Olympus Fluoview1000 for viewing, which was carried out within 2 h after microinjection for peptide since we were concerned that the peptide would degrade over extended periods of time. Measurements of cells that have been microinjected with α-synuclein were monitored over a longer period of 24 h to allow the proteins to fully aggregate.
Oxidation Stress Experiments
Oxidative stress studies of SK–N–SH cells were carried out by adding 1 mM CoCl2 to untreated or α-synuclein transfected cells overnight at 37 °C as described.24 20 µg total cell lysate was then loaded to 12% SDS gel. Levels of PLCβ1 and α-synuclein were analyzed by Western blot analysis. For solution studies, 4 µM fresh purified α-synuclein monomers were mixed with the same amount of purified PLCβ1 or peptide and incubated at room temperature for 30 min and treated by 1 mM CoCl2 for 15 min followed by addition of SDS sample buffer and loaded on 15% SDS gel.
RESULTS
α-Synuclein Forms Oligomers in Cells Which Is Reversed by PLCβ1
Autosomal dominant Parkinson’s disease and Lewy body dementia can be caused by triplication of the α-synuclein gene suggesting that the high cellular levels of α-synuclein promotes disease states.25,26 Since PLCβ1 binds to α-synuclein in cells, we reasoned that down-regulation of PLCβ1 by siRNA would promote α-synuclein aggregation. We followed aggregation of mCherry-α-synuclein in PC12 cells by confocal imaging over 72 h (Figure 1), and found that down-regulation of PLCβ1 leads to an increase in the number and size of visible α-synuclein aggregates. We note that these aggregates were widely dispersed throughout the cell, with the exception of the nucleus which appears to have lower levels of the protein.
Figure 1.
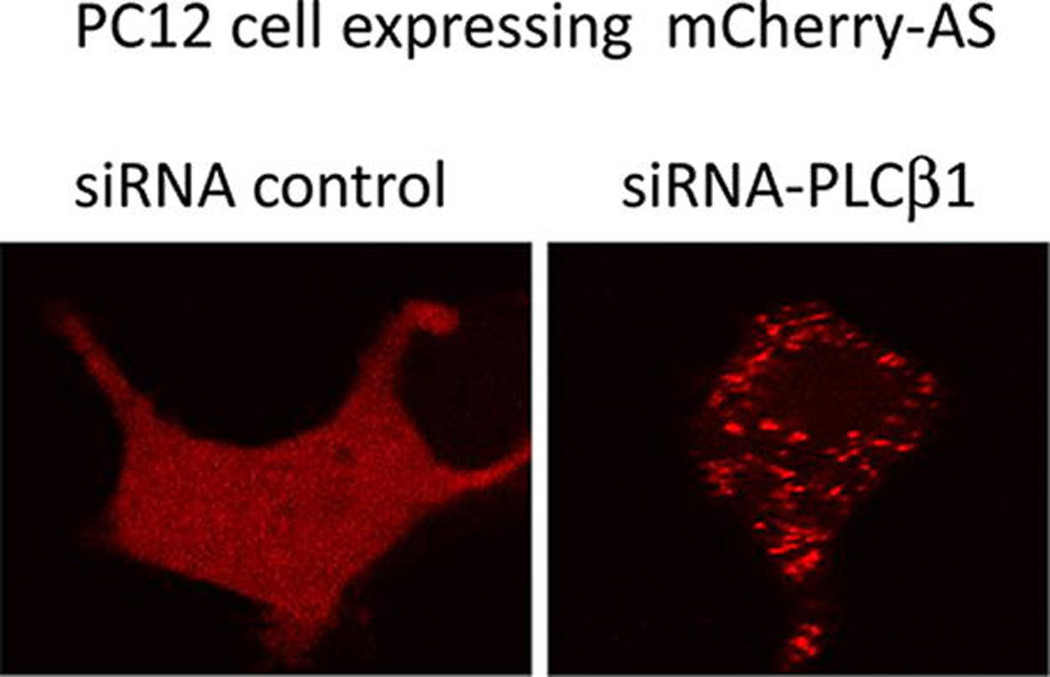
Down-regulation of PLCβ1 promotes α-synuclein aggregation. PC12 cells expressing mCherry-α-synuclein were transfected with siRNA control or siRNA(PLCβ1). Down-regulation of PLCβ1 was estimated to be ~60% by Western blot analysis. The images are representative of n = 19 and n = 23 images, respectively.
To further investigate the effect of PLCβ1 on α-synuclein aggregation, we placed a small, fluorescent label (Alexa488) on the N-terminus of purified α-synuclein and microinjected the labeled protein into HEK293β1 cells that can be induced to overexpress PLCβ1 upon addition of tetracycline.19 In untreated cells, the microinjected α-synuclein was widely distributed throughout the cells similar to the broad localization seen for mCherry-α-synuclein (Figure 2 upper panels). When the cells were induced to overexpress PLCβ1, less α-synuclein aggregation was visibly seen (Figure 2 lower panels). Together for the results found with mCherry-α-synuclein, our studies suggest that the presence of PLCβ1 decreases large scale aggregation of α-synuclein.
Figure 2.
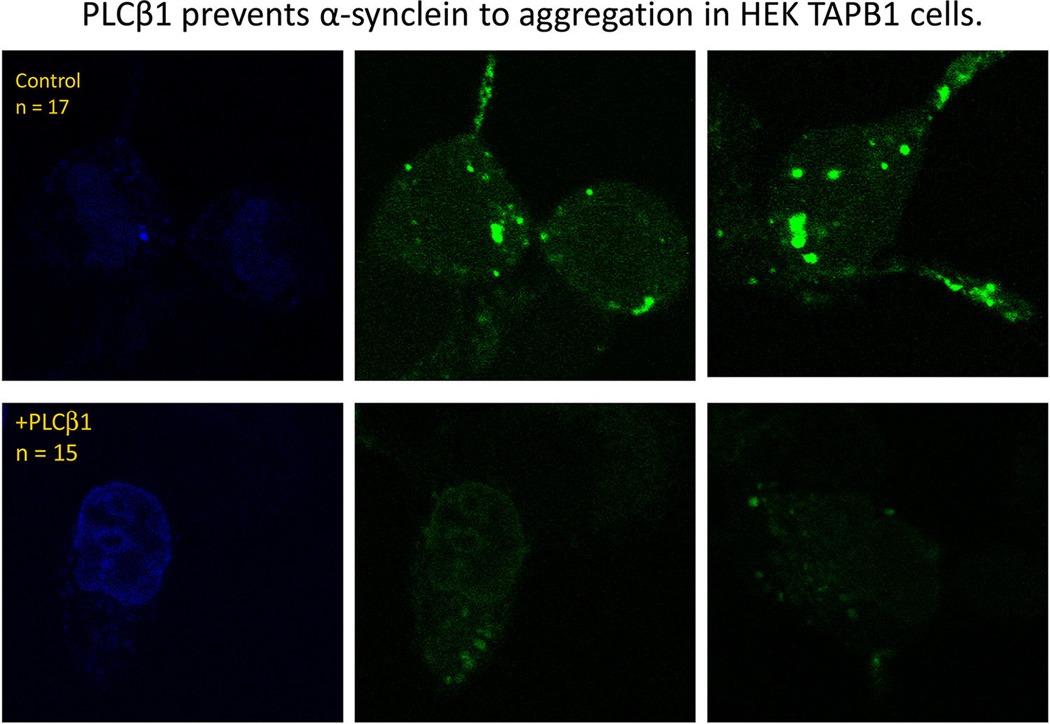
PLCβ1 prevents α-synuclein aggregation in HEKβ1 cells. Untreated HEK293 cells (top panels) or tetracycline treated cells that overproduce PLCβ1 (bottom panels) were microinjected with 2 µM Alexa488-α-synuclein mixed with trace amounts of DAPI. Presented in the figure are typical cells showing the nucleus as seen by DAPI (left panels) and Alexa488-α-synuclein after 24 h of incubation at 37 °C.
To quantify α-synuclein aggregation, we viewed association by Förster Resonance Energy Transfer (FRET) as previously described.23 For these studies, we transfected either HEK293β1 cells or PC12 cells (a neuronal-like cell line) with eGFP-α-synuclein and mCherry-α-synuclein. We then measured the change in FRET when PLCβ1 expression was either induced in HEK293β1 cells or down-regulated in PC12 cells. In uninduced HEK293β1 cells, we find the values of GFP-α-synuclein/mCherry-α-synuclein FRET are broadly distributed over a large range of values suggesting the presence of several aggregated states. However, when the level of PLCβ1 was raised by treatment of the cells tetracycline, the level of FRET was reduced to a mean value similar to negative controls (Figure 3A).
Figure 3.
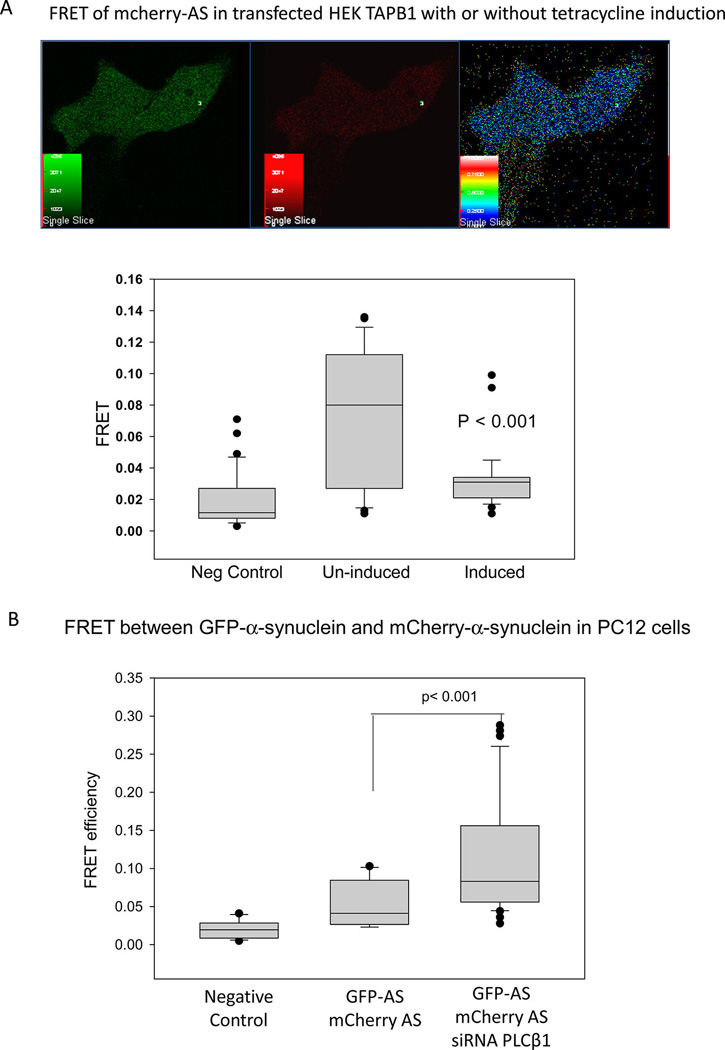
PLCβ1 expression modules α-synuclein oligomerization as seen by FRET. A. Change in FRET between eGFP-α-synuclein and mCherry-α-synuclein in untreated HEK293β1 cells and in cells induced to overexpress PLCβ1. Top – An uninduced HEK293β1 cells imaged in the GFP or green (left) channel, the mCherry channel red (middle), and FRET (right) channel. The bottom graph shows the range of FRET values for free eGFP and mCherry where the mean FRET = 0.019 ± 0.003 (n = 33) (left); for eGFP-α-synuclein/mCherry-α-synuclein in uninduced HEK293β1 cells FRET = 0.076 ± 0.008 (n = 30); and HEK293β1 cells induced to overexpress PLCβ1, FRET = 0.032 ± 0.003 (n = 33). B. Change in FRET between eGFP-α-synuclein and mCherry-α-synuclein in PC12 cells where the negative control on the left is free GFP and mCherry (mean FRET = 0.020 ± 0003, n = 14); the middle sample is for GFP-α-synuclein/mCherry-α-synuclein (mean FRET = 0.052 ± 0.007, n = 17); and the right sample is for PC12 cells expressing GFP-α-synuclein/mCherry-α-synuclein that have been transfected with siRNA(PLCβ1) where FRET = 0.119 ± 0.0126 (n = 23).
In a second series of studies, we transfected PC12 cells with the eGFP-α-synuclein, mCherry-α-synuclein, and siRNA-(PLCβ1). We found that the mean value of GFP-α-synuclein/mCherry-α-synuclein FRET significantly increased when PLCβ1 was down-regulated as compared to controls (Figure 3B). These results suggest that loss of PLCβ1 increases α-synuclein self-association.
Oxidation Promotes α-Synuclein Aggregation and Loss of PLCβ
There is evidence that oxidation is directly related to the onset and severity of Alzheimer’s disease, and oxidation promotes aggregation of purified α-synuclein.27 To this end, we assessed the increase in α-synuclein aggregation in cells under oxidative conditions. This was accomplished by monitoring the increase in FRET between eGFP-α-synuclein/mCherry-α-synuclein in transfected cells from a human neuronal cell line (SK-N-SH) when subjected to oxidative stress. As shown in Figure 4, cells subjected to overnight treatment with 1 mM CoCl224 show a significant increase in FRET, suggesting α-synuclein aggregation.
Figure 4.
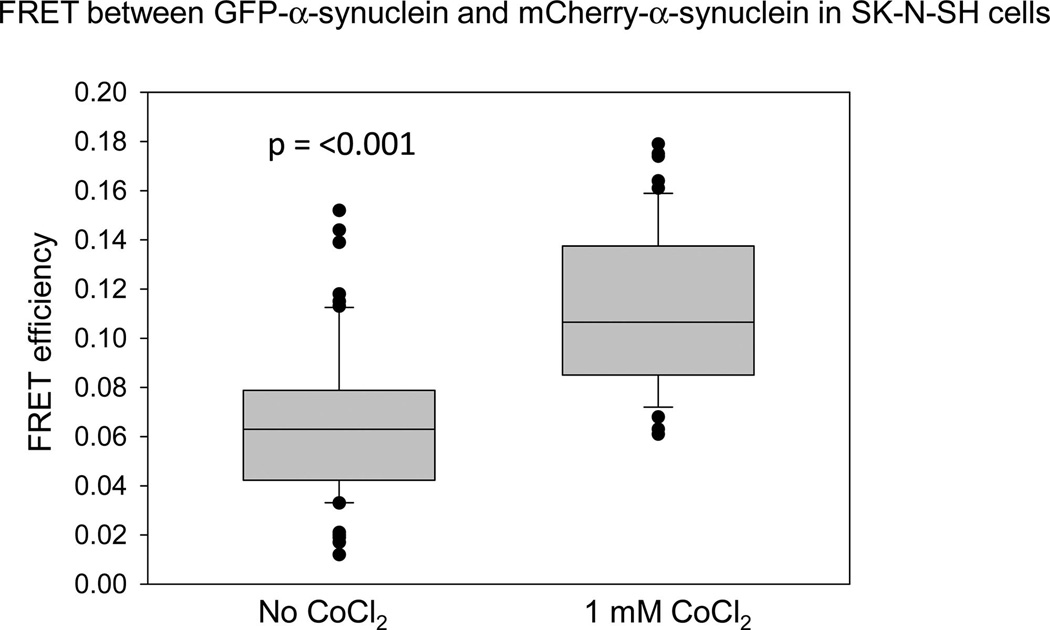
Oxidation promotes α-synuclein oligomerization in cells. SKN-SH cells transfected with GFP-α-synuclein and mCherry-α-synuclein were either untreated or treated overnight with 1 mM CoCl2 before imaging.
We determined whether oxidation changes the levels of PLCβ1 and α-synuclein by running the extracts of cells that were subjected to oxidation on denaturing SDS gels and visualizing the amount of protein relative to controls by both coomassie staining and Western blot analysis. We find that CoCl2 treatment significantly reduces the cellular level of PLCβ1, but surprisingly has little effect on the amount of α-synuclein in untransfected SK-H-SH cells or cells transfected with α-synuclein (Figure 5A,B). Thus, α-synuclein does not protect PLCβ1 from degradation by oxidative stress resulting in a loss in this binding partner.
Figure 5.
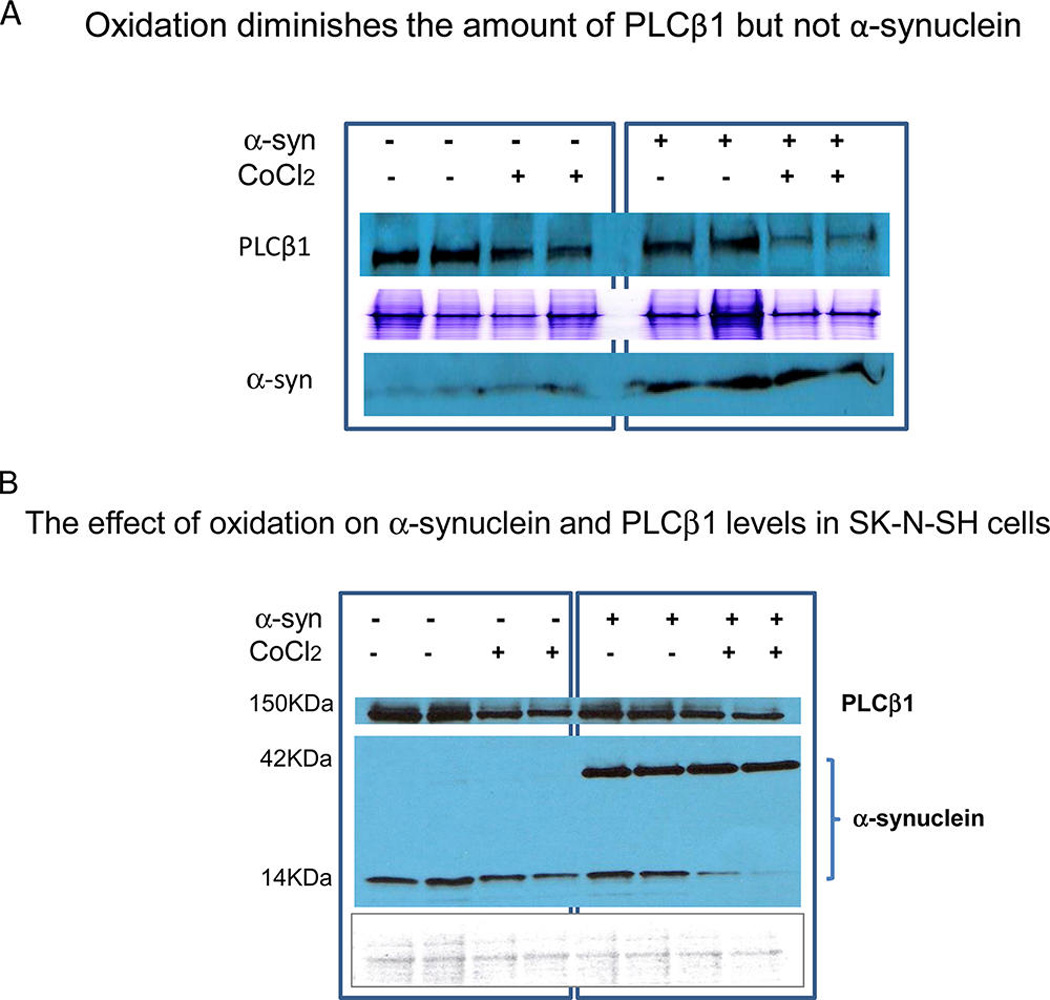
Oxidation diminishes the amount of PLCβ1 but not α-synuclein. A. Untransfected or α-synuclein-transfected SK-N-SH cells were incubated overnight without or with 1 mM CoCl2 and the levels of α-synuclein and PLCβ1 are visualized by Western blotting on a denaturing gel. The middle panel is a coomassie stained gel to show protein loading. B. Similar study carried out at high α-synuclein expression levels showing the increase in α-synuclein tetramers at the expense of monomers during oxidative stress. The bottom panel is a Ponceau stained gel to show protein loading.
We note that the decrease of the levels of PLCβ1 due to oxidation appears more pronounced in the presence of α-synuclein for reasons that are not clear. Additionally, we note that SK-H-SH cells, being a neuronal cell line, has endogenous α-synuclein that runs as a monomer but increases to a tetramer when overexpressed. The oligomerization of α-synuclein into tetramers has been observed by other groups.5,28,29
PLCβ1 Peptide That Blocks α-Synuclein-PLCβ Association Reduces Aggregation
We have previously found that α-synuclein binds to PLCβ1and PLCβ2 protecting the enzymes from degradation by the calcium dependent protease calpain.16 We synthesized a peptide with a PLCβ1 sequence (residues 733–746) surrounding the calpain cleavage site that we identified on PLCβ2 mass spectrometry (Cys743). We first tested whether this peptide would interfere with the association between purified PLCβ1 and α-synuclein. This study was done by covalently labeling PLCβ1 with a FRET donor (CPM), and labeling α-synuclein with a nonfluorescent FRET acceptor (Dabcyl). Without peptide, a strong reduction in CPM-PLCβ1 fluorescence was seen that leveled off by 20 nM Dabcyl-α-synuclein, which is indicative of protein association. However, in the presence of peptide, only a minor reduction in fluorescence was seen up to 50 nM Dabcyl-α-synuclein showing that the peptide inhibits PLCβ1-α-synuclein association (Figure 6A).
Figure 6.
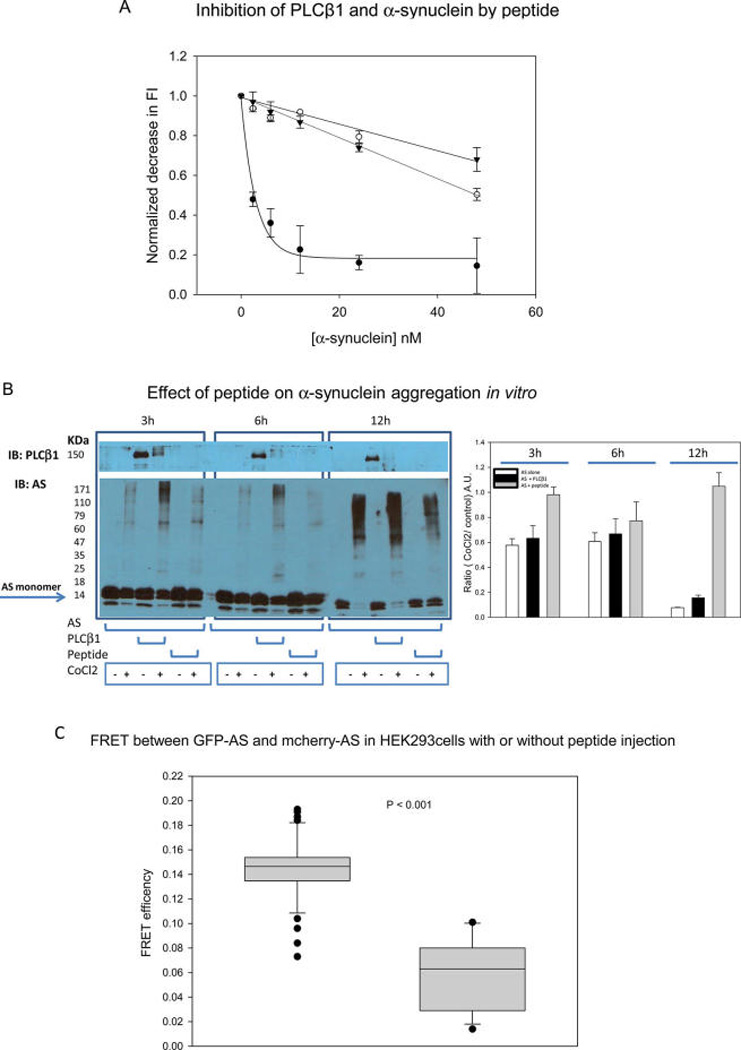
Effect of peptide on PLCβ-α-synuclein association. A. PLCβ1 was labeled with CPM and its association to Dabcyl-α-synuclein was monitored by the decrease in CPM fluorescence due to FRET from the CPM donors to nonfluorescent Dabcyl acceptors where data were taken in the absence (●) and presence of 200 µM (○) and 500 µM (▼) peptide. B. Aggregation of purified α-synuclein (4 µM) with addition of 1 mM CoCl2 at 3, 6, and 12 h in the absence or presence of equimolar amounts of PLCβ1 or peptide and loaded onto a 15% SDS gel. The graph on the right shows the ratio of the monomer α-synuclein band intensities of CoCl2-treated over untreated protein. C. Change in GFP-α-synuclein/mCherry-α-synuclein FRET in HEK293 cells in control cells and ones microinjected with peptide were n = 10 for both sets of samples.
We then tested whether the peptide could reduce aggregation of α-synuclein induced by oxidation. This study was done by subjecting purified α-synuclein to oxidation by CoCl2 in the absence or presence of either PLCβ1 or peptide, and monitoring aggregation on 15% SDS gels. We find that CoCl2 treatment produces visible α-synuclein aggregation (Figure 6B). CoCl2 also reduces the amount of native PLCβ1 and we find more prominent α-synuclein aggregates in the presence of PLCβ1. However, in the presence of peptide, α-synuclein aggregation was reduced as clearly seen at longer treatment times. These results support the idea that the availability of PLCβ1 binding sites, either through the whole enzyme or simply by a peptide mimic of the binding site, reduces α-synuclein aggregation as promoted by oxidative stress.
To determine whether the peptide could interfere with α-synuclein association in cells, it was microinjected into PC12 and HEK293 cells transfected with GFP α-synuclein and mCherry-α-synuclein. The FRET values for the data taken in PC12 cells were the same within error, most likely due to their relatively high endogenous levels of PLCβ1 and α-synuclein. However, microinjection of the peptide into HEK293β1 cells reduced the amount of FRET from 0.15 ±0.004 to 0.06 ±0.008 suggesting that the peptide reduces α-synuclein aggregation (Figure 6C).
DISCUSSION
In this study, we have found that the cellular levels of PLCβ1 can modulate self-association of α-synuclein. In parallel, down-regulation of PLCβ increases the propensity of α-synuclein to aggregate. Importantly, oxidative stress results in a relative lowering of the cellular concentration of PLCβ1 promoting in α-synuclein aggregation. Thus, the link between oxidative stress and α-synuclein aggregation may be due to the loss of protein partners.
Despite intense interest, the cellular function of α-synuclein is largely unknown. One approach used to glean insight into α-synuclein function was the generation of knockout mice. While these mice were viable, the lipid composition of endosomes and other subcelluar organelles were altered30,31 and they showed neurological abnormalities32 that are difficult to understand on a mechanistic level. Using more molecular approaches, others have looked for α-synuclein binding partners. While many potential interaction proteins have been uncovered, our lab found that PLCβ has one of the strongest affinities for α-synuclein15 and these proteins associate in cells.16 In cells, α-synuclein prevents PLCβ1 degradation by calpain and raises the intracellular level of the enzyme.16 α-Synuclein also inhibits PLCβ activity and this inhibition can be overcome by G proteins.15 This latter finding correlates well with the observation that α-synuclein moves from the plasma membrane to the cytosol with Gαq activation.33 Additionally, we have carried out in vitro FRET studies showing that α-synuclein prevents Alexa488-PLCβ1 from interacting with Alexa546-C3PO, which is a member of the RNA induced silencing complex (RISC) and binds to a similar site as Gαq19 (Sahu, Guo, and Scarlata, unpublished results).
Overexpression of α-synuclein has been shown to cause inclusions and neuronal death,34 but our results suggest that loss of binding partners due to oxidation may also play a role. PLCβ1, like α-synuclein, is highly expressed in neuronal tissue, and since PLCβ1 binds α-synuclein in cells, it is not surprising that increasing the level of PLCβ reduces α-synuclein self-association and aggregation as seen using fluorescence imaging methods. However, it is surprising that oxidation has different effects on the cellular levels of the two proteins. In cells, oxidation increases α-synuclein self-association as seen by FRET and by the transition from monomers to tetramers as seen by native gels (Figure 5), and in previous studies using other biophysical methods.5,6,29 The different effects of oxidation on α-synuclein and PLCβ1 levels may be attributed to their different mechanisms of down-regulation. PLCβ1 is down-regulated by ubiquitination which is promoted by protein damage whereas α-synuclein is down-regulated by chaperone-assisted autophagy.13 It is likely that oxidative stress reduces the level of many other α-synuclein binding partners increasing the effective level of unbound α-synuclein and further promoting aggregation.
Since the integrity of PLCβ1 is sensitive to oxidation, we used a PLC-derived peptide that blocks α-synuclein/PLCβ association, to determine whether α-synuclein aggregation could be prevented. We found that this peptide was very effective in preventing α-synuclein aggregation. The sequence of this peptide, residues 743–766 of PLCβ1, was based on previous studies where we found that α-synuclein protects PLCβ2 from calpain cleavage at Cys743. Interestingly, this sequence is mimicked in the residues 45–53 of α-synuclein as shown in Figure 7. This finding suggests that α-synuclein may have internal association site(s). We speculate that PLCβ1 and this peptide may replace this internal association site to prevent α-synuclein self-association. In any case, the identification of this peptide and its potential for preventing α-synuclein aggregation under basal or oxidative conditions offers a basis for new targets for neurodegenerative diseases.
Figure 7.
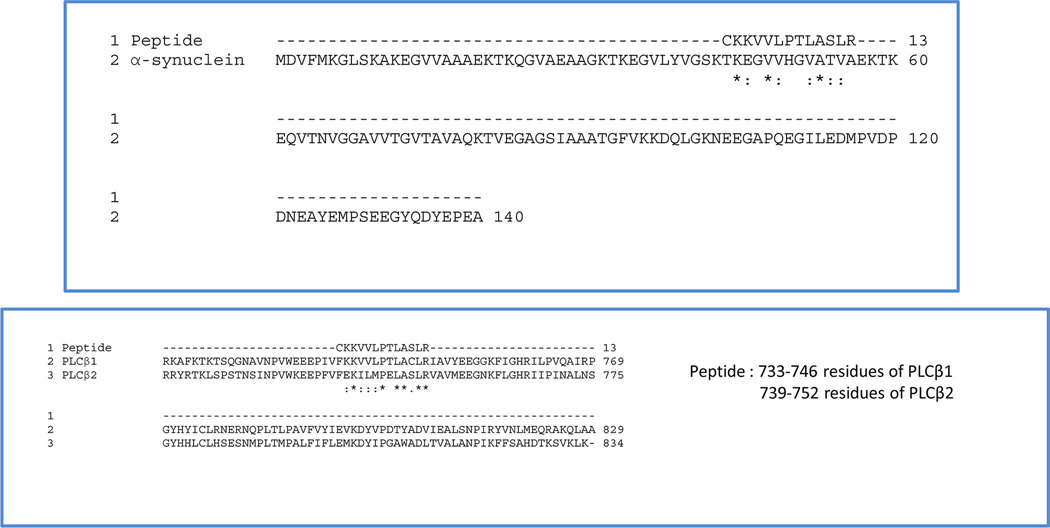
Alignment of the peptide to the sequence of α-synuclein (top) and comparison of the putative a-synuclein binding site of PLCβ1 and PLCβ1 (right).
ACKNOWLEDGMENTS
We would like to thank Urszula Golebiewska and Finly Philip for their helpful comments and to Narindra Kalipiprasada and Parijat Sengupta for their technical assistance.
Funding
This work was supported by NIH GM053132.
ABBREVIATIONS
- PLC
mammalian inositide-specificphospholipase C
- Dabcyl SE
4-dimethylaminophenylazophenyl-4-sulfonyl chloride succinylester
- CPM
7-diethylamino-3-(4-maleimidylphenyl)-4-methylcoumarin
- HEK293β1 cells
human embryonic kidney 293 cells containing PLCβ1 DNA regulated by a tetracycline promoter
- eGFP
enhanced green fluorescent protein
- mCherry
monomeric cherry fluorescent proteins
- FRET
Föster resonance energy transfer
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J. Neurosci. Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 2.Lundvig D, Lindersson E, Jensen PH. Pathogenic effects of α-synuclein aggregation. Mol. Brain Res. 2005;134:3–17. doi: 10.1016/j.molbrainres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 3.George JM. The synucleins. Genome Biol. 2002;3:3002.3001–3002.3006. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayanan V, Scarlata S. Membrane binding and self-association of alpha-synuclein. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 5.Bartels T, Choi JG, Selkoe DJ. [agr]-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U. S. A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsian A, Racette B, Zhang ZH, Chakraverty S, Rundle M, Goate A, Perlmutter JS. Mutation, sequence analysis, and association studies of alpha-synuclein in Parkinson’s disease. Neurology. 1998;51:1757–1759. doi: 10.1212/wnl.51.6.1757. [DOI] [PubMed] [Google Scholar]
- 9.Maguire-Zeiss KA, Short DW, Federoff HJ. Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Mol. Brain Res. 2005;134:18–23. doi: 10.1016/j.molbrainres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha- synuclein nitration in synucleinopathy lesions [In Process Citation] Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 11.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 12.Ancolio K, Alves da Costa C, Ueda K, Checler F. Alpha-synuclein and the Parkinson’s disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci. Lett. 2000;285:79–82. doi: 10.1016/s0304-3940(00)01049-1. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 14.Lee FJ, et al. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 15.Narayanan V, Guo Y, Scarlata S. Fluorescence studies suggest a role for a-synuclein in the phosphatdiylinositol lipid signaling pathway. Biochemistry. 2005;44:462–470. doi: 10.1021/bi0487140. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Rosati B, Scarlata S. α-synuclein increases the cellular level of phospholipase Cβ1. Cell. Signalling. 2012;24:1109–1114. doi: 10.1016/j.cellsig.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suh P, Park J, Manzoli L, Cocco L, Peak J, Katan M, Fukami K, Kataoka T, Yun S, Ryu S. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008;41:415–434. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- 18.Rebecchi M, Pentylana S. Structure, function and control of phosphoinositide-specific phospholipase C. Phys. Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 19.Philip F, Guo Y, Aisiku O, Scarlata S. Phospholipase Cβ1 is linked to RNA interference of specific genes through translin-associated factor X. FASEB J. 2012;26:4903–4913. doi: 10.1096/fj.12-213934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Runnels LW, Scarlata S. Determination of the affinities between heterotrimeric G protein subunits and their phospholipase C- b effectors. Biochemistry. 1999;38:1488–1496. doi: 10.1021/bi9821519. [DOI] [PubMed] [Google Scholar]
- 21.Dowal L, Provitera P, Scarlata S. Stable association between G alpha(q) and phospholipase C beta 1 in living cells. J. Biol. Chem. 2006;281:23999–24014. doi: 10.1074/jbc.M512330200. [DOI] [PubMed] [Google Scholar]
- 22.Golebiewska U, Johnston JM, Devi L, Filizola M, Scarlata S. Differential Response to Morphine of the Oligomeric State of μ-Opioid in the Presence of δ-Opioid Receptors. Biochemistry. 2011;50:2829–2837. doi: 10.1021/bi101701x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowal L, Elliott J, Popov S, Wilkie TM, Scarlata S. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta-1. Biochemistry. 2001;40:414–421. doi: 10.1021/bi001923+. [DOI] [PubMed] [Google Scholar]
- 24.Dai R-P, Yu F-X, Goh S-R, Chng H-W, Tan Y-L, Fu J-L, Zheng L, Luo Y. Histone 2B (H2B) Expression Is Confined to a Proper NAD+/NADH Redox Status. J. Biol. Chem. 2008;283:26894–26901. doi: 10.1074/jbc.M804307200. [DOI] [PubMed] [Google Scholar]
- 25.Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Ann. Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 26.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Baptista M, Miller D, Blancate J, Hardy J, Gwinn-Hardy K. a-Synuclein locus triplication casues Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 27.Paik SR, Shin HJ, Lee JH, Chang CS, Kim J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 1999;340:821–828. [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trexler AJ, Rhoades E. N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012;21:601–605. doi: 10.1002/pro.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golovko MY, Rosenberger TA, Feddersen S, Færgeman NJ, Murphy EJ. α-Synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J. Neurochem. 2007;101:201–211. doi: 10.1111/j.1471-4159.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- 32.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 33.Leng Y, Chase TN, Bennett MC. Muscarininc receptor stimulation induces translocation of an alpha-synuclein oligomer from plasma membrane to a light vesicles fraction in cytoplasm. J. Biol. Chem. 2001;276:28212–28218. doi: 10.1074/jbc.M011121200. [DOI] [PubMed] [Google Scholar]
- 34.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]