

Abstract
Cardiovascular disease (CVD) represents the most significant cause of death in postmenopausal women. Advanced glycation end products (AGEs) are formed by nonenzymatic modification of proteins, lipids, and nucleic acids by glucose. This review focuses on the contribution of AGEs and their receptors to the development of CVD in menopause. Advanced glycation end products circulate and activate the proinflammatory endothelial cell surface receptor called RAGE, bind to the extracellular matrix of the cardiovascular system, or bind to the circulating anti-inflammatory soluble form of RAGE (sRAGE). Data emerging from human and animal studies suggest that AGEs and both receptors (RAGE and sRAGE) are implicated in the pathophysiology of CVD. Particular emphasis has been given to the role of AGE–RAGE axis in oxidative stress, inflammation, endothelial cell toxicity, and progression of atherosclerosis in menopause. Data accruing from human and animal studies suggest that RAGE expression level and circulating sRAGE level are associated with estradiol and are correlated with CVD risk factors, such as adiposity, dyslipidemia, insulin resistance, diabetes, and metabolic syndrome. By recognizing the impact of AGEs on atherosclerosis, pharmacological strategies targeting the AGE–RAGE pathway hold therapeutic potential for CVD in menopausal women.
Keywords: cardiovascular disease, advanced glycation end products, RAGE, sRAGE, atherosclerosis, menopause
Introduction
The natural loss of ovarian function gives rise to menopause, a phase in a woman’s life that is characterized by loss of reproductive competence. During this period, women acquire an increased risk of cardiovascular disease (CVD), osteoporosis, cognitive dysfunction, diabetes, dyslipidemia, and mortality.1 Cardiovascular disease is recognized as the most significant cause of death in postmenopausal women.1 In addition to the traditional CVD risk factors such as smoking, hypertension, obesity, and dyslipidemia, advanced glycation end products (AGEs) have been recently recognized for their contribution to CVD. Most of AGEs’ biological effects are mediated through the receptor RAGE, present on the cell membrane of several tissues including the cardiovascular system.2 The activation of RAGE generates oxidative stress and modulates cellular and tissue fate. On the other hand, the soluble form of RAGE (sRAGE) acts as a decoy by binding AGEs thus preventing them from binding to the proinflammatory RAGE receptor.3,4 Data accruing from human and animal studies suggest that RAGE expression level and circulating sRAGE level are associated with estradiol and are correlated with CVD risk factors. This review underscores the implications of AGEs and their receptors in CVD in menopause with a focus on adiposity, insulin resistance, dyslipidemia, and hormone replacement therapy.
Pharmacology of AGEs and Their Receptors
The nonenzymatic modification of proteins, lipids, and nucleic acids by glucose produces AGEs, such as N∊-carboxymethyl-lysine (CML), pyrraline, and pentosidine.2 Advanced glycation end products endogenously accumulate in the serum and tissues with aging and at an accelerated rate in diabetes, insulin resistance, obesity, metabolic syndrome, hypoxia, and oxidative stress.5 The extracellular effect of AGEs includes modification of the structural integrity of the vessel wall and the underlying basement membranes by inducing cross-linking of matrix proteins.6 Additionally, AGEs exist in diet and get absorbed by the gastrointestinal system.7 Food high in protein and fat, such as fast-food meals, is rich in absorbable AGEs, influencing serum and tissue AGEs’ concentration.8–11 Such dietary AGEs can also contribute to increased oxidant stress and inflammation.8
RAGE is a member of the immunoglobulin superfamily of receptors.12 The human RAGE gene is located in the major histocompatibility complex locus on chromosome 6.13 Engagement of RAGE in intracellular signaling leads to reactive oxygen species production via activation of the proinflammatory transcription factor nuclear factor κB and Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. This leads to the production of proinflammatory cytokines (such as interleukin [IL] 1, IL-6, and IL-8), chemokines, apoptosis regulators (such as bcl-2 and Fas), adhesion molecules (such as vascular cell adhesion molecule 1 and intercellular adhesion 1), and activation of macrophages and platelets.6,14 Interestingly, activation of RAGE induces a positive feedback loop by increasing its own expression (Figure 1).6,15,16
Figure 1.
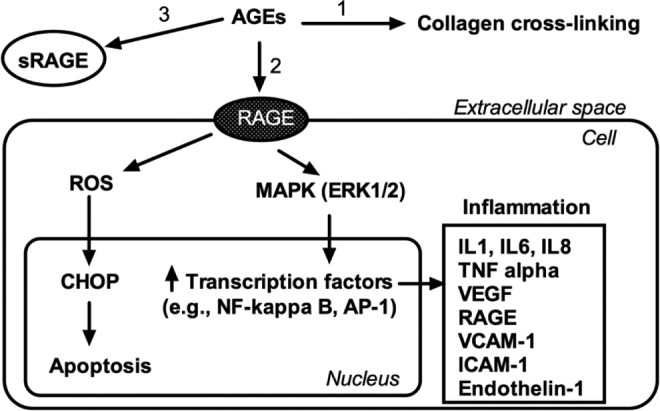
Schematic diagram of the pathogenetic effects of AGEs. They could damage cellular structures via formation of cross-links between key molecules in the extracellular matrix, for example, collagen (1). The interaction with the cell membrane receptor RAGE induces inflammation and apoptosis (2). The circulating receptor soluble form of RAGE (sRAGE) acts as decoy by binding the circulating AGEs, thus conferring a protective role (3). AGEs indicates advanced glycation end products; RAGE, receptor for AGE; NF-kB, nuclear factor kappa B; AP-1, activator protein-1; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase. CHOP, C/EBP homologous protein; IL, interleukin; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor; VCAM-1, vascular cell adhesion protein 1; ICAM-1, intercellular adhesion.
sRAGE lacks both the cytosolic and transmembrane domains and is a product of either splicing of RAGE gene and/or cleavage of the membrane-bound RAGE by a disintegrin and metallopeptidase 10 and matrix metalloproteinases.3,4 sRAGE receptors circulate and act as decoy by binding the circulating AG, thus competitively inhibiting AGE–RAGE interaction and its downstream proinflammatory signaling.3 Therefore, sRAGE is often considered a “good” receptor.
Role of AGEs and Their Receptors in Vascular Dysfunction
Atherosclerosis is not only attributed to high fat diet, diabetes, obesity, smoking, and hypertension17 but also to products containing sugars, which may provide substrates for the nonenzymatic glycation reaction, leading to the formation of AGEs.8 The deposition of AGEs in blood vessels contributes to CVD5,18 by activating RAGE signaling causing chronic activation of the inflammatory processes resulting in accelerated atherosclerosis.14,19,20
The relationship between sRAGE plasma levels and atherosclerosis (in the presence or absence of coronary artery disease) has been studied but the results are contentious. The divergence in the results could be due to difference in ethnicity of the study participants as well as major confounding variables such as diabetes and renal failure.21–23 Nondiabetic participants with hypertension and coronary artery disease were found to have lower levels of plasma sRAGE when compared to healthy participants.24–26
Recently, Falcone et al3 showed that patients with coronary artery disease presenting with peripheral artery disease have lower sRAGE levels than patients with coronary artery disease without peripheral artery disease (615 vs 766 pg/mL, respectively; P = .02). Additionally, that study demonstrated that stable atherosclerotic lesions in different vascular districts are inversely correlated with circulating sRAGE levels.3 These results suggest that stable atherosclerotic lesions in different vascular districts are inversely related to the soluble decoy receptor sRAGE. Additionally, others have demonstrated that a low plasma sRAGE level was significantly related to endothelial dysfunction in nondiabetic patients.4 Decreased plasma sRAGE concentration was a predictor of cardiovascular events, suggesting its role in atherothrombosis,4 thus the authors speculated that sRAGE could be a potentially protective agent against vascular complications.4 sRAGE can act as an important inhibitor of vascular inflammatory responses generated in branches and curvatures of the arterial tree.27 In a mouse model, treatment with sRAGE in the region of atherosclerotic plaque attenuated the development of plaque formation and markedly attenuated monocyte–endothelial cell adhesion.28 In that study, RAGE expression was suppressed following treatment with sRAGE. This suggest that sRAGE exerts antiatherogenic effects by blocking the activation of the RAGE signaling pathway and may thus be a potential therapeutic target for the prevention of atherosclerosis. Of note, a 12-week moderate intensity aerobic exercise program has been shown to significantly increase circulating sRAGE levels in women with type 2 diabetes resulting in atherosclerosis risk reduction.29 Altogether, most of these data reveal that sRAGE actions result in an antiatherogenic effect.
On the other hand, several studies challenged the protective effect of sRAGE. For instance, elevated sRAGE levels in women aged 65 and older were associated with an increased risk of cardiovascular death in the Women’s Health and Aging Study I, a population-based study designed to evaluate the causes and course of physical disability in older disabled women living in a community.30 However, since this study involved moderately to severely disabled community-dwelling women, their results may not be translated to young, healthier women.30 In a 5-year prospective study, Japanese participants with type 2 diabetes had higher CVD events as serum sRAGE levels increased (P = .046) and serum sRAGE levels were independently associated with CVD (P = .034) after adjusting for conventional coronary risk factors.22
Studies have been performed on prevalent genetic variants of RAGE and CVD. Similar to other studies,31,32 Hofmann et al33 found no association between RAGE genotype Gly82Ser and CVD in the Framingham offspring study. Bansal et al34 investigated the association between RAGE gene polymorphisms (-374T/A, -429T/C, and G82S) with macrovascular complications in Indian diabetic patients. Their results showed that -429T/C polymorphism was associated with macrovascular complications; however, -374T/A polymorphism conferred protection, and G82S polymorphism did not show any association.
Association Between AGE–RAGE Axis and Risk Factors for CVD in Menopause
Interestingly, the AGE–RAGE axis has a relationship with CVD risk factors. Koyama et al35 demonstrated that plasma sRAGE levels were significantly and inversely correlated with body mass index, blood pressure, glucose intolerance, triglyceride, hemoglobulin A1c, and insulin resistance index even in the absence of diabetes.35 The authors also reported that sRAGE was inversely associated with atherosclerosis in carotid and femoral arteries, regardless of diabetes status.35 Sebekova et al,36 in a study including 59% females (mean age of 33), demonstrated that plasma sRAGE and CML levels decreased with increasing number of metabolic syndrome risk factors, in particular waist circumference.36 Additionally, in a study by Norata et al,37 plasma sRAGE levels were negatively and significantly correlated with body mass index and waist/hip circumference in nondiabetic healthy participants.37 These correlations were mainly observed in women (n = 55, mean age of 62).
Insulin Resistance
Menopause coincides with an increase in several comorbidities that include insulin resistance.38,39 Two decades ago, Lindheim et al40 demonstrated that insulin resistance is prevalent in healthy postmenopausal women. Further, Lindheim et al41 assessed insulin sensitivity in pre- (n = 18) and postmenopausal (n = 10) women after randomization to either oral estrogen (0.625 mg conjugated equine estrogen) or estrogen with progesterone therapy (0.625 mg conjugated equine estrogen/10 mg progestin) for 6 months. The authors concluded that there was a mild degree of insulin resistance in healthy postmenopausal women and that estrogen appeared to improve insulin sensitivity, while added progestin attenuated this beneficial effect.41
Since oxidative stress generation and inflammation are also associated with insulin resistance, AGE–RAGE axis plays a role in the pathogenesis of insulin resistance and subsequently the development of diabetes.42 The Leeds family study43 demonstrated a link between polymorphism of the RAGE gene and insulin resistance. Additionally, regardless of obesity and diabetes status, Tan et al44 have demonstrated that the circulating level of AGEs is associated with insulin resistance.44 Further, glycated albumin (a source of AGEs) could be involved in the modulation of insulin signaling and hence in the generation of insulin resistance in skeletal muscle cells.45 The effects of methylglyoxal (precursor of AGEs) on insulin signaling pathway have been investigated in a rat model of insulin resistance.46 The authors reported an increase in endogenous methylglyoxal accumulation, which impaired insulin-signaling pathway and decreased insulin-stimulated glucose uptake by adipose tissue.46 It was recently reported in a nonobese mouse model that methylglyoxal-AGEs were identified as a nontraditional risk factor for insulin resistance in nonobese mice independent of overnutrition.47 Interestingly, the phenotypic shift of weight gain, adiposity, and insulin resistance observed in mice treated with an isocaloric diet in which AGEs were largely substituted for by synthetic methylglyoxal derivatives of a single protein almost mirrored the metabolic syndrome in humans.47 Advanced glycation end products could also alter insulin sensitivity via an indirect mechanism involving the dysregulation of adipokines and cytokines in adipocytes,48 macrophages,49 and endothelial cells.48 Taken together, these considerations place AGE–RAGE in the center of biochemical and molecular stresses that characterize insulin resistance, a mediator of CVD in menopausal women.
Lipids
The decline in estradiol following the menopausal transition leaves the vasculature vulnerable to CVD risk factors, such as lipids.50 Dyslipidemia in menopause is characterized by an increase in low-density lipoprotein (LDL) levels and a decline in HDL.51,52 In Healthy Women Study, total and LDL cholesterol increased and HDL and HDL2 cholesterol declined among premenopausal women who ceased menstruating at least 1 year compared to age-matched premenopausal women who continued menstruating.53 Interestingly, both The Los Angeles Atherosclerosis Study and the SWAN Heart Women demonstrated that the antiatherogenic effect of HDL diminishes in women around the age of menopause54 and it was suggested that it is possibly related to changes in the lipoprotein subclass profile observed during the menopausal transition.55
Data support the involvement of AGE–RAGE axis in dyslipidemia. Lopes-Virella et al56 reported that oxidized LDL and AGE-modified LDL levels in circulating immune complexes were significantly associated with progression and increased levels of carotid intima–media thickness in Diabetes Control and Complications trial participants.56 Later, the same group reported that levels of AGE-modified LDL in circulating immune complexes were independent predictors for the development and progression of atherosclerosis over a 12-year period in patients with type 1 diabetes.57 Turk et al58 demonstrated a positive correlation between serum level of methylglyoxal adducts and LDL-cholesterol and a positive correlation between urinary levels of methylglyoxal adducts and serum triglycerides in diabetic patients.58
Cholesterol-lowering statin therapy reduces morbidity and mortality from CVD in diabetic patients. Simultaneously, statin treatment suppresses the AGE–RAGE axis by reducing serum levels of AGEs and by lowering RAGE expression in patients with type 2 diabetes.59 Atovastatin treatment in patients with hypercholesterolemia and type 2 diabetes also showed an increase in serum sRAGE levels.60,61 In diabetic rats, atovastatin treatment downregulated the RAGE expression.62 These studies suggest that statins may prevent the development of RAGE-mediated pathogenesis.63 On the other hand, Collaborative Atorvastatin Diabetes Study trial results showed that long-term, daily treatment with atovastatin had no effect on sRAGE values.64
Given the relationship between “menopause and dyslipidemia” and given the association between “AGE–RAGE system and dyslipidemia,” it is reasonable to hypothesize that there is a possible involvement for the proinflammatory AGE–RAGE system in the pathogenesis of dyslipidemia in menopause, a major risk factor for CVD (Figure 2).
Figure 2.

The relation between cardiovascular disease risk factors and advanced glycation end products. The postmenopausal changes in adiposity, lipid metabolism, and insulin resistance indicate a shift toward a proatherogenic state. The findings of relationships between CVD risk factors and the AGE–RAGE axis at the serum and cellular levels provide a framework for explaining the role of such axis in the development of CVD. Thus, AGEs increase oxidative stress, which during the menopausal transition is already heightened as a consequence of greater production of reactive oxygen species with a decline in antioxidant defense. Oxidative stress causes endothelial dysfunction, leading eventually to atherosclerosis. AGE indicates advanced glycation end product CVD, cardiovascular disease; RAGE, receptor for AGE.
Menopausal Hormone Therapy and AGE–RAGE Axis
Up to date, the data on the impact of menopausal hormone therapy on AGE–RAGE axis are quite limited but studies suggest an involvement of estradiol in AGE–RAGE signaling. Six months of estrogen treatment (estradiol valerate 2 mg/daily) resulted in significant decrease in AGEs (such as a non-fluorescent glycation crosslink NFC-1) in vaginal epithelial tissues of postmenopausal women when compared to placebo.65 These findings suggest a possible anti-inflammatory and risk-reducing role of estrogens in postmenopausal women.66 Additionally, 10 nmol/L of 17β-estradiol, predominantly found in the circulation of premenopausal women, induced the expression of RAGE in in vitro cultured human endothelial cells.67 This indicates that estrogens might actually be the trigger that fuels CVD by inducing the expression of the proinflammatory molecule RAGE.68 Therefore, administration of synthetic estrogens to postmenopausal women might further increase the level of proinflammation leading to the progression of various cardiovascular complications via enhancing the expression of RAGE.65
Pullerits et al69 evaluated the effect of menopausal hormone therapy (2 mg of estradiol + 1 mg of noretisterone acetate) on circulating sRAGE levels in postmenopausal women with rheumatoid arthritis in a prospective 2-year randomized, single blinded, and controlled study. The results indicated that menopausal hormone therapy decreased circulating sRAGE levels. The increase in serum estradiol was associated with the decline in sRAGE levels.69 Moreover, higher sRAGE levels were associated with elevated levels of bone resorption and bone turnover markers at baseline, whereas the decrease in sRAGE levels paralleled with diminished concentration of these molecules indicating a possible role of sRAGE in bone/cartilage metabolism.69 These data indicate that estrogens might be directly involved in the regulation of sRAGE levels, but studies are needed to provide insights into the clinical significance of menopausal hormone therapy in sRAGE regulation and vice versa.
Pharmacological Strategies Targeting AGE-Induced Pathophysiological Mechanisms
Recognizing the impact of AGEs on atherosclerosis, pharmacological strategies targeting reduction in the pathophysiological mechanisms caused by AGEs are being developed (Table 1).70–88 The precise chemical product responsible for in vivo AGE formation has not been identified yet and therefore the development of specific inhibitors is quite challenging.70 This section will discuss some of the strategies since the blockade of AGEs’ formation, obstruction of AGEs with RAGE interaction, and/or suppression of the RAGE downstream pathway represent promising therapeutic strategies for CVD.
Table 1.
Pharmacological Strategies Targeting Pathophysiological Mechanisms Caused by AGEs on Endothelial Cells.
| General Description | Mechanism of Action | Compound Name (examples) |
|---|---|---|
| Inhibitors of AGE formation de novo | Trap reactive carbonyl intermediates and block the formation of AGEs | Aminoguanidine, pyridoxamine,85 cinnamic acid,74 and isoferulic acid74 |
| Inhibitors of AGE cross-link | Destruct collagen cross-linking structure resulting in overcoming of artery stiffening | Phenylthiazol, DPTC chloride, ALT-711, and LR-9073 |
| Compounds that downregulate RAGE expression | Suppressors of ROS generation | Analogue of prostacyclin-beraprost86 or forskolin activator of adenylate cyclase,86 angiotensin II-converting enzyme inhibitors, angiotensin II receptor type 1 blocker,87 red grape skin extract88 |
| Compounds that remove excess metal ions | Chelate | Triethylenetetramine84 |
Abbreviations: AGE, advanced glycation end product; ROS, reactive oxygen species; DPTC chloride, 4,5-dimethyl-3-phenyl-acyl-thiazole; ALT-711, chloride, 4,5-dimethyl-3-(2-oxo-2-phenyl-ethyl)-thiazole; LR-90, 4,402-chloro-phenyl-ureide; RAGE, receptor for AGE.
AGE formation inhibitors
Aminoguanidine: Aminoguanidine was demonstrated in vitro to prevent the formation of AGE-modified apolipoprotein A-1.71 Aminoguanidine treatment increased arterial elasticity and decreased vascular AGE accumulation as well as the severity of atherosclerotic plaques in streptozotocin-induced diabetic rats with diabetic nephropathy.72 Aminoguanidine was studied in humans and thus it holds a promise in the prevention of CVD in menopausal women. For instance, Aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II) was a randomized, double-blind, and placebo-controlled trial comparing 2 dose levels of aminoguanidine with placebo on the progression of nephropathy in 599 type 2 diabetic patients (72% male and 28% female) with renal disease from 84 centers in the United States and Canada. The primary end point was time to doubling of serum creatinine concentration. Secondary end points included the effect of aminoguanidine on time to all-cause mortality, such as cardiovascular morbidity and mortality, end-stage renal disease.
Cinnamic acid: In vitro results demonstrated that cinnamic acid significantly inhibited the formation of AGEs.73 Cinnamic acid also significantly decreased the protein carbonyl content and increased the level of protein thiol.73
Isoferulic acid: In vitro studies have been shown to markedly suppress the formation of fructosamine (an Amadori product clinically used as an indicator for short-term control of blood sugar in diabetic patients) as well as AGEs.74 The production of CML, an precursor of AGEs formation, was also inhibited by isoferulic acid both in fructose- and glucose-induced glycation.74 At a concentration of 5 mmol/L, it significantly inhibited the formation of CML by 47.0% in BSA/fructose and 21.9% in BSA/glucose system.74 Isoferulic acid might be a new promising antiglycation agent for the prevention of CVD in menopausal women.
Pyridoxamine: It is a broad inhibitor of advanced glycation. In diabetic rats, pyridoxamine inhibited the progression of retinopathy, attenuated the accumulation of AGEs in aortic collagen,75 and decreased hyperlipidemia.76 The safety and tolerability of pyridoxamine in patients with overt nephropathy and type 1/type 2 diabetes have been reported; pyridoxamine showed a beneficial effect on the progression of renal disease.77 The potential concern with this agent is its relationship with the vitamin B family, which has demonstrated some serious adverse drug events such as stroke and myocardial infarction in diabetic patients with kidney disease.78
Inhibitor of AGE cross-link
Alagebrium (formerly known as ALT-711): It was the first drug candidate to be clinically tested for the purpose of breaking the cross-links caused by AGEs. In rats, alagebrium was found to reverse erectile dysfunction and preserve vasodilatation.79 This action was associated with a restoration in penile neuronal nitric oxide synthase content and a reduction in serum and penile tissue AGE levels.79 Alagebrium has been studied in humans.80,81 A clinical trial (42% males and 58% females, mean ages in the 60s) has shown that patients who received alagebrium experienced statistically significant reduction in arterial pulse pressure and an increase in large artery compliance compared to those who received placebo.80 Moreover, alagebrium improved endothelial dysfunction in male patients with isolated systolic hypertension as well.81
Chelators
Chelators are compounds that remove excess metal ions such as iron and copper. Examples of chelators are EDTA, triethylenetetramine, and penicillamine. Several recent studies demonstrated therapeutic benefits of chelators for diabetic cardiovascular and renal disease in humans.82,83 Thus, chelation therapy deserves serious consideration as a clinical tool for prevention and treatment of CVD in menopausal women.
Conclusion
Menopausal women have increased risk for CVD. AGEs and their receptors contribute to an abnormal atherogenic milieu. In this review, we provided data that place the AGE–RAGE system in the center of biochemical and molecular stresses that characterize CVD in postmenopausal women. Data emerging from human studies suggest that levels of RAGE ligands and/or sRAGE in the circulation correlate with CVD risk factors. Interestingly, polymorphisms in the gene encoding RAGE may hold promise for the identification of postmenopausal women who are vulnerable to CVD. The intricate interaction between steroid hormones and the AGE–RAGE system could further explain the increased incidence of cardiovascular events in menopausal women. Up to date, data on the effect of HT on the AGE–RAGE axis are limited and equivocal. Further studies are needed to understand the clinical significance of menopausal HT in RAGE and sRAGE regulation. Finally, therapeutic antagonism of RAGE-dependent signaling provides a new target for the prevention of the deleterious consequences of oxidative stress and inflammation, particularly associated with the increased risk of CVD in the menopausal years.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: American Society for Reproductive Medicine, Ferring Pharmaceuticals, and University of Vermont Internal Grant to ZM.
References
- 1. Roger VL, Go AS, Lloyd-Jones DM, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125 (22):e2–e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piperi C, Adamopoulos C, Dalagiorgou G, Diamanti-Kandarakis E, Papavassiliou AG. Crosstalk between advanced glycation and endoplasmic reticulum stress: emerging therapeutic targeting for metabolic diseases. J Clin Endocrinol Metab. 2012;97 (7):2231–2242. [DOI] [PubMed] [Google Scholar]
- 3. Falcone C, Bozzini S, Guasti L, et al. Soluble RAGE plasma levels in patients with coronary artery disease and peripheral artery disease. Scientific World Journal. 2013;2013:584–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiang KH, Huang PH, Huang SS, Wu TC, Chen JW, Lin SJ. Plasma levels of soluble receptor for advanced glycation end products are associated with endothelial function and predict cardiovascular events in nondiabetic patients. Coron Artery Dis. 2009;20 (4):267–273. [DOI] [PubMed] [Google Scholar]
- 5. Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev. 2013;19(1):49–63. [DOI] [PubMed] [Google Scholar]
- 6. Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med(Berl). 2009;87 (3):235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uribarri J, Woodruff S, Goodman S, et al. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110(6):911–16.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uribarri J, Cai W, Peppa M, et al. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62 (4):427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldberg T, Cai W, Peppa M, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104 (8):1287–1291. [DOI] [PubMed] [Google Scholar]
- 10. Vlassara H, Cai W, Crandall J, et al. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci USA. 2002;99 (24):15596–15601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vlassara H, Cai W, Goodman S, et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: role of the antiinflammatory AGE receptor-1. J Clin Endocrinol Metab. 2009;94 (11):4483–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt AM, Vianna M, Gerlach M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267 (21):14987-14997. [PubMed] [Google Scholar]
- 13. Sugaya K, Fukagawa T, Matsumoto K, et al. Three genes in the human MHC class III region near the junction with the class II gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics. 1994;23 (2):408-419. [DOI] [PubMed] [Google Scholar]
- 14. Schmidt AM, Hori O, Chen JX, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96 (3):1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107 (9):1058–10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramasamy R, Yan SF, Schmidt AM. Advanced glycation endproducts: from precursors to RAGE: round and round we go. Amino Acids. 2012;42 (4):1151–11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuzan A, Chwiłkowska A, Kobielarz M, Pezowicz C, Gamian A. Glycation of extracellular matrix proteins and its role in atherosclerosis [in Polish]. Postepy Hig Med Dosw (Online). 2012;66:804–809. [DOI] [PubMed] [Google Scholar]
- 18. Ramasamy R, Schmidt AM. Receptor for advanced glycation end products (RAGE) and implications for the pathophysiology of heart failure. Curr Heart Fail Rep. 2012;9 (2):107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt AM, Hasu M, Popov D, et al. Receptor for advanced glycation end products (AGEs) has a central role in vessel wall interactions and gene activation in response to circulating AGE proteins. Proc Natl Acad Sci USA. 1994;91 (19):8807–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidt AM, Stern DM. Receptor for age (RAGE) is a gene within the major histocompatibility class III region: implications for host response mechanisms in homeostasis and chronic disease. Front Biosci. 2001;6:D1151–D1160. [DOI] [PubMed] [Google Scholar]
- 21. Mahajan N, Malik N, Bahl A, Dhawan V. Receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in non-diabetic subjects with pre-mature coronary artery disease. Atherosclerosis. 2009;207 (2):597–602. [DOI] [PubMed] [Google Scholar]
- 22. Fujisawa K, Katakami N, Kaneto H, et al. Circulating soluble RAGE as a predictive biomarker of cardiovascular event risk in patients with type 2 diabetes. Atherosclerosis. 2013;227 (2):425–428. [DOI] [PubMed] [Google Scholar]
- 23. Selvin E, Halushka MK, Rawlings AM, et al. sRAGE and risk of diabetes, cardiovascular disease, and death. Diabetes. 2013;62 (6):2116–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Falcone C, Emanuele E, D’Angelo A, et al. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler Thromb Vasc Biol. 2005;25 (5):1032–1037. [DOI] [PubMed] [Google Scholar]
- 25. Falcone C, Buzzi MP, Bozzini S, et al. ; TALENT Investigators. Relationship between sRAGE and eotaxin-3 with CRP in hypertensive patients at high cardiovascular risk. J Nephrol. 2013;26 (1):144–151. [DOI] [PubMed] [Google Scholar]
- 26. Geroldi D, Falcone C, Emanuele E, et al. Decreased plasma levels of soluble receptor for advanced glycation end-products in patients with essential hypertension. J Hypertension. 2005;23 (9):1725–1729. [DOI] [PubMed] [Google Scholar]
- 27. Frommhold D, Kamphues A, Hepper I, et al. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010;116 (5):841–849. [DOI] [PubMed] [Google Scholar]
- 28. Ha CH, Kim S, Chung J, et al. Inhibitory effect of soluble RAGE in disturbed flow-induced atherogenesis. Int J Mol Med. 2013;32 (2):373–380. [DOI] [PubMed] [Google Scholar]
- 29. Choi KM, Han KA, Ahn HJ, et al. Effects of exercise on sRAGE levels and cardiometabolic risk factors in patients with type 2 diabetes: a randomized controlled trial. J Clin Endocrinol Metab. 2012;97 (10):3751–3758. [DOI] [PubMed] [Google Scholar]
- 30. Semba RD, Ferrucci L, Sun K, et al. Advanced glycation end products and their circulating receptors predict cardiovascular disease mortality in older community-dwelling women. Aging Clin Exp Res. 2009;2 (2):182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pulkkinen A, Viitanen L, Kareinen A, Lehto S, Laakso M. Gly82Ser polymorphism of the receptor of advanced glycation end product gene is not associated with coronary heart disease in Finnish nondiabetic subjects or in patients with type 2 diabetes. Diabetes Care. 2000;23 (6):864. [DOI] [PubMed] [Google Scholar]
- 32. Hudson BI, Stickland MH, Grant PJ. Identification of polymorphisms in the receptor for advanced glycation end products (RAGE) gene: prevalence in type 2 diabetes and ethnic groups. Diabetes. 1998;47 (7):1155–1157. [DOI] [PubMed] [Google Scholar]
- 33. Hofmann MA, Yang Q, Harja E, et al. The RAGE Gly82Ser polymorphism is not associated with cardiovascular disease in the Framingham offspring study. Atherosclerosis. 2005;182 (2):301–305. [DOI] [PubMed] [Google Scholar]
- 34. Bansal S, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Association of RAGE gene polymorphism with circulating AGEs level and paraoxonase activity in relation to macro-vascular complications in Indian type 2 diabetes mellitus(T2DM) patients. Gene. 2013;526 (2):325–330. [DOI] [PubMed] [Google Scholar]
- 35. Koyama H, Shoji T, Yokoyama H, et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25 (12):2587–2593. [DOI] [PubMed] [Google Scholar]
- 36. Sebeková K, Krivošíková Z, Gajdoš M. Total plasma N∊-(carboxymethyl)lysine and sRAGE levels are inversely associated with a number of metabolic syndrome risk factors in non-diabetic young-to-middle-aged medication-free subjects. Clin Chem Lab Med. 2014;52 (1):139–149. [DOI] [PubMed] [Google Scholar]
- 37. Norata GD, Garlaschelli K, Grigore L, et al. Circulating soluble receptor for advanced glycation end products is inversely associated with body mass index and waist/hip ratio in the general population. Nutr Metab Cardiovasc Dis. 2009;19 (2):129–134. [DOI] [PubMed] [Google Scholar]
- 38. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119 (5 suppl 1):S10–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindheim SR, Presser SC, Ditkoff EC, Vijod MA, Stanczyk FZ, Lobo RA. A possible bimodal effect of estrogen on insulin sensitivity in postmenopausal women and the attenuating effect of added progestin. Fertil Steril. 1993;60 (4):664–667. [DOI] [PubMed] [Google Scholar]
- 41. Lindheim SR, Buchanan TA, Duffy DM, et al. Comparison of estimates of insulin sensitivity in pre- and postmenopausal women using the insulin tolerance test and the frequently sampled intravenous glucose tolerance test. J Soc Gynecol Investig. 1994;1 (2):150–154. [DOI] [PubMed] [Google Scholar]
- 42. Unoki H, Yamagishi S. Advanced glycation end products and insulin resistance. Curr Pharm Des. 2008;14 (10):987–989. [DOI] [PubMed] [Google Scholar]
- 43. Sullivan CM, Futers TS, Barrett JH, Hudson BI, Freeman MS, Grant PJ. RAGE polymorphisms and the heritability of insulin resistance: the Leeds family study. Diab Vasc Dis Res. 2005;2 (1):42–44. [DOI] [PubMed] [Google Scholar]
- 44. Tan KC, Shiu SW, Wong Y, Tam X. Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes Metab Res Rev. 2011;27 (5):488–492. [DOI] [PubMed] [Google Scholar]
- 45. Miele C, Riboulet A, Maitan MA, et al. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J Biol Chem. 2003;278 (48):47376–47387. [DOI] [PubMed] [Google Scholar]
- 46. Jia X, Wu L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol Cell Biochem. 2007;306 (1-2):133–139. [DOI] [PubMed] [Google Scholar]
- 47. Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA. 2012;109 (39):15888–15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uchida Y, Ohba K, Yoshioka T, Irie K, Muraki T, Maru Y. Cellular carbonyl stress enhances the expression of plasminogen activator inhibitor-1 in rat white adipocytes via reactive oxygen species-dependent pathway. J Biol Chem. 2004;279 (6):4075–4083. [DOI] [PubMed] [Google Scholar]
- 49. Fan X, Subramaniam R, Weiss MF, Monnier VM. Methylglyoxal-bovine serum albumin stimulates tumor necrosis factor alpha secretion in RAW 264.7 cells through activation of mitogen-activating protein kinase, nuclear factor kappaB and intracellular reactive oxygen species formation. Arch Biochem Biophys. 2003;409 (2):274–286. [DOI] [PubMed] [Google Scholar]
- 50. Matthews KA, Crawford SL, Chae CU, et al. Are changes in cardiovascular disease risk factors in midlife women due to chronological aging or to the menopausal transition? J Am Coll Cardiol. 2009;54 (25):2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Berg G, Mesch V, Boero L, et al. Lipid and lipoprotein profile in menopausal transition. Effects of hormones, age and fat distribution. Horm Metab Res. 2004;36 (4):215–220. [DOI] [PubMed] [Google Scholar]
- 52. Derby CA, Crawford SL, Pasternak RC, Sowers M, Sternfeld B, Matthews KA. Lipid changes during the menopause transition in relation to age and weight: the Study of Women's Health Across the Nation. Am J Epidemiol. 2009;169 (11):1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matthews KA, Meilahn E, Kuller LH, Kelsey SF, Caggiula AW, Wing RR. Menopause and risk factors for coronary heart disease. N Engl J Med. 1989;321 (10):641–646. [DOI] [PubMed] [Google Scholar]
- 54. Fan AZ, Dwyer JH. Sex differences in the relation of HDL cholesterol to progression of carotid intima–media thickness: the Los Angeles Atherosclerosis Study. Atherosclerosis. 2007;195 (1):e191–e196. [DOI] [PubMed] [Google Scholar]
- 55. Woodard GA, Brooks MM, Barinas-Mitchell E, Mackey RH, Matthews KA, Sutton-Tyrrell K. Lipids, menopause, and early atherosclerosis in Study of Women's Health Across the Nation Heart women. Menopause. 2011;18 (4):376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lopes-Virella MF, Hunt KJ, Baker NL, et al. Levels of oxidized LDL and advanced glycation end products-modified LDL in circulating immune complexes are strongly associated with increased levels of carotid intima–media thickness and its progression in type 1 diabetes. Diabetes. 2011;60 (2):582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hunt KJ, Baker N, Cleary P, et al. ; DCCT/EDIC Research Group. Oxidized LDL and AGE-LDL in circulating immune complexes strongly predict progression of carotid artery IMT in type 1 diabetes. Atherosclerosis. 2013;231(2):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Turk Z, Cavlović-Naglić M, Turk N. Relationship of methylglyoxal-adduct biogenesis to LDL and triglyceride levels in diabetics. Life Sci. 2011;89 (13-14):485–490. [DOI] [PubMed] [Google Scholar]
- 59. Jinnouchi Y, Yamagishi S, Takeuchi M, et al. Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin Exp Med. 2006;6 (4):191–193. [DOI] [PubMed] [Google Scholar]
- 60. Quade-Lyssy P, Kanarek AM, Baiersdörfer M, Postina R, Kojro E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J Lipid Res. 2013;54 (11):3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu L, Peng WH, Wang W, Wang LJ, Chen QJ, Shen WF. Effects of atorvastatin on progression of diabetic nephropathy and local RAGE and soluble RAGE expressions in rats. J Zhejiang Univ Sci B. 2011;12 (8):652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Santilli F, Bucciarelli L, Noto D, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43 (9):1255–1262. [DOI] [PubMed] [Google Scholar]
- 63. Tam HL, Shiu SW, Wong Y, Chow WS, Betteridge DJ, Tan KC. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis. 2010;209 (1):173–177. [DOI] [PubMed] [Google Scholar]
- 64. Colhoun HM, Betteridge DJ, Durrington P, et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: an analysis from the CARDS trial. Diabetes. 2011;60 (9):2379–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mukhopadhyay S, Mukherjee TK. Bridging advanced glycation end product, receptor for advanced glycation end product and nitric oxide with hormonal replacement/estrogen therapy in healthy versus diabetic postmenopausal women: a perspective. Biochim Biophys Acta 2005;1745 (2):145–155. [DOI] [PubMed] [Google Scholar]
- 66. Merhi Z. Advanced glycation end-products: pathway of potentially significant pathophysiological and therapeutic relevance for metabolic syndrome in menopausal women. J Clin Endocrinol Metab. 2014;99 (4):1146–1148. [DOI] [PubMed] [Google Scholar]
- 67. Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycosylation end products is induced by the glycation product themselves and TNF-alpha through nuclear factor kappa B and by 17-beta-estradiol through Sp1 in human vascular endothelial cells. J Biol Chem. 2000;275 (33):25781–25790. [DOI] [PubMed] [Google Scholar]
- 68. Mukherjee TK, Nathan L, Dinh H, Reddy ST, Chaudhuri G. 17-epiestriol, an estrogen metabolite, is more potent than estradiol in inhibiting vascular cell adhesion molecule 1 (VCAM1) mRNA expression. J Biol Chem. 2003;278 (14):11746–11752. [DOI] [PubMed] [Google Scholar]
- 69. Pullerits R, d'Elia HF, Tarkowski A, Carlsten H. The decrease of soluble RAGE levels in rheumatoid arthritis patients following hormone replacement therapy is associated with increased bone mineral density and diminished bone/cartilage turnover: a randomized controlled trial. Rheumatology (Oxford). 2009;48 (7):785–790. [DOI] [PubMed] [Google Scholar]
- 70. Merhi Z. Advanced glycation end products and their relevance in female reproduction. Hum Reprod. 2014;29 (1):135–145. [DOI] [PubMed] [Google Scholar]
- 71. Bucala R, Makita Z, Vega G, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. 1994;91 (20):9441–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hu C, Cong XD, Dai DZ, Zhang Y, Zhang GL, Dai Y. Argirein alleviates diabetic nephropathy through attenuating NADPH oxidase, Cx43, and PERK in renal tissue. Naunyn Schmiedebergs Arch Pharmacol. 2011;383 (3):309–319. [DOI] [PubMed] [Google Scholar]
- 73. Adisakwattana S, Sompong W, Meeprom A, Ngamukote S, Yibchok-Anun S. Cinnamic acid and its derivatives inhibit fructose-mediated protein glycation. Int J Mol Sci. 2012;13 (2):1778–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Meeprom A, Sompong W, Chan CB, Adisakwattana S. Isoferulic acid, a new anti-glycation agent, inhibits fructose- and glucose-mediated protein glycation in vitro. Molecules. 2013;18 (6):6439–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chang KC, Liang JT, Tsai PS, Wu MS, Hsu KL. Prevention of arterial stiffening by pyridoxamine in diabetes is associated with inhibition of the pathogenic glycation on aortic collagen. Br J Pharmacol. 2009;157 (8):1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Degenhardt TP, Alderson NL, Arrington DD, et al. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2000;61 (3):939–950. [DOI] [PubMed] [Google Scholar]
- 77. Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27 (6):605–614. [DOI] [PubMed] [Google Scholar]
- 78. Shepler B, Nash C, Smith C, Dimarco A, Petty J, Szewciw S. Update on potential drugs for the treatment of diabetic kidney disease. Clin Ther. 2012;34 (6):1237–1246. [DOI] [PubMed] [Google Scholar]
- 79. Usta MF, Kendirci M, Gur S, et al. The breakdown of preformed advanced glycation end products reverses erectile dysfunction in streptozotocin-induced diabetic rats: preventive versus curative treatment. J Sex Med. 2006;3 (2):242–250. [DOI] [PubMed] [Google Scholar]
- 80. Kass DA, Shapiro EP, Kawaguchi M, et al. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104 (13):1464–1470. [DOI] [PubMed] [Google Scholar]
- 81. Zieman SJ, Melenovsky V, Clattenburg L, et al. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens. 2007;25 (3):577–583. [DOI] [PubMed] [Google Scholar]
- 82. Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes. 2012;61 (3):549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cooper GJ. Selective divalent copper chelation for the treatment of diabetes mellitus. Curr Med Chem. 2012;19 (17):2828–2860. [DOI] [PubMed] [Google Scholar]
- 84. Frizzell N, Baynes JW. Chelation therapy for the management of diabetic complications: a hypothesis and a proposal for clinical laboratory assessment of metal ion homeostasis in plasma. Clin Chem Lab Med. 2014;52:69–75. [DOI] [PubMed] [Google Scholar]
- 85. Ahmad S, Shahab U, Baig MH, et al. Inhibitory effect of metformin and pyridoxamine in the formation of early, intermediate and advanced glycation end-products. PLoS One. 2013;8 (9):e72128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yamagishi S, Amano S, Inagaki Y, Okamoto T, Takeuchi M, Makita Z. Beraprost sodium, a prostaglandin I2 analogue, protects against advanced glycation end products-induced injury in cultured retinal pericytes. Mol Med. 2002;8 (9):546–550. [PMC free article] [PubMed] [Google Scholar]
- 87. Meerwaldt R, van der Vaart MG, van Dam GM, et al. Clinical relevance of advanced glycation endproducts for vascular surgery. Eur J Vasc Endovasc Surg. 2008;36 (2):125–131. [DOI] [PubMed] [Google Scholar]
- 88. Jariyapamornkoon N, Yibchok-anun S, Adisakwattana S. Inhibition of advanced glycation end products by red grape skin extract and its antioxidant activity. BMC Complement Altern Med. 2013;13:171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]