

Abstract
Aim: Gastric cancer is a major health problem and current treatment lacks lasting effect. Targeted therapy for gastric cancer with specific genetic background is in urgent need. Methods: We have studied The Cancer Genomic Atlas (TCGA) and The Genomics of Drug Sensitivity in Cancer (GDSC) databases to reveal genes with high frequency of mutation and possible sensitive compound against such gene mutation. In vitro studies were conducted to validate the in silico findings. Results: CDKN2A is frequently mutated in gastric cancer, revealed in TCGA database. CDK4/6 inhibitor PD-0332991 was sensitive in cancer cells with CDKN2A mutation, revealed in GDSC database. In vitro studies showed that PD-0332991 could selectively inhibit proliferation of gastric cancer cell with CDKN2A mutation. PD-0332991 could also inhibit cell invasion, migration, and colony formation of gastric cancer cell with CDKN2A mutation. PD-0332991 induced cell cycle arrest but not apoptosis. PD-0332991 inhibited xenograft gastric cancer mouse model. Conclusion: Gastric cancer with CDKN2A mutation is sensitive to CDK4/6 inhibitor. PD-0332991 is a potential therapeutic agent for gastric cancer.
Keywords: Gastric cancer, cell cycle, CDKN2A, CDK4/6
Introduction
Gastric cancer is a major health problem ranking the third most lethal cancer worldwide and one of the top leading cause of cancer-related death in China [1]. Disease at early stage usually lacks specific symptoms and patients admitted are always at later stage and treatment efficacy is usually compromised [2]. Surgical removal with chemotherapy is the currently the standard treatment of gastric cancer. However, many cases still progress or metastasize and succumb [3]. Therefore, novel treatment for gastric cancer is in urgent need.
Targeted therapy for cancer is currently proven effective in a variety of cancers [4]. The basis of developing potential targeted drugs is the insight of specific genomic or genetic alteration of a certain cancer type. Cancers with high frequency of mutation of certain genes are generally addicted to the according pathway and compounds that target this addiction are selectively sensitive in this very entity. Thus, global and insightful understanding of cancer genomics is requisite to discover effective drugs. The Cancer Genomic Atlas (TCGA) is collaborative studies that used next-generation sequencing techniques in a variety of cancers including gastric cancer and offers unprecedented insights into the genomic landscape of gastric cancer [5,6]. The TCGA gastric cancer projects propose a molecular classification dividing gastric cancer into four subtypes: tumours positive for Epstein-Barr virus, which display recurrent PIK3CA mutations, extreme DNA hypermethylation, and amplification of JAK2, CD274 (also known as PD-L1) and PDCD1LG2 (also known as PD-L2); microsatellite unstable tumours, which show elevated mutation rates, including mutations of genes encoding targetable oncogenic signalling proteins; genomically stable tumours, which are enriched for the diffuse histological variant and mutations of RHOA or fusions involving RHO-family GTPase-activating proteins; and tumours with chromosomal instability, which show marked aneuploidy and focal amplification of receptor tyrosine kinases [5].
The Genomics of Drug Sensitivity in Cancer (GDSC) project is a collaboration between the Cancer Genome Project at the Wellcome Trust Sanger Institute (UK) and the Center for Molecular Therapeutics, Massachusetts General Hospital Cancer Center (USA), and is funded by the Wellcome Trust [7]. The project screens the sensitivity of a panel of compounds in a variety of cancer cells and all cells are genotyped for common cancer mutations. In the current study, we used these two major databases to identify CDK4/6 inhibitor as a potential drug for gastric cancer with CDKN2A mutation and conducted in vitro and in vivo studies to confirm this surmise.
Materials and methods
Data mining of TCGA and GDSC databases
As the initial TCGA global analysis on gastric cancer was published and as per the latest publication guidelines of TCGA, our reproduction of the data had no limitations or restrictions [5]. The TCGA gastric cancer database analysed on the cBioPortal platform [8,9] contained in all 258 tumours with all of the following somatic data (complete tumours): mutations; putative copy-number alterations; mRNA expression data presented by Z-scores detected with microarray, with RNA-seq, or the mRNA/miRNA expression within all genes; and protein/phosphoprotein level detected with reverse phase protein array (RPPA). We confirm that reproduction and publication of the GDSC data and figures complied with the organization [10]. We started by searching compounds with significant selectivity for CDKN2A mutation. The scatter plots and the Mann-Whitney-Wilcoxon (MWW) tests were generated and computed via the GDSC online platform.
Cell culture and reagents
Human NCI-SNU-5 and NCI-N87 gastric cancer cells were acquired from ATCC. The COSMIC database was used to determine the genetic status of CDKN2A of the cells. Both cells were cultured in RPMI 1640 (PAA, Germany) with 10% foetal bovine serum (FBS) (PAA). CDK4/6 inhibitor PD-0332991 (Palbociclib) was purchased from Selleck Chemicals.
Western blotting
Blotting was performed as per previous reports [11,12]. Total protein of lysates was extracted and loaded onto 10% sodium dodecyl sulphate polyacrylamide gel for electrophoresis. The separated proteins were transferred to nitrocellulose membrane. The membranes were blocked for 1 h with 5% non-fat milk. Primary antibodies of CDKN2A, CDK4, CDK6, and GAPDH were all purchased from Abcam then added and membranes were kept at 4°C overnight. Horseradish-coupled secondary antibodies were used to detect the bands.
Proliferation assay
Briefly, cells were seeded in 96-well plates and were treated with single dose of PD-0332991 or control. Cells were then cultured for 72 h and subjected to formalin fixation. After several washes, cells were dyed using crystal violet, which were later dissolved using methanol. Proliferation was profiled by absorption at 540 nm read on plate reader.
Migration and invasion assay
Migration and invasion assay sere performed according to previous reports [11]. Briefly, cells were seeded in the top chamber of the Transwell inserts, which were coated with Matrigel for invasion assay and uncoated for migration assay. Complete medium was added to the lower chamber and cells were then incubated. Inserts were harvested and cells penetrated the membrane were stained with crystal violet and were counted microscopically. The average number of cells in three high power fields was calculated. All assays were done in triplicates.
Colony formation assay
Soft agar assay was used to study the ability of colony formation of cells. Briefly, 1,000 cells were resuspended in complete medium mixed with 0.4% Noble agar. The mixture was placed on top of the similar mixture consisting of complete medium and 0.6% of agar on 60-mm plates. Complete medium containing treatment and control drug were added at the upmost layer and were changed every 3 days. After 2 weeks of culture, plates were stained with crystal violet for and colonies were counted microscopically.
Xenograft model
Twelve male BALB/c athymic nude mice which were 6 weeks of age were bred in licensed SPF (special pathogen-free) grade laboratory. Mice were randomly divided into 2 groups. A total of 1.5×107 cells were injected subcutaneously at the left axilla of each mouse. PD0332991 was suspended in sodium lactate solution (Sigma-Aldrich) and administered by oral gavage daily at 150 mg/kg for 28 days. Tumour size was calculated with the formula, Length × Width2 × 0.5236.
Statistical analysis
Comparisons between groups were analysed with the 2-tailed Student’s t-test. The P value of <0.05 was accepted as statistically significant.
Results
CDKN2A is frequently mutated in gastric cancer
With reproduction of the TCGA gastric cancer cohort, we identified high frequency of involvement of CDKN2A pathway (Figure 1A), In particular, mutation and loss of heterozygosity comprised up to 38% of cases in gastric cancer (Figure 1A). Besides CDKN2A, CDK4, CDK6, and CCND1 showed significant tendency for co-occurrence in gastric cancer, indicating a possible redundant role of CDK4 and CDK6 (Figure 1B). Most mutations of CDKN2A were non-synonymous (Figure 1C). Reproduction of GDSC database showed that CKD4/6 inhibitor PD-0332991 was a selective inhibitor for cancer cells with CDKN2A inhibitor (Table 1; Figure 1D). We then studied whether loss of CDKN2A impacted on survival by reproduction of the TCGA database. We found that overall survival and disease-free period was not associated with altered CDKN2A gene set, including CDKN2A, CDK4, CDK6, CCND1, and RB1 (Figure 2A, 2B). Overall survival and disease-free period was not associated with altered CDKN2A either (Figure 2C, 2D). Here we showed that CDK4/6 inhibitor PD-0332991 could be a promising therapeutic agent for gastric cancer with CDKN2A mutation, which occurred at high frequency in gastric cancer.
Figure 1.
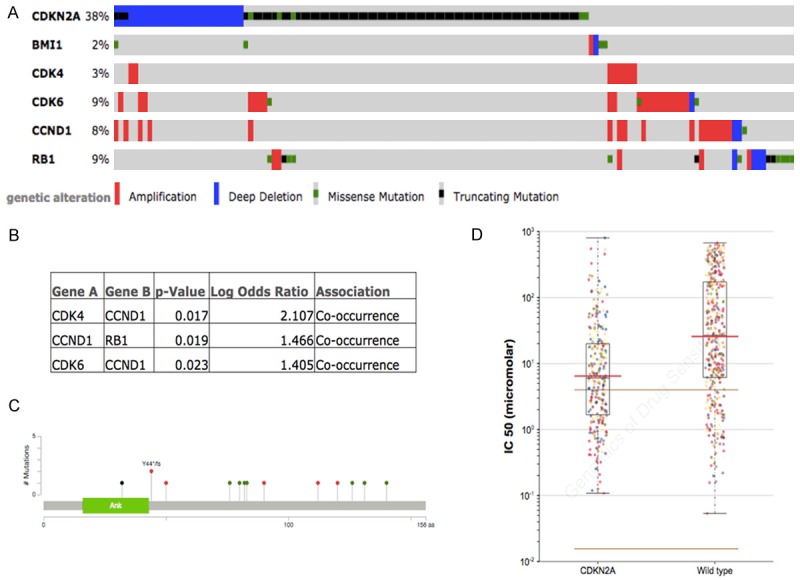
CDKN2A gene set is altered in gastric cancer reproduced from TCGA database. (A) High frequency of CDKN2A mutation in gastric cancer along with amplification in CDK4/6 and CCND1; (B) Mutual exclusivity analysis revealing significant association between CDK4, CDK6, CCND1, and RB1, with a trend of co-occurrence; (C) Mutations in CDKN2A in gastric cancer in TCGA with green showing missense, red showing truncating, and black showing inframe mutation; Reproduction of GDSC database showed (D) CDK4/6 inhibitor, PD-0332991 is sensitive in tumour cells with mutated CDKN2A.
Table 1.
Top 10 sensitive drugs/compounds in tumours with CDKN2A mutation reproduced from the GDSC database
| Drug | Drug Target | Effect | P-value | No. of mutations |
|---|---|---|---|---|
| PD-0332991 | CDK4/6 | 0.418 | 4.03E-23 | 262 |
| Dasatinib | ABL, SRC, KIT, PDGFR | 0.331 | 4.68E-12 | 120 |
| Cytarabine | DNA synthesis | 0.742 | 1.75E-10 | 278 |
| WH-4-023 | SRC family, ABL | 0.316 | 1.28E-09 | 120 |
| AZD-0530 | SRC, ABL1 | 0.333 | 2.57E-08 | 121 |
| RDEA119 | MEK1/2 | 0.371 | 0.00000011 | 274 |
| CI-1040 | MEK1/2 | 0.552 | 0.000000137 | 278 |
| 17-AAG | HSP90 | 0.4 | 0.000000408 | 278 |
| Docetaxel | Microtubules | 0.373 | 0.000000765 | 278 |
| PD-0325901 | MEK1/2 | 0.456 | 0.00000133 | 274 |
Figure 2.
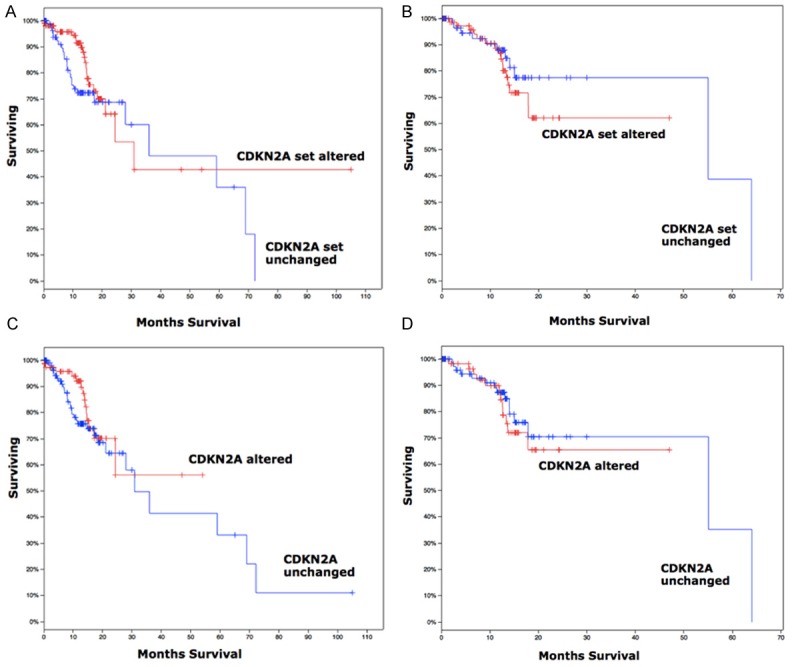
Clinical outcome of gastric cancer patients reproduced from TCGA database showing (A) overall survival and (B) disease-free period was not associated with altered CDKN2A gene set (CDKN2A, CDK4, CDK6, CCND1, and RB1); (C) overall survival and (D) disease-free period was not associated with altered CDKN2A either.
PD0332991 selectively inhibits gastric cancer with CDKN2A mutation
We first studied the effect of PD-0332991 in vitro using 2 gastric cell lines with different CDKN2A status. In line with the COSMIC data, NCI-SNU-5 and NCI-N87 gastric cancer cells had mutated and wild type CDKN2A, respectively (Figure 3A). Accordingly, levels of CDK4 and CDK6 were inhibited by PD-0332991 in CDKN2A mutated cells whereas the basal level of CDK4/6 in CDKN2A wild type cells was low (Figure 3A). We found that PD-0332991 selectively inhibited the growth of gastric cancer cells with CDKN2A mutation (Figure 3B, 3C). Further, we showed that PD-0332991 selectively inhibited cell migration and invasion of gastric cells with CDKN2A (Figure 4A, 4B). PD-0332991 also selectively inhibited the anchorage-independent growth of gastric cancer cells (Figure 4C). As CDKN2A was essential in cell cycle control, we showed that PD-0332991 induced cell population in G1 phase in gastric cancer cells with CDKN2A mutation (Figure 5A-C). We also showed that cell apoptosis was not significantly different between cells treated with PD-0332991 or control (Figure 5D-F). Finally, we tested the efficacy of PD-0332991 in mouse xenograft models implanted with gastric cancer cells with CDKN2A mutation. We found that PD-0332991 significantly inhibited tumour growth of gastric cancer cells with CDKN2A mutation (Figure 6A, 6B).
Figure 3.
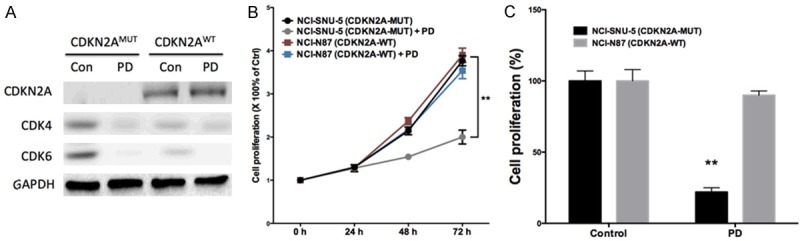
CDK4/6 inhibitor PD-0332991 selectively inhibited gastric cancer cells with CDKN2A mutation. (A) PD-0332991 inhibited both CDK4 and CDK6 in gastric cancer cells; (B) PD-0332991 selectively inhibited growth of gastric cancer cells with CDKN2A mutation at (C) 72 h of treatment at 5 μM (n = 3; *P < 0.05; **P < 0.01).
Figure 4.
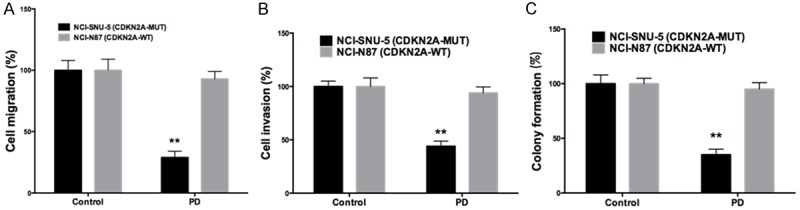
CDK4/6 inhibitor PD-0332991 selectively inhibited motility of gastric cancer cells with CDKN2A mutation by suppressing (A) cell migration and (B) cell invasion, and inhibited anchorage-independent growth by suppressing (C) colony formation, all treated at 5 μM (n = 3; **P < 0.01).
Figure 5.

CDK4/6 inhibitor PD-0332991 induced cell cycle arrest of gastric cancer cells with CDKN2A mutation. Compared with (A) control, (B) PD-0332991 induced (C) higher population of cells in G1 phase; Whereas compared with (D) control, (E) PD-0332991 did not induce (C) more apoptosis of cells (n = 3; NS = not significant; **P < 0.01).
Figure 6.
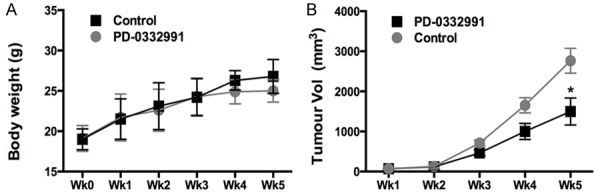
CDK4/6 inhibitor PD-0332991 inhibited gastric cell growth in vivo; A. Xenograft tumour model showing comparable body weight between mice treated with control or PD-0332991; B. On week 5, tumour volume of PD-0332991 treated mice were significantly smaller that control (n = 6; *P < 0.05).
Discussion
Stomach cancer or gastric cancer is cancer developing from the lining of the stomach. Early symptoms may include heartburn, upper abdominal pain, nausea and loss of appetite. Later signs and symptoms may include weight loss, yellow skin, vomiting, difficulty swallowing, and blood in the stool among others [13]. The cancer may spread from the stomach to other parts of the body, particularly the liver, lungs, bones, lining of the abdomen and lymph nodes.
In the current study, we have identified that CDK4/6 inhibitor PD-0332991 is a selective inhibitor for gastric cancer with CDKN2A mutation. CDKN2A is a gene, which in humans is located at chromosome 9, position 21. It codes for several proteins, including p16 and p14arf [14]. Both act as tumour suppressors, and CDKN2A has been extensively studied in association with melanoma and pancreatic cancer [14,15]. p16 was originally found in an “open reading frame of 148 amino acids encoding a protein of molecular weight 15,845 comprising four ankyrin repeats. p16Ink4A is named after its molecular weight and its role in inhibiting CDK4. p16 is an inhibitor of cyclin dependent kinases such as CDK4 and CDK6 [16]. These latter kinases phosphorylate retinoblastoma protein (pRB), which eventually results in progression from G1 phase to S phase. p16 plays an important role in cell cycle regulation by decelerating cells progression from G1 phase to S phase, and therefore acts as a tumour suppressor that is implicated in the prevention of cancers, notably melanoma, oropharyngeal squamous cell carcinoma, cervical cancer, and oesophageal cancer [17]. p16 can be used to improve the histological diagnostic accuracy of CIN3. The CDKN2A gene is frequently mutated or deleted in a wide variety of tumours.
p14ARF is induced in response to elevated mitogenic stimulation, such as aberrant growth signaling from MYC and Ras [18]. It accumulates mainly in the nucleolus where it forms stable complexes with NPM or Mdm2. These interactions allow p14ARF to act as a tumour suppressor by inhibiting ribosome biogenesis or initiating p53-dependent cell cycle arrest and apoptosis, respectively [19]. p14ARF is an atypical protein, in terms of its transcription, its amino acid composition, and its degradation: it is transcribed in an alternate reading frame of a different protein, it is highly basic, and it is polyubiquinated at the N-terminus. Both p16INK4a and p14ARF are involved in cell cycle regulation. p14ARF inhibits mdm2, thus promoting p53, which promotes p21 activation, which then binds and inactivates certain cyclin-CDK complexes, which would otherwise promote transcription of genes that would carry the cell through the G1/S checkpoint of the cell cycle. Loss of p14ARF by a homozygous mutation in the CDKN2A (INK4A) gene will lead to elevated levels in mdm2 and, therefore, loss of p53 function and cell cycle control.
Frequent amplifications of cell cycle mediators (CCNE1, CCND1 and CDK6) suggest the potential for therapeutic inhibition of cyclin-dependent kinases in gastric cancer. In our study, we showed that inhibition of CDK4/6 could be a promising therapeutic approach in gastric cancer. CDK4/6 is elements downstream of the CDKN2A signaling axis and play critical roles in cell cycle progression. Accordingly in our study, we identified cell cycle arrest but not apoptosis induced by CDK4/6 inhibitor.
In summary, we have incorporated two major databases and have showed that gastric cancer harbor high frequency of CDKN2A mutation and loss. Gastric cancer with CDKN2A mutation is sensitive to CDK4/6 inhibitor. PD-0332991 is a potential therapeutic agent for gastric cancer.
Acknowledgements
This study was supported in part by The Natural Science Grant of Ningbo, Zhejiang, No. 2013A610204.
Disclosure of conflict of interest
None.
References
- 1.Deng K, Yang L, Hu B, Wu H, Zhu H, Tang C. The Prognostic Significance of Pretreatment Serum CEA Levels in Gastric Cancer: A Meta-Analysis Including 14651 Patients. PLoS One. 2015;10:e0124151. doi: 10.1371/journal.pone.0124151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu DH, Tang L, Dong H, Dong Z, Zhang L, Fu J, Su X, Zhang T, Fu H, Han L, Xie L, Chen H, Qian Z, Zhu G, Wang J, Ye Q, Zhang J, Yin X, Zhang X, Ji J, Ji Q. Oncogenic HER2 fusions in gastric cancer. J Transl Med. 2015;13:116. doi: 10.1186/s12967-015-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu X, Li Z, Li Z, Jia Y, Shan F, Ji X, Bu Z, Zhang L, Wu A, Ji J. Hyperthermic intraperitoneal chemotherapy plus simultaneous versus staged cytoreductive surgery for gastric cancer with occult peritoneal metastasis. J Surg Oncol. 2015;111:840–847. doi: 10.1002/jso.23889. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes E, Ferreira JA, Peixoto A, Lima L, Barroso S, Sarmento B, Santos LL. New trends in guided nanotherapies for digestive cancers: a systematic review. J Control Release. 2015;209:288–307. doi: 10.1016/j.jconrel.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, Stratton MR, Benes C, McDermott U, Garnett MJ. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding G, Feng C, Jiang H, Ding Q, Zhang L, Na R, Xu H, Liu J. Combination of Rapamycin, CI-1040, and 17-AAG Inhibits Metastatic Capacity of Prostate Cancer via Slug Inhibition. PLos One. 2013;8:e77400. doi: 10.1371/journal.pone.0077400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng C, Wu Z, Guo T, Jiang H, Guan M, Zhang Y, Wen H, Ding Q. BLCA-4 expression is related to MMP-9, VEGF, IL-1α and IL-8 in bladder cancer but not to PEDF, TNF-α or angiogenesis. Pathol Biol (Paris) 2012;60:e36–e40. doi: 10.1016/j.patbio.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Lim H, Park YS, Lee JH, Son DH, Ahn JY, Choi KS, Kim DH, Choi KD, Song HJ, Lee GH, Jung HY, Kim JH, Yook JH, Kim BS. Features of Gastric Carcinoma with Lymphoid Stroma Associate with Epstein-Barr Virus. Clin Gastroenterol Hepatol. 2015 doi: 10.1016/j.cgh.2015.04.015. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Helgadottir H, Hoiom V, Tuominen R, Jonsson G, Mansson-Brahme E, Olsson H, Hansson J. CDKN2A mutation-negative melanoma families have increased risk exclusively for skin cancers but not for other malignancies. Int J Cancer. 2015;137:2220–6. doi: 10.1002/ijc.29595. [DOI] [PubMed] [Google Scholar]
- 15.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA Australian Pancreatic Cancer Genome Initiative. Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Diez E, Quereda V, Bellutti F, Prchal-Murphy M, Partida D, Eguren M, Kollmann K, Gomez de Cedron M, Dubus P, Canamero M, Martinez D, Sexl V, Malumbres M. Cdk4 and Cdk6 cooperate in counteracting the INK4 family of inhibitors during murine leukemogenesis. Blood. 2014;124:2380–2390. doi: 10.1182/blood-2014-02-555292. [DOI] [PubMed] [Google Scholar]
- 17.Yaswen P, MacKenzie KL, Keith WN, Hentosh P, Rodier F, Zhu J, Firestone GL, Matheu A, Carnero A, Bilsland A, Sundin T, Honoki K, Fujii H, Georgakilas AG, Amedei A, Amin A, Helferich B, Boosani CS, Guha G, Ciriolo MR, Chen S, Mohammed SI, Azmi AS, Bhakta D, Halicka D, Niccolai E, Aquilano K, Ashraf SS, Nowsheen S, Yang X. Therapeutic targeting of replicative immortality. Semin Cancer Biol. 2015 doi: 10.1016/j.semcancer.2015.03.007. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humbey O, Pimkina J, Zilfou JT, Jarnik M, Dominguez-Brauer C, Burgess DJ, Eischen CM, Murphy ME. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008;68:9608–9613. doi: 10.1158/0008-5472.CAN-08-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veneziano L, Barra V, Lentini L, Spatafora S, Di Leonardo A. p14 Prevents Proliferation of Aneuploid Cells by Inducing p53-Dependent Apoptosis. J Cell Physiol. 2015 doi: 10.1002/jcp.24976. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]