

Abstract
Background/Aim
The Akt signaling pathway mediates a potent anti-apoptotic signal in pancreatic cancer and inhibition of this pathway has become an attractive mechanism to increase the efficacy of traditional chemotherapies. Autophagy is a lysosomal catabolic pathway by which eukaryotic cells recycle macromolecules and organelles. Although autophagy may function as a survival mechanism under metabolic stress conditions, it also serves as an alternate route to programmed cell death distinct from apoptosis. In the present study, we examined the role of autophagy in Akt-mediated regulation of cell death in pancreatic cancer.
Materials and Methods
Mia-PaCa-2 and PANC-1 human pancreatic cancer cell lines were used in our experiments. The small-molecule inhibitor A-443654 was used to inhibit Akt, and rapamycin was used to inhibit mTOR. Autophagy was inhibited with Chloroquine and 3-methyladenine. Autophagy was assessed by immunoblotting for Light Chain-3 (LC-3) processing as well as fluorescence microscopy for autophagosome formation following transfection with a LC-/GFP construct. Cell death was determined by fluorescence-activated cell sorting (FACS) with quantitation of the sub-G0 content.
Results
Inhibition of either Akt or mTOR induced autophagy; inhibition of Akt but not of mTOR led to traditional caspase-mediated apoptosis. When autophagy was inhibited, cell death was abrogated following Akt, but not mTOR, inhibition.
Conclusion
The Akt signaling pathway regulates both autophagy and apoptosis through divergent pathways; mTOR mediates autophagy signaling but appears to be un-involved in cell death. Autophagy appears to play a role in the regulation of cell survival by Akt, but only when proximal signaling pathways not involving mTOR are simultaneously activated.
Keywords: Pancreatic cancer, AKT, mTOR, autophagy
Pancreatic cancer is the most lethal of all malignancies due both to the typically late presentation following the establishment of metastatic disease as well as the profound resistance to the cytotoxic effect of traditional chemotherapy. Apoptosis is considered the primary mechanism of chemotherapy-induced cell death. However, apoptosis is just one mechanism regulating the balance of cell survival and cell death following exposure to noxious extracellular stimuli. Autophagy is a broadly-conserved catabolic process in which bulk cytoplasmic proteins as well as organelles are degraded by the formation of autophagosomes with subsequent fusion to lysosomes for recycling of non-essential macromolecules to preserve cellular energy stores (1). Although originally described as a cell survival response induced by metabolic starvation, it has recently been recognized that the autophagy pathway can also mediate cell death (2). There is further emerging evidence to suggest a central role of autophagy in cancer, both as a survival mechanism triggered by hypoxia but also potentially as a mediator of stress-induced cell death (3, 4). The specific mechanisms that regulate this dichotomous effect of autophagy are unclear, though length and magnitude of cell stress have been postulated (5, 6). While the signaling pathways for autophagosome assembly are well-established, the pathways that sense and initiate autophagy are unclear.
The investigation of the biochemical mechanisms responsible for the inherent apoptotic resistance of pancreatic cancer has identified Akt, a serine-threonine kinase, as a potential cause. Our group as well as others have shown that Akt is constitutively activated in human pancreatic cancer cell lines as well as human tumor specimens (7-9). This activation is associated with resistance to chemotherapy-induced apoptosis; inhibiting Akt has been shown to increase the apoptotic effect of chemotherapy in pancreatic cancer (10-12). mTOR, a downstream target of autophagy, has been shown to regulate autophagy, however it remains unclear whether the autophagy initiates or inhibits cell death (13, 14). Fujiwara et al. have suggested that induction of autophagy by mTOR inhibition in malignant glioma augments cell death event as the cytotoxic effect of radiation therapy was enhanced (15). However, Takeuchi et al. demonstrated that while directly inhibiting either Akt or mTOR was sufficient to induce autophagy, only Akt inhibition led to cell death in malignant glioma cells (16). Conversely, Aoki et al. inhibited the Akt/mTOR pathways in glioma cells and noted the induction of autophagy but without any concomitant cell death (17). These studies suggest the involvement of divergent pathways that independently regulate autophagy and apoptosis may be both cell-specific as well as dependent on additional inducers of apoptosis (such as chemotherapy or radiation therapy) or simultaneous activation of other signaling pathways, such as ERK.
We, therefore, tested the hypothesis that inhibition of both Akt and mTOR would initiate autophagy in pancreatic cancer, and that furthermore, these events would subsequently lead to the induction of cell death independently of apoptosis.
Materials and Methods
Reagents and cell lines
All chemical reagents were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA) unless otherwise specified. The experimental small-molecule Akt inhibitor A-443654 was provided by Abbott Laboratories (Abbott Park, IL, USA) (18). The mTOR inhibitor rapamycin was purchased from Calbiochem (EMD Chemicals; Merck KGaA, Darmstadt, Germany). The pan-Caspase inhibitor ZVAD-fmk was purchased from MBL International (Woburn, MA, USA). Monoclonal antibodies used were: Akt, phospho-Akt, phospho-GSK, PARP, and LC3 (Cell Signaling; Beverly, MA, USA); caspase 3 (Biosource; Camarillo, CA, USA), and a polyclonal antibody to actin (Santa Cruz Biotechnology; Santa Cruz, CA, USA).The MiaPaca-2 and PANC-1 cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, sodium pyruvate, nonessential amino acids, L-glutamine, vitamins, penicillin, and streptomycin. Cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Western blotting
Following treatments, cells were harvested by trypsination, washed with PBS and re-suspended in lysis buffer. Lysates were resolved by SDS-PAGE on either 10% or 15% acrylamide gels. Separated proteins were then electrophoretically-transferred to 0.2-mm nitrocellulose membranes (Schleicher & Schuell; Keene, NH, USA), probed overnight with primary antibodies, and developed using species-specific secondary antisera. Immunoreactivity was detected by the enhanced chemiluminescence technique (Amersham; Piscataway, NJ). For LC-3 immunoblots, relative band intensity was determined using Quantity One software (BioRad Labs, Hercules, CA).
Cell-cycle analysis with flow Cytometry
1 × 106 cells per treatment were plated in 100-mm2 plates and allowed to recover overnight. Following treatments, cells were trypsinized, washed in PBS, and re-suspended in 150 μL of FACSMAX solution (Genlantis, San Diego, CA, USA) and incubated for 10 min on ice to maximally-disperse the cells. They were then fixed by adding ice-cold 70% ethanol dropwise and kept at 4° C. Prior to analysis, cells were washed with PBS, then incubated with RNAse A (200 μg/ml) for 30 min at 37° C and then stained with propridium iodide (10 μg/ml). The cells were then analyzed using either a Beckman Coulter Counter Epics XL flow cytometer (Beckman Coulter, Miami, FL) or a Becton Dickenson FACscan (BD Biosciences, San Jose, CA, USA) to determine the DNA fractions and the Sub-G0 cell population was quantified using Flowjo software (Treestar, inc., Ashland, OR, USA).
Live cell fluorescence microscopy for LC-3
MiaPaca-2 cells and Panc-1 cells were transfected to overexpress eGFP-LC-3 using Lipofectin Reagent (Gibco BRL/Invitrogen; Carlsbad, CA, USA) and stable clones were isolated as we have previously described (19). These cells were then seeded in 35-mm2 glass bottom dishes (WillCo Wells, BV; Amsterdam, the Netherlands), or in Ibidi 8-chamber ultra-thin culture slides (Integrated BioDiagnostics; Munich, Germany) and allowed to recover for 48 h under standard culture conditions. Culture medium was then changed to include the treatment drugs and cells were imaged using a IX-71 Delta Vision (Applied Precision; Issaquah, WA, USA) inverted fluorescent microscope with a ×60 1.40 NA oil objective and FITC filter (excitation, 480 nm; emission, 535 nm). During imaging the cells were kept at 37° C with an ASI 400 air stream incubator (Nevtek; Williamsville, VA, USA).
Results
Cellular effects of Akt and mTOR inhibition
MiaPaca-2 and PANC-1 cells were treated with increasing concentrations of the small-molecule Akt inhibitor (A-443654) and analysis of cell lysates by western blotting showed a dose-dependent decrease in phosphorylated GSK-3, a downstream target of activated Akt (Figure 1A). Given the significant inhibition of Akt at 50 nM of A-443654, this dose was used for all future studies in both MiaPaCa-2 and PANC-1 cells unless otherwise specified. To evaluate for the induction of autophagy, we used two established methods, either immunoblotting for the processing of LC3-I to LC3-II or live cell fluorescent microscopy in the eGFP-LC-3 stably transfected cells. The former biochemical assay evaluates the early phases of protein assembly into the autophagosome, while the latter assay provides cellular confirmation through the identification of the “punctae” of intense LC-3 fluorescence as a result of autophagosome maturation. MiaPaCa-2 and PANC-1 cells were treated with either the Akt inhibitor A-443654 (50 nM) or Rapamycin (2 μM) over a short time course and cell lysates analyzed by immuno-blotting for LC3 processing. It was noted that these cells demonstrated a significant basal level of autophagy, manifested by the presence of the processed LC3-II form. However, following treatment, a clear shift was seen from the LC3-I (un-lipidated) to the LC3-II (lipidated) fractions over both cell lines. This was quantified by measuring the individual band densities and calculating the ratio of LC3-II to LC3-I for each time-point and plotting the percent change from baseline over time (Figure 1B and C). Interestingly, PANC-1 was more resistant to the induction of LC3 processing by both agents, perhaps related to the higher constitutive activation of Akt (20).
Figure 1.

(A) Effect of small-molecule Akt inhibitor, A-443654, across a modest dose range (10–50 nM) on phosphorylated GSK (p-GSK) in MiaPaca-2 or PANC-1 cells, effect of A-443654 on processing of LC3-I to LC3-II determined by immunoblot and then quantified over 60 min of treatment in MIA-PaCa-2 cells (B) or PANC-1 cells (C).
Live cell fluorescence microscopy was used to evaluate the cellular response of the cells stably-expressing eGFP-LC3 following treatment with A-443654 or rapamycin. At baseline, GFP-LC3 was distributed throughout the cytoplasm as well as associated with a small number of punctae, which have been shown to correspond to autophagosomes (Figure 2A). Following treatment with either A-443654 or rapamycin, the diffuse cytoplasmic staining was lost and the number of punctae dramatically increased (Figure 2A). To clearly demonstrate the induction of autophagy in these cells, we co-treated cells with 3-methyladenine (3-MA; 10 mM), an inhibitor of the early stages of autophagy initiation. In both cell lines, the changes in LC3 distribution following Akt or mTOR inhibition were blunted, with the maintenance of the diffuse cytoplasmic distribution and abrogation of autophagosome formation (Figure 2B). Furthermore, the induction of autophagy was similar between Akt inhibition and mTOR inhibition, as was the ability of 3-MA to inhibit the effect of both agents.
Figure 2.

(A) Single-cell immunofluorescence of GFP-LC3 in MIA-PaCa-2 and PANC-1 cells after inhibition of Akt (A-443654) or mTOR (rapamycin), and (B) effect of inhibition of autophagy by co-treatment with 3-MA after cells after inhibition of Akt (A-443654) or mTOR (rapamycin) with blunting of the autophagic punctae development.
Akt inhibition but not mTOR inhibition induces caspase activation and cell death
To characterize the cellular effect of Akt and mTOR inhibition, Mia-PaCa-2 cells were treated with A-443654 or rapamycin for a range of time-points and analyzed for the induction of cell death. AKT inhibition, but for not mTOR inhibition, induced apoptosis following 24 h of treatment (Figure 3A). Experiments carried out at 72 h failed to demonstrate any effect on apoptosis following mTOR inhibition (data not shown). A-443654 effectively induced cleavage of caspase-3, though no effect was observed following treatment with rapamycin (Figure 3B). The pan-caspase inhibitor ZVad-fmk specifically blocked cleavage of caspase-3 induced by both A-443654 and paclitaxel, which served as a positive control (Figure 3B). Similarly, only A-443654-but not rapamycin-treatment initiated PARP cleavage (data not shown).
Figure 3.
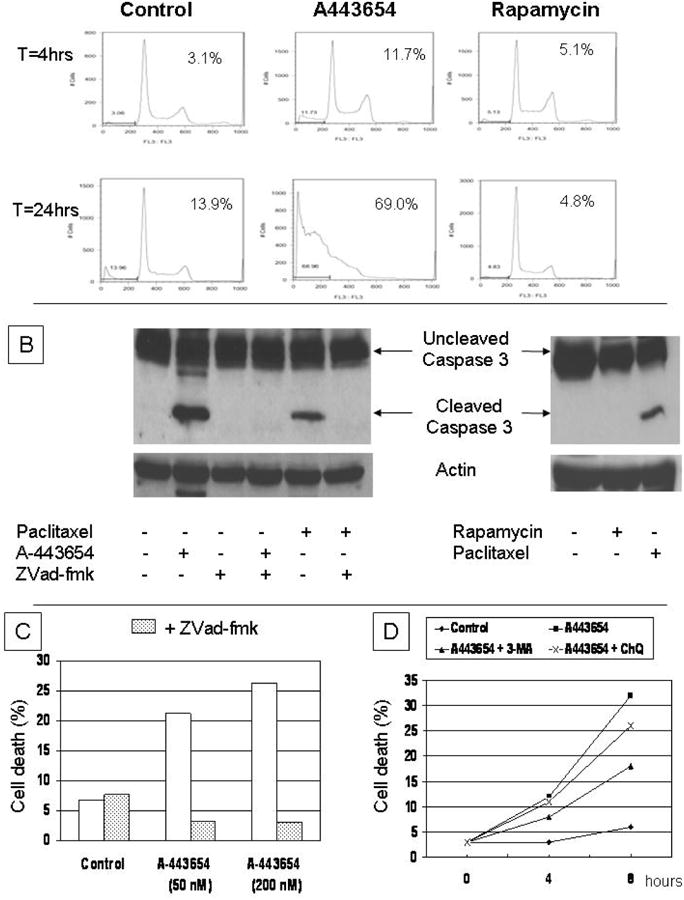
(A) Flow cytometry of MIA-PaCa-2 cells at 4 and 24 h after treatment with either A-443654 or rapamycin with increase in sub-G0 fraction following only A-443654 treatment with notation of amount of sub-G0 cells. (B) Immunoblot of caspase-3 in MIA-PaCa-2 cells after the indicated treatment with A-443654, ZVad-fmak, rapamycin or paclitaxel. (C) Quantification of cell death by flow cytometry in MIA-PaCa-2 cells following treatment with A-443654 (50 nM or 200 nM) in the absence or presence of ZVad-fmk, and (D) quantitation of cell death in MIA-PaCa-2 cells following treatment with A-443654 (50 nM) in the absence or presence of either 3-MA or chloroquine (ChQ).
We then evaluated whether inhibition of caspase activation (using ZVad-fmk) or autophagy (using 3-MA or chloroquine) altered the induction of cell death following Akt inhibition. MiaPaCa-2 cells were treated with A-443654 (8 h, 50 or 200 nM) in the absence or presence of ZVad-fmk. Inhibition of caspase activation by ZVad-fmk completely abrogated the induction of cell death mediated by A-443654 treatment (Figure 3C), but had no effect on the induction of autophagy (data not shown). To explore the role of autophagy in the cellular response to Akt inhibition, we co-treated cells with A-443654 and either chloroquine or 3-MA to inhibit autophagy. At 8 hours treatment, there was a modest but incomplete inhibition of the A-443654-induced cell death achieved by either 3-MA or chloroquine (Figure 3D). Therefore, we propose an AKT/mTOR signaling pathway that regulates autophagy, but an mTOR-independent pathway for which inhibition mediates caspase-dependent apoptosis. Furthermore, the autophagy induced by the Akt/mTOR pathway plays an early protective role in cell survival following Akt inhibition but is unable to suppress a long-term apoptotic stimulus (Figure 4).
Figure 4.

Representation of proposed signaling pathways of Akt and mTOR in regulation of autophagy and apoptotic cell death in pancreatic cancer.
Discussion
We showed that Akt inhibition induces both autophagy and then subsequent apoptotic cell death in pancreatic cancer cells, while mTOR inhibition induces autophagy but without a cytotoxic effect. Furthermore, the autophagy induced following Akt inhibition is required for the full cell death response as inhibition of autophagy blunted the apoptotic response. While Akt has been investigated as a target in cancer therapy, inhibition does not consistently induce cell death though autophagy is consistently observed (21). Although mTOR is thought to be the downstream effector of autophagy signaling mediated by Akt, it was interesting to note that inhibition of those two points in the same pathway caused such divergent effects on apoptosis. While Akt inhibition rapidly and universally progressed to apoptosis, inhibition of mTOR was followed-out to 3 days with no effect on the cell viability noted. This is in contrast to a study by Dai et al. in which mTOR inhibition with rapamycin induced apoptosis and inhibited cell growth (22). Using the mTOR inhibitor CCI-779, Asano et al. may have identified the disparities in results targeting mTOR in pancreatic cancer; wild type p53 and c-Jun responsiveness may be required for cytotoxicity mediated by mTOR inhibition (23). Due to the frequent mutation of p53, these data may explain why two clinical studies of mTOR inhibition have failed to demonstrate any efficacy of this therapy (24).
When autophagy was simultaneously inhibited in the setting of Akt inhibition, we observed a decrease in apoptosis. This implies that in the setting of Akt inhibition, autophagy augments cell death. The role of autophagy in treatment-induced cell death in cancer remains controversial. Jeong et al. performed similar studies examining the role of Akt inhibition to evaluate the role of autophagy in non-small lung cancer (25). Using GSK690693 to inhibit Akt and CCI-779 to inhibit mTOR, autophagy inhibition using 3-MA significantly increased the cytotoxicity of combined therapy suggesting a cytoprotective role for the autophagy that occurs following either Akt or mTOR inhibition. Nearly identical results were observed by Lamoureux and Zoubeidi evaluating the Akt inhibitor AZD5363 in prostate cancer in which autophagy inhibition enhanced cell death (26). Using the PKC-d inhibitor rottlerin to inhibit the PI3K/Akt/mTOR pathway in pancreatic cancer stem cells, Singh et al observed increased apoptosis when genetic silencing of key mediators blocked autophagy (27).
However, there are also data to support the active role of autophagy in the cell death mediated by Akt inhibition. Using triptolide to inhibit Akt in pancreatic cancer, Mujumdar et al. observed a caspase-dependent cell death as well as a caspase-independent, autophagic cell death (28). This is quite similar to our data, as autophagy inhibition with 3-MA did not completely block all of the cell death mediated by A-443654. However, simultaneous apoptosis may not be required for autophagic cell death associated with Akt inhibition. Ling et al. examined the biochemical and cellular events following treatment of non-small cell lung cancer cells with elisidepsin, a natural cyclic peptide of the kahalaldie family (29). Elisidepsin was a potent inhibitor of the Akt/mTOR signaling pathway but did not induce caspase activation, PARP cleavage or traditional apoptosis, however induction of autophagy and cell death was observed. Furthermore, inhibition autophagy with 3-MA partially inhibited the elisidepsin-mediated cell death, suggesting a pure autophagic mode of cell death associated with Akt inhibition.
These conflicting data regarding the role of autophagy in cell death mediated by Akt inhibition may be related to underlying genetic variables (such as p53 status), the length and severity of the induction of autophagy, or the underlying addiction of the cell to Akt-mediated survival signals. However, as newer agents become available to either activate or inhibit autophagy, these can then be used to augment the cytotoxic effect of various agents that induce apoptosis.
References
- 1.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2012;10(9):1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–83. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol. 2012;4(9):a008763. doi: 10.1101/cshperspect.a008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226(2):255–73. doi: 10.1002/path.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibson SB. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy. 2010;6(7):835–7. doi: 10.4161/auto.6.7.13335. [DOI] [PubMed] [Google Scholar]
- 7.Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89(11):2110–5. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23(53):8571–80. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 9.Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87(4):470–6. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 10.Fahy BN, Schlieman M, Virudachalam S, Bold RJ. AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003;89(2):391–7. doi: 10.1038/sj.bjc.6601037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22(21):3243–51. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 12.Ng SSW, Tsao MS, Chow S, Hedley DW. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res. 2000;60(19):5451–5. [PubMed] [Google Scholar]
- 13.Wang SY, Yu QJ, Zhang RD, Liu B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int J Biochem Cell Biol. 2011;43(9):1263–6. doi: 10.1016/j.biocel.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 14.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8(9):528–39. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 15.Fujiwara K, Iwado E, Mills GB, Sawaya R, Kondo S, Kondo Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int J Oncol. 2007;31(4):753–60. [PubMed] [Google Scholar]
- 16.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 17.Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72(1):29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 18.Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, de Jong R, Han EK, Li T, Stoll VS, Powlas JA, Oleksijew A, Mitten MJ, Shi Y, Guan R, McGonigal TP, Klinghofer V, Johnson EF, Leverson JD, Bouska JJ, Mamo M, Smith RA, Gramling-Evans EE, Zinker BA, Mika AK, Nguyen PT, Oltersdorf T, Rosenberg SH, Li Q, Giranda VL. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4(6):977–86. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 19.Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, Gandour-Edwards R, Chuang FY, Bold RJ, Kung HJ. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69(2):700–8. doi: 10.1158/0008-5472.CAN-08-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W, Ju JH, Lee KM, Nam K, Oh S, Shin I. Protein kinase B/Akt1 inhibits autophagy by down-regulating UVRAG expression. Exp Cell Res. 2013;319(3):122–33. doi: 10.1016/j.yexcr.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Degtyarev M, De Mazière A, Klumperman J, Lin K. Autophagy, an Achilles' heel AKTing against cancer? Autophagy. 2009;5(3):415–8. doi: 10.4161/auto.5.3.7827. [DOI] [PubMed] [Google Scholar]
- 22.Dai ZJ, Gao J, Ma XB, Kang HF, Wang BF, Lu WF, Lin S, Wang XJ, Wu WY. Antitumor effects of rapamycin in pancreatic cancer cells by inducing apoptosis and autophagy. Int J Mol Sci. 2012;14(1):273–85. doi: 10.3390/ijms14010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The rapamycin analog CCI-779 is a potent inhibitor of pancreatic cancer cell proliferation. Biochem Biophys Res Commun. 2005;331(1):295–302. doi: 10.1016/j.bbrc.2005.03.166. [DOI] [PubMed] [Google Scholar]
- 24.Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeong EH, Choi HS, Lee TG, Kim HR, Kim CH. Dual Inhibition of PI3K/Akt/mTOR Pathway and Role of Autophagy in Non-Small Cell Lung Cancer Cells. Tuberc Respir Dis (Seoul) 2012;72(4):343–51. doi: 10.4046/trd.2012.72.4.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013;9(7):1119–20. doi: 10.4161/auto.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh BN, Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharmacol. 2012;84(9):1154–63. doi: 10.1016/j.bcp.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Mujumdar N, Mackenzie TN, Dudeja V, Chugh R, Antonoff MB, Borja-Cacho D, Sangwan V, Dawra R, Vickers SM, Saluja AK. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology. 2010;139(2):598–608. doi: 10.1053/j.gastro.2010.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling YH, Aracil M, Zou Y, Yuan Z, Lu B, Jimeno J, Cuervo AM, Perez-Soler R. PM02734 (elisidepsin) induces caspase-independent cell death associated with features of autophagy, inhibition of the Akt/mTOR signaling pathway, and activation of death-associated protein kinase. Clin Cancer Res. 2011;17(16):5353–66. doi: 10.1158/1078-0432.CCR-10-1948. [DOI] [PubMed] [Google Scholar]