

Abstract
Purpose
Aurora kinases are key regulators of mitotic events. Dysfunction of these kinases can cause polyploidy and chromosomal instability, a contributor to tumorigenesis. MK-5108 is a potent inhibitor of Aurora A kinase that has shown preclinical potent activity in malignancies of breast, cervical, colon, ovarian, and pancreatic origin. We sought to assess the preclinical efficacy of MK-5108 in a panel of non-small-cell lung cancer cell lines as a single agent and in combination with cisplatin and docetaxel.
Methods
Eleven lung cancer cell lines were studied. Growth inhibition by MK-5108 was assessed with short- and long-term MTT assays. Cell cycling was measured by flow cytometry. Immunoblotting was used to determine targeted activity of MK-5108 on Aurora A and downstream effects (TACC3 and Plk1). Efficacy of combination studies performed with cisplatin and docetaxel was evaluated by median effect analysis.
Results
All cell lines demonstrated sustained growth inhibition following MK-5108 at varying nanomolar concentrations. MK-5108 induced G2/M accumulation, polyploidy, and apoptosis (increased sub-G1/PARP cleavage). Levels of Aurora A, TACC3, and Plk1 diminished. Concurrent treatment of MK-5108 with cisplatin or docetaxel synergistically inhibited cell growth with the docetaxel combination performing better. When administered sequentially, treatment with docetaxel first followed by MK-5108 exhibited greater growth inhibition than the inverse; yet concurrent treatment remained superior.
Conclusions
MK-5108 has potent anti-proliferative activity in lung cancer cell lines alone and in combination with chemotherapies. Determining how best to integrate Aurora inhibitors into current lung cancer treatment regimens would be beneficial.
Keywords: MK-5108, Aurora kinase, Lung cancer, Developmental therapeutics
Introduction
The human Aurora kinase family consists of three highly conserved serine/threonine protein kinases, designated A, B, and C (Lok et al. 2010). Aurora A and B are ubiquitously expressed and involved in cell proliferation in many cell types, while Aurora C is commonly found in testicular tissue and does not regulate mitosis in somatic or tumor cells (Marumoto et al. 2005; Lok et al. 2010; Shimomura et al. 2010). The biology of Aurora kinases is still in its infancy, particularly with regard to their mechanism of action, as the identification of their substrates and correlating function is ongoing (Sardon et al. 2010). Generally, Aurora kinases serve as key regulators of mitotic events such as centrosome maturation/separation, mitotic entry, microtubule spindle assembly, chromosome assembly/segregation, mitotic checkpoint activation, and cytokinesis (Lok et al. 2010; Carvajal et al. 2006; Marumoto et al. 2005). Dysfunction of Aurora kinases can cause polyploidy and chromosomal instability, a contributor to tumorigenesis. Inhibition or depletion of Aurora A has been shown to result in chromosome misalignment, and consequently, the accumulation of cells in G2/M phase and depleted levels of Aurora B leads to polyploidy (Shimomura et al. 2010; Lok et al. 2010). Such inhibitory effects on cell viability as well as evidence linking over-expression to malignancy have made both Aurora A and B attractive anticancer drug targets (Carvajal et al. 2006). Aurora A expression in tumors is often associated with poor histological differentiation and poor prognosis (Barr and Gergely 2007). Compared to Aurora B, Aurora A has been more consistently implicated in tumorigenesis as its over-expression is often linked to an amplification of chromosome 20q13.2 encoding the AURKA gene (Gautschi et al. 2008; Lok et al. 2010). The role of Aurora B in tumorigenesis is less clear, and while typically not amplified in tumors, studies now suggest the involvement of additional factors associated with Aurora B in the process of malignant transformation (Lok et al. 2010; Gautschi et al. 2008; Kanda et al. 2005). For instance, in one study, elevated Aurora B activity facilitated Rasmediated cell transformation by enhancing oncogenic signaling and promoting aneuploidy (Kanda et al. 2005).
Aurora A and B have been evaluated preclinically as therapeutic targets in various tumor types including bladder, breast, colon, liver, lung, ovary, and pancreas (Lok et al. 2010; Carvajal et al. 2006). The early clinical development of small molecular inhibitors of Aurora kinases initially targeted all three subtypes to varying degrees— hesperidin primarily inhibits Aurora B, ZM447439 targets both Aurora A and B, and VX680 (MK-0457) inhibits all three Aurora subtypes (Barr and Gergely 2007; Lok et al. 2010; Carvajal et al. 2006). These agents have been shown to cause phenotypes associated with Aurora B inhibition, such as diminished levels of phosphorylated histone H3 (p-HH3) on serine 10, inhibited cytokinesis, polyploidy, and apoptosis in cell-based assays (Carvajal et al. 2006). VX680 was the first compound to be clinically evaluated in patients with previously treated solid tumors and hematological malignancies. Although it reached phase II, studies were discontinued due to treatment-related cardiac toxicities (Carvajal et al. 2006; Lok et al. 2010; Gautschi et al. 2008; Bebbington et al. 2009). A new class of orally available and selective molecular inhibitors was led by the development of MLN8054 and subsequently alisertib (MLN8237), both selective Aurora A inhibitors that showed promising antitumor activity in vitro and in vivo (Gautschi et al. 2008; Gorgun et al. 2010). Recently, a phase III trial of MLN8237 was initiated in patients with relapsed or refractory peripheral T cell lymphoma (Pai and Albaugh 2012).
Another highly selective Aurora A kinase inhibitor with potent antitumor activity is MK-5108 (aka VX-689). MK-5108 can selectively inhibit Aurora A and the proliferation of cultured human tumor cell lines (breast, cervix, colon, ovary, pancreas) and xenograft tumors (Shimomura et al. 2010). When used in combination with docetaxel, MK-5108 enhanced its antitumor activity in vivo without increasing toxicity (Shimomura et al. 2010). MK-5108 was also found to enhance lymphoma cell death when used in combination with the histone deacetylase inhibitor vorinostat in vitro (Kretzner et al. 2011). A phase I dose-escalation and pharmacodynamic trial was completed in 2011 for MK-5108 as monotherapy and in combination with docetaxel in patients with advanced and/or refractory solid tumors (Minton et al. 2010). Stable disease was observed in seven patients on monotherapy and four on combination therapy, and partial response was noted in two patients receiving the combination (Minton et al. 2010).
Given Shimomura et al.’s in vitro findings of MK-5108’s potent activity as a single agent and synergistic interaction with docetaxel, we hypothesize that this agent will have a similar effect in lung cancer. The location of Aurora A’s chromosomal locus (20q13.2) is a region identified as one of the most frequent genetic aberrations in lung cancer, suggesting that it may be involved in lung cancer tumorigenesis (Gu et al. 2007). Previous studies report non-small-cell lung cancer (NSCLC) histological subtypes showed different levels of Aurora A over-expression at both transcript and protein levels (Lo Iacono et al. 2011). Aurora A expression was significantly up-modulated in moderately and poorly differentiated lung cancers, specifically in squamous and large cell carcinomas (Lo Iacono et al. 2011). In this study, we evaluate the in vitro anticancer activity of MK-5108 in a panel of eleven NSCLC cell lines as a single agent and in combination with the standard lung cancer chemotherapies cisplatin and docetaxel.
Methods
Cell lines and reagents
Eleven NSCLC cell lines from American Type Culture Collection (Manassas, VA) were studied: A427, A549, Calu-1, H358, H460, H727, H1355, H1650, H1666, H1975, and HCC827 (Online Resource 1). They were maintained at 37 °C in 5 % CO2 in RPMI 1640 medium (Gibco, Grand Island, NY) supplemented with 1 × penicillin/streptomycin, 1× L-glutamine, 1× MEM vitamin solution (Invitrogen, Carlsbad, CA), and 10 % heat-inactivated fetal bovine serum (FBS) (JR Scientific, Inc., Woodland, CA). GMP-grade MK-5108 Aurora A kinase inhibitor was provided by Merck, Inc. (Whitehouse Station, NJ, USA). The drug was dissolved in dimethyl sulfoxide and stored at −20 °C in 10 mM stock aliquots and further diluted in RPMI media for in vitro study. Infusion-grade cisplatin and docetaxel were also diluted in media before use.
Growth inhibition assays
Short-term MTT
Cells were seeded at 1 × 103–4 × 103 cells/well in 96-well flat-bottom plates and allowed to attach overnight prior to treatment. Cells were treated with MK-5108 for 72 h (0.01–20 μM) in six replicates including an untreated control. Cells were then assayed by MTT (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s protocol, and dose–response curves for triplicate experiments were generated via Prism software (GraphPad, La Jolla, CA) to assess 50 % inhibitory concentration (IC50) values.
Long-term MTT
To further determine the anti-proliferative effect following treatment, cells were seeded in 24-well plates at 1 × 103–4 × 103 cells/well and treated 24 h later with MK-5108 (0.031–0.625 μM) for 72 h. The drug was subsequently washed out, and cells were further incubated in fresh media for various durations (due to inherent differences in growth rate) to allow for repopulation: 4 days for A549, Calu-1, H460, H727, and H1975; 5 days for HCC827 and H1650; 6 days for H1355; 7 days for A427 and H1666; and 10 days for H358. Cell density was assessed by MTT when the untreated control approached confluence. Dose–response curves for triplicate experiments were generated to assess IC50 values.
Flow cytometry
To assess cell cycle effects following single-agent treatment with MK-5108, cells were seeded in 100-mm dishes (1.5 × 105–5 × 105 cells) and treated at 0.4 μM for 6, 12, 24, 48, and 72 h, after which time they were harvested and stored at −20 °C until staining with propidium iodide (Roche, Indianapolis, IN) as described previously (Chinn et al. 2012). Cell lines used to assess apoptosis via sub-G1 content for the MK-5108 + docetaxel combination were treated for 24 h at 0.5 μM MK-5108 and 1 nM docetaxel. Cellular DNA content was measured by a Becton–Dickinson FACScan and CellQuest software (BD Biosciences, San Jose, CA). Cell cycle distribution, sub-G1, and polyploidy populations were analyzed and quantified using Flowing Software (Turku Centre for Biotechnology, Turku, Finland).
Cell synchronization for mitotic accumulation
To analyze the protein levels of Aurora A and its substrates, a single thymidine block followed by release into nocodazole was used to synchronize cells and trap them in mitosis. Cells were initially plated in 2-mM thymidine-containing RPMI media, incubated for 17 h, washed twice with 1 × PBS, and incubated in fresh media for 3 h for release. The media was then replaced by 100 ng/ml nocodazole-containing RPMI, and cells were incubated for 24 h. Rounded cellular morphology indicated the presence of mitotic cells.
Immunoblotting
A time course experiment was conducted in unsynchronized cells to evaluate the targeted effect of MK-5108 treatment at 0.4 μM over time and to correlate Aurora A and p-HH3 levels with G2/M accumulation as observed by flow cytometry. The representative cell lines, H1975, H460, H1335, and Calu-1 were plated at 2 × 106–3.5 × 106 in 150-cm2 dishes and treated for/harvested at the follow times: 6, 12, 24, 72 h. As previously described (Chinn et al. 2012), SDS-PAGE was performed with 72 μg of protein per sample. Levels of phosphorylated Aurora A (p-Aur-A) (Th288) (C39D8, #3079), Aurora A/AIK (1G4, #4718), p-HH3 (Ser10) (D2C8, #3377), and histone H3 (D1H2, #4499) (Cell Signaling Technology, Danvers, MA) were determined. β-actin (AC-74, #A2228) (Sigma-Aldrich) was used as a loading control.
For PARP (F-2 sc-8007, Santa Cruz Biotechnology, Santa Cruz, CA) cleavage analysis, cells exhibiting high sub-G1 content were plated at a concentration of 5 × 105–1.2 × 106 in 100-mm dishes, treated with 0.4 μM MK-5108, and harvested after 6, 24, and 72 h of exposure.
To assess the targeted activity of MK-5108, H460, and Calu-1, cells were plated at 1 × 106–4 × 106 cells in 150-cm2 dishes according to the synchronization protocol described previously. Following the 24-h nocodazole exposure, cells were treated with MK-5108 at 0.25, 0.5, and 1 μM for 1 h and harvested. Unsynchronized and synchronized untreated controls were included. SDS-PAGE was performed with 35–70 μg of protein per sample, and levels of p-Aur-A and the Aurora A substrates, p-TACC3 (Ser558) (D8H10, #8842), and p-Plk1 (Thr210) (#5472) (Cell Sign-aling Technology, Danvers, MA) were determined.
For combination treatment studies, H460 and A549 cells (unsynchronized) were plated and treated 24 h later with 0.5 μM MK-5108 and 1 nM docetaxel as single agents and in combination. Cell line samples were harvested following 24 and 72 h of exposure and evaluated for PARP cleavage.
Combination therapy study
To assess the interaction of MK-5108 with either cisplatin or docetaxel, median effect analysis was used (Chou and Talalay 1984). Cells were seeded in 96-well plates at 1 × 103–5 × 103 cells/well and concurrently exposed to MK-5108 and cisplatin/docetaxel for 72 h. Single-agent doses and combination treatments with both agents were performed based on singleagent IC50 values (x) fixed to a constant ratio (0.25x, 0.5x, 1.0x, 2.0x). The cell line-specific IC50 doses (1x) ranged from 0.25 to 10 μM for MK-5108, 2.5–10 μM for cisplatin, and 2.5–80 nM for docetaxel. Triplicate wells were used for single-agent treatment controls and six replicates for combination treatments and the untreated control. Surviving cell populations were quantified by MTT, and combination indexes (CI) were generated.
The MK-5108 + docetaxel combination was further explored in another cell proliferation assay using a fixed MK-5108 dose (0.5 μM) and a dose range of 0.25–20 nM docetaxel for the combination and single-agent docetaxel control. Cell lines were treated for 72 h and quantified by MTT. Dose–response curves were generated.
Sequenced treatment schedules were performed for the MK-5108 + docetaxel combination to compare their anti-proliferative efficacy with that of concurrent treatments. H460 and Calu-1 cells were plated at 6 × 103–8 × 103 cells per 35-mm dish. Cells were treated with MK-5108 (0.1 μM) for 24 h, docetaxel (0.75 nM) was added to the media for another 24 h, and the treated media was removed at 72 h from the time of the first drug addition. Cells were incubated in fresh media for an additional 72 h (until the untreated control approached confluence), at which time the remaining viable cell population was quantified by MTT. The same procedure was conducted for the drug sequence of docetaxel first followed by MK-5108. Concurrent treatment and single-agent controls were included. Comparative plots of relative growth were generated.
Statistical analysis
All data were analyzed following a minimum of triplicate experiments. Prism software (GraphPad, La Jolla, CA) was used to generate all plots and conduct one-way analysis of variance (ANOVA) and multiple comparison post-tests (Tukey’s). Using median effect analysis as originally described by Chou and Talalay (Chou and Talalay 1984), CI values were formally generated with CalcuSyn software (Biosoft, Cambridge, UK) to determine whether the combination treatments were synergistic (CI < 1). ED50 and ED75 values were reported.
Results
Anti-proliferative activity of MK-5108 in NSCLC cell lines
We evaluated the single-agent anti-proliferative activity of the novel Aurora A kinase inhibitor MK-5108 in a panel of eleven NSCLC cell lines using a short-term and long-term growth inhibition assay. Baseline expression levels of total and phosphorylated Aurora A in unsynchronized, log-phase cells are shown in Fig. 1a. All of the cell lines tested demonstrated sensitivity to varying degrees following MK-5108 treatment in 72-h MTT assays with IC50 values ranging from 0.25 μM to greater than 10 μM (Fig. 1b). The H358, H1355, and H460 cell lines were most responsive to treatment (IC50 ≈ 0.25 μM), whereas the A427, H1666, H1975, and A549 cell lines were moderately sensitive (IC50 ≈ 0.625–2.5 μM). The Calu-1, HCC827, H1650, and H727 cell lines required a concentration of 10 μM or greater to achieve an IC50. No immediate correlations were apparent between expression of Aurora A and response to MK-5108.
Fig. 1.
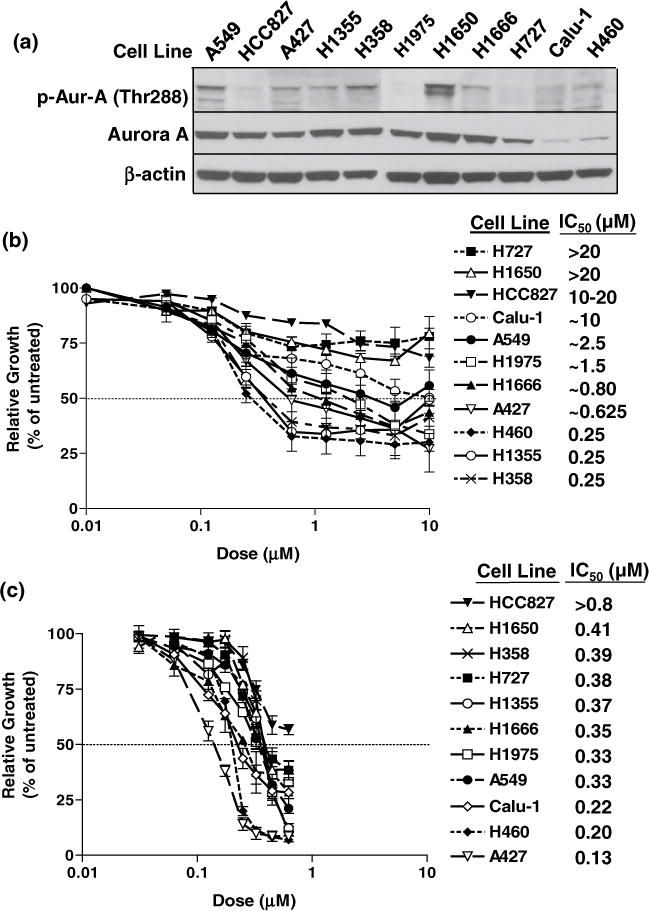
a Immunoblots of baseline Aurora A levels in unsynchronized, log-phase NSCLC cells. Phosphorylated and total Aurora A did not appear to correlate with the different inhibitory effects of MK-5108. b Short-term growth inhibition analysis of MK-5108 treatment. Cells were treated for 72 h (0.01–20 μM) and assayed by MTT. H358, H1355, and H460 cells were the most responsive to MK-5108 (IC50 = 0.25 μM). A427, H1666, H1975, and A549 cells were moderately sensitive (IC50 = 0.625–2.5 μM), while the remaining four cell lines (Calu-1, HCC827, H1650, H727) were less responsive (IC50 ≥ 10 μM). c Long-term growth inhibition analysis of MK-5108. Cells were treated for 72 h (0.031–0.625 μM), incubated in fresh media, and assayed by MTT. Growth curves show sustained growth inhibition at IC50 values ranging from 0.13 to >0.8 μM. Cell lines and IC50 values are listed in order of sensitivity from low to high
A key element in assessing agent efficacy is the ability of cells to recover and repopulate after removal of the drug. We therefore used a long-term growth assay to further define the anti-proliferative effect of MK-5108. Cells were treated for 72 h with continuous exposure to MK-5108 (0.031–0.625 μM) followed by a drug-free incubation period. All of the cell lines tested demonstrated sensitivity at nanomolar doses (0.13 to greater than 0.8 μM) much lower than those used in the short-term assay (Fig. 1c). A427 cells responded with the greatest sensitivity to MK-5108, while HCC827 was the least responsive of the panel (Fig. 1c). Most of the cell lines performed similarly in degrees of sensitivity among the short- and long-term assays. The cell lines that appeared to be less responsive (H727, H1650, HCC827, and Calu-1) in the short-term assay showed delayed re-growth and lower IC50 values in the long-term assay (Fig. 1b, c).
Cell cycle effects of MK-5108
Cell cycling analysis via flow cytometry (Fig. 2a) was performed on the cell line panel following treatment with 0.4 μM MK-5108. Polyploid cell populations and sub-G1 content were examined (Figs. 2b, 3). G2/M phase accumulation was evident in all cell lines by 6 h. In HCC827 cells, G2/M accumulation appeared to peak prior to the 12-h time point (Fig. 2a). Three cell lines (A549, H1975, and H358) exhibited the greatest G2/M accumulation at 12 h, while Calu-1, H460, and A427 cells required 24 h; however, reentry into the cell cycle for these six cell lines was evident by 72 h. H1666, H1355, and H727 cell lines showed a gradual increase in G2/M accumulation through 48 h, while H1650 cells appeared to remain in G2/M over the 72-h period (Fig. 2a).
Fig. 2.
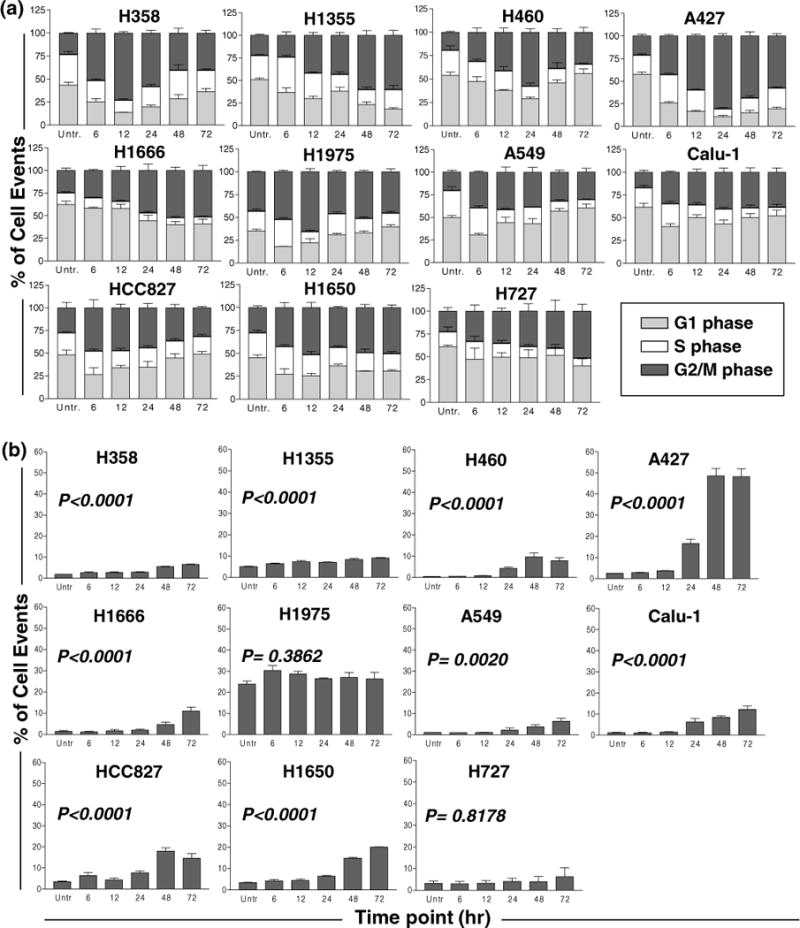
a Flow cytometric analysis of cell cycle effects (at 6, 12, 24, 48, and 72 h). Treatment with 0.4 μM MK-5108 resulted in G2/M accumulation in the NSCLC cell line panel. Accumulation peaked between 6 and 24 h for the HCC827, A549, H1975, H358, Calu-1, H460, and A427 cells, yet cells resumed cycling by 48 h. H1666, H1355, H727, and H1650 cells exhibited either a gradual increase or sustained arrest in G2/M. b Flow cytometric assessment of polyploidy for 0.4 μM MK-5108 treatment. DNA content greater than 2N was quantified. Polyploidy increased over the 72-h period relative to the untreated control in most cell lines, although A427 cells exhibited the greatest increase concomitantly with cell cycle reentry following G2/M arrest. All cell line graphs are grouped based on similar short-term growth assay IC50 values from left to right by row (most to least sensitive)
Fig. 3.
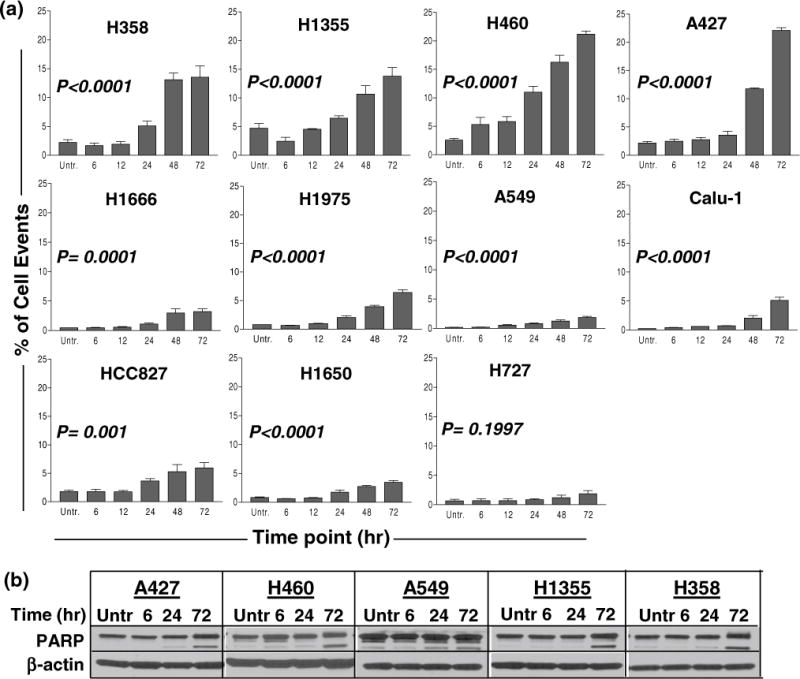
Apoptotic effects of MK-5108 treatment at 0.4 μM. a Sub-G1 content, indicative of apoptosis, was determined by flow cytometry. The greatest increase was observed in four cell lines (A427, H460, H1355, and H358). Cell line graphs are grouped based on similar short-term growth assay IC50 values from left to right by row (most to least sensitive). b Corresponding immunoblots of representative cell lines (chosen based on their varying sub-G1 content) show PARP cleavage by 72 h, correlating with their increased sub-G1 content. A549 cells showed a minimal sub-G1 increase, yet PARP cleavage was evident
Polyploidy was assessed by quantifying the cellular subpopulation with greater than 2N DNA content. For the majority of the cell lines, the increase in polyploidy was significant over time relative to the untreated cells (Fig. 2b). A427, one of the most sensitive cell lines, exhibited the largest increase following treatment with 0.4 μM MK-5108 at 48 and 72 h, the time at which cells also begin to reenter the cell cycle (Fig. 2a, b). This pattern is also evident in the HCC827 and H460 cells (Fig. 2a, b). The relatively constant population of polyploid cells for H1975 may be inherent (P = 0.3862), while H727 did not show a significant increase, possibly due to its innately slow growth rate (Fig. 2b).
Apoptotic effects of MK-5108
MK-5108-induced apoptosis was measured by sub-G1 content and PARP cleavage. ANOVA tests indicated a significant difference in sub-G1 DNA content over time and relative to the untreated in the majority of the cell lines (Fig. 3a), especially in A427, H460, H1355, and H358. Multiple comparisons in post-test showed a significant increase in sub-G1 content by the 72-h time point in all cell lines except H727, which was less responsive to treatment. PARP cleavage was also evident by 72 h in the cell lines tested (Fig. 3b).
Targeted activity of MK-5108
As Aurora A kinase activity is dependent on autophosphorylation at threonine 288 (Littlepage et al. 2002; Satinover et al. 2004), we evaluated the effect of 0.4 μM MK-5108 on p-Aur-A levels in addition to p-HH3, an indicator of mitotic cells, over a 72-h time period (Goto et al. 2002). Four unsynchronized cell lines (H460, Calu-1, H1975, H1355) were selected for this evaluation (Fig. 4a). Immunoblots showed little to no p-Aur-A levels at any of the time points compared to the synchronized untreated positive control (data not shown), thereby indicating the need for cell synchronization to detect p-Aur-A. Treatment with MK-5108 induced a time-dependent increase in Aurora A expression (Fig. 4a) in a pattern consistent with the G2/M accumulation observed in Fig. 2a, peaking at 12–24 h in H460, Calu-1, and H1975, and by 72 h in H1355. This effect was also evident in p-HH3 expression (Fig. 4a) due to the increase in mitotic cells as a result of Aurora A inhibition (Pollard and Mortimore 2009).
Fig. 4.
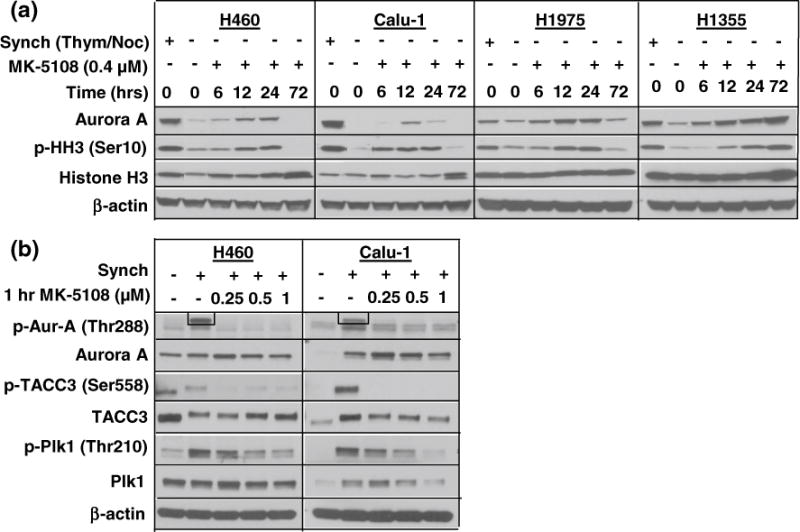
a Correlating Aurora A expression with effect of MK-5108 on cell cycling. Unsynchronized cells (representative cell lines shown) treated at 0.4 μM exhibited increased total Aurora A and p-HH3 levels in a manner consistent with G2/M accumulation over a 72-h period. b Targeted and downstream activity of MK-5108. Synchronized cells were treated for 1-h at three doses. P-Aur-A levels diminished (note: boxed band indicates Aurora A position) at all doses while total Aurora A and Plk1 decreased dose-dependently. P-TACC3 reduction was also evident
To assess the ability of MK-5108 to inhibit the activation and function of Aurora A in NSCLC cells, H460 and Calu-1 cells were synchronized (by thymidine), trapped in mitosis (by nocodazole), and treated with MK-5108 at doses of 0.25, 0.5, and 1 μM. Figure 4b indicates diminished p-Aur-A in both treated synchronized cell lines compared to the untreated (synchronized) control. We further validated target inhibition by evaluating downstream effects of MK-5108 treatment at the same three doses by examining phosphorylation levels of the Aurora A substrates TACC3 (Ser558) and Plk1 (Thr210) (Fig. 4b), which have been found to be highly expressed in NSCLC (Jung et al. 2006; Wolf et al. 1997). TACC3 is a mitotic protein that modulates microtubule stabilization at the spindle poles while Plk1 regulates mitotic progression. Both are dependent on Aurora A for activation (LeRoy et al. 2007; Macurek et al. 2008; Lu and Yu 2009; Yao et al. 2012). P-TACC3 was consistently diminished at all three MK-5108 doses, whereas p-Plk1 diminished in a dose-dependent fashion (Fig. 4b).
Combinatorial effects of MK-5108 and chemotherapeutic agents
Combination studies of MK-5108 with cisplatin and docetaxel were conducted on the NSCLC cell line panel. Cells were exposed to single-agent doses and to four concurrent treatments of MK-5108 and cisplatin or docetaxel based on the single-agent IC50 values (x) fixed to constant ratios (0.25x, 0.5x, 1x, and 2x) over a 72-h period. CI values for the concurrent treatments were formally generated via CalcuSyn. Following the MK-5108 and cisplatin combination, the H1975 cell line demonstrated synergistic activity at the ED50 (CI < 0.9), while the remaining 10 cell lines performed additively or sub-additively for the combination (Online Resource 2). The MK-5108 and docetaxel combination, however, exhibited greater synergy when comparing the CI values (Online Resource 3). Seven cell lines, which were relatively sensitive to single-agent MK-5108, possessed a CI <1 for an ED50 (Online Resource 3). A549 cells responded more synergistically at the ED75, and the HCC827 cell line, which had minimal response to MK-5108 alone (Fig. 1), was synergistic at both the ED50 and ED75 (Online Resource 3).
To better evaluate the efficacy of the MK-5108-docetaxel drug combination, we treated the cell lines concurrently for 72 h with a fixed dose of 0.5 μM MK-5108 and a range of 0.25–20 nM for docetaxel. As seen in Fig. 5a, the concurrent combination treatment synergistically enhanced cell growth inhibition between 0.5 and 2 nM for most cell lines (except for H727; H358 data not shown) compared to the single-agent treatments. Beyond this dose range, the combination performed similarly to docetaxel alone.
Fig. 5.
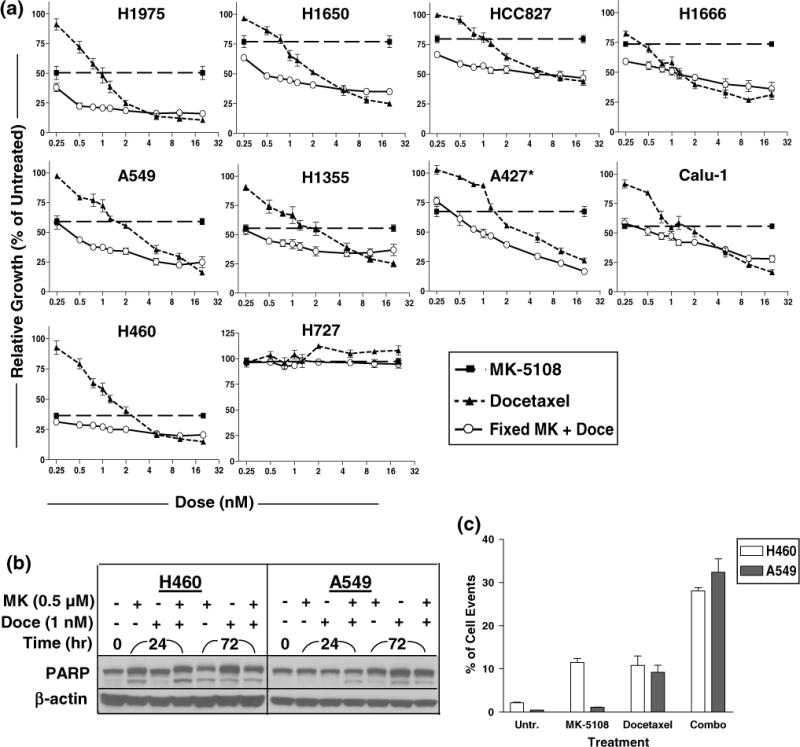
a Long-term growth inhibition assays of combined treatment with MK-5108 (MK) + docetaxel (Doce). Cells were concurrently treated for 72 h with a fixed dose of MK (0.5 μM) and a dose range (0.25–20 nM) for Doce and assayed by MTT. Combined treatment exhibited the greatest effect between 0.5 and 2 μM in most cell lines. *A427: a lower dose (0.125 μM) of MK was used for this cell line due to its sensitivity. b, c Cytotoxicity assessment of concurrent combined treatment with 0.5 μM MK + 1 nM Doce. Increased PARP cleavage and sub-G1 content were best observed at 24 h compared to the single-agent treatments for both H460 and A549 cell lines
To assess the cytotoxicity of combined treatment, H460 and A549 cells were treated with 0.5 μM MK-5108 and 1 nM docetaxel as single agents and concurrently for the combination. Flow cytometry and immunoblotting analysis showed enhanced apoptosis of combined treatment compared to the single-agent treatments as indicated by elevated sub-G1 content and PARP cleavage at 24 h post-treatment for both cell lines (Fig. 5b, c).
We then tested whether a sequenced drug treatment schedule (at sub-IC50 value doses) was more effective than concurrent drug treatment using the long-term growth inhibition assay (Fig. 6a). T tests indicated a significant difference between the drug treatment sequences (Fig. 6b). For both H460 and Calu-1 cells, treatment with docetaxel for the first 24 h followed by MK-5108 exhibited greater sustained growth inhibition (H460 ≈ 32 %, Calu-1 ≈ 69 % cells remaining) than treatment with MK-5108 followed by docetaxel (H460 ≈ 45 %, Calu-1 ≈ 98 %) (Fig. 6b). Between the two cell lines, H460 was more sensitive to treatment with docetaxel first (Fig. 6a). However, concurrent treatment of MK-5108 and docetaxel demonstrated greater sustained growth inhibition (H460 ≈ 20 %, Calu-1 ≈ 56 %) than either sequencing scheme in both cell lines (Fig. 6a).
Fig. 6.
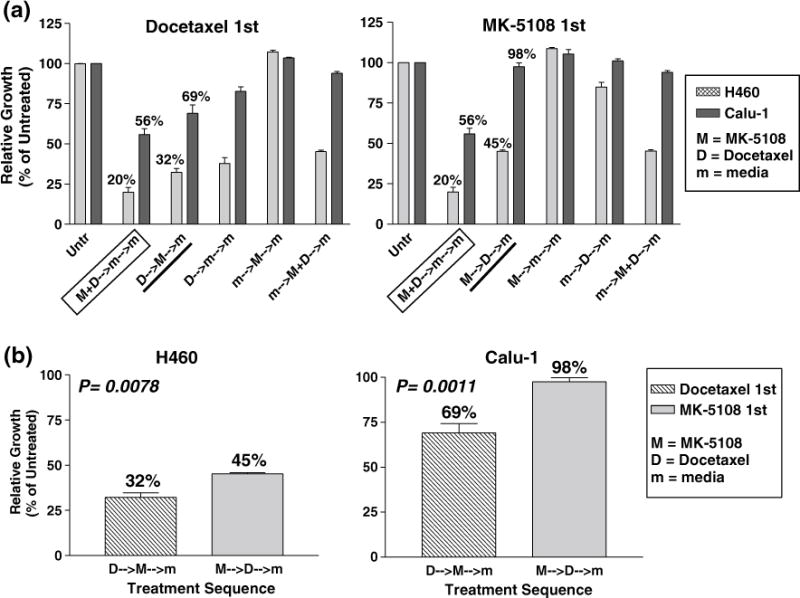
Long-term growth inhibition assay for sequencing treatment with MK-5108 (0.1 μM) and docetaxel (0.75 nM). a Concurrent treatment with both drugs still appears to induce the greatest sustained growth inhibition than either sequential treatment in H460 and Calu-1 cells. b These graphs are derived from those in part (a). Treatment with docetaxel for the first 24 h followed by MK-5108 the next day was more effective in both cell lines as indicated by the significant difference in relative growth
Discussion
The aim of the present study was to evaluate the preclinical efficacy of the selective Aurora A inhibitor MK-5108 in a panel of NSCLC cell lines as a single agent and in combination with lung cancer chemotherapies cisplatin and docetaxel. In most of the cell lines tested, MK-5108 treatment resulted in sustained cell growth inhibition at nanomolar doses, increased cell death, accumulation of cells in G2/M, inhibition of Aurora A activity, and increased histone H3 phosphorylation, consistent with studies on MK-5108 in other cancer cell types and the activity of previous Aurora A-selective inhibitors (Manfredi et al. 2007; Gorgun et al. 2010; Shimomura et al. 2010; Shan et al. 2012). The varying sensitivities to MK-5108 observed in the NSCLC cell lines did not appear to correlate with cell line-specific basal Aurora A levels. However, it should be noted that Aurora A expression is highly dependent on cell cycle progression in all NSCLC lines we examined, and thus, Aurora A expression in unsynchronized cells is likely a reflection of relative G2/M phase accumulation. Off-target effects were previously evaluated, and MK-5108 was confirmed to have high selectivity for Aurora A over 233 other protein kinases (Shimomura et al. 2010). Aurora A expression alone may merely be insufficient as a biomarker for MK-5108 drug sensitivity. Other studies have also reported either limited or no correlations between Aurora A/B inhibitor sensitivity and Aurora protein levels alone (Hook et al. 2012; Vidarsdottir et al. 2012; Chen et al. 2013). In breast cancer cells, it was reported that only a combination of high Aurora A and B levels, BRCA2 mutation, and p53 defects exhibited greater sensitivity toward Aurora inhibition by ZM447439 (Vidarsdottir et al. 2012). Additional molecular factors that influence sensitivity and resistance to MK-5108 and other Aurora kinase inhibitors are yet to be identified. A recent study implicated TPX2, a co-activator of Aurora A as a therapeutic biomarker for MK-8745, also an Aurora A inhibitor, in non-Hodgkin lymphoma (Chowdhury et al. 2012). Downstream factors of Aurora A may serve as better potential markers of drug sensitivity. The status of the tumor suppressor p53, a substrate of Aurora A, has been reported to affect cell fate in colon cancer cells following treatment with MK-8745 (Nair et al. 2012). Aurora A inhibition curtailed cell growth overall, yet induced apoptosis (wild-type p53) or polyploidy (null/mutated p53) depending on cellular p53 status (Nair et al. 2012). The majority of our NSCLC cell line panel harbors p53 defects.
Further validation of MK-5108 target inhibition of Aurora A was conducted via examining Aurora A substrates TACC3 and Plk1. TACC3 is a mitotic protein involved in the organization and stabilization of microtubules at the spindle poles and is dependent on Aurora A for phosphorylation at serine 558 and localization to the centrosomes (LeRoy et al. 2007). Plk1 is a kinase that regulates mitotic progression and requires phosphorylation at threonine 210 by Aurora A to promote mitotic entry (Macurek et al. 2008; Lu and Yu 2009). We observed diminished levels of p-TACC3 and dose-dependent reduction of p-Plk1 in the treated cell lines. Both TACC3 and Plk1 have also been implicated as cancer therapeutic targets (Plk1 inhibitors have been evaluated) as studies have shown that depletion of these critical mitotic proteins leads to in vivo tumor regression via mitotic arrest and consequent apoptosis (Yao et al. 2012; Lu and Yu 2009). Elevated levels of these substrates have also been previously detected in NSCLC, and TACC3 was reported as a potential prognostic marker since its expression was found to correlate with p53 expression (Wolf et al. 1997; Jung et al. 2006). Further studies exploring the interaction between Aurora A and these substrates in lung cancer could provide a better understanding for biomarker validation and potential combination studies with inhibitors of these targets.
The different cell cycle effects observed in the NSCLC cell lines, namely a transient or gradual mitotic accumulation over time was also noted in a study of uterine leiomyosarcoma cell lines treated with MK-5108 (Shan et al. 2012). Our data showed that following 72 h of treatment with MK-5108 at nanomolar doses, sustained growth inhibition was achieved, which may suggest that cells that were able to escape mitotic arrest at this juncture may not be viable due to chromosomal instability. This effect (mitotic slippage) was best observed in the A427 cells which exhibited a marked increase in polyploidy and cell death concomitantly with cell cycle reentry. Mitotic slippage and resulting multi-nucleation may either lead to cell death through apoptotic or non-apoptotic mechanisms or merely senescence (Blagosklonny 2007). We observed mitotic slippage and increased cell death to a lesser extent in the H460, H358, and HCC827 cells. Alternatively, cells may also die during mitotic arrest via apoptosis possibly due to depletion of short-lived anti-apoptotic proteins (Blagosklonny 2007), which may explain the progressive G2/M increase (H1355 cells) or prolonged G2/M arrest (H1650 cells) accompanied by increased sub-G1 content in our results.
With regard to anti-mitotic therapeutic strategies, past preclinical studies have indicated that MK-5108 can enhance the activity of chemotherapies, particularly the taxanes (Lok et al. 2010; Hata et al. 2005; Tanaka et al. 2007; Mazumdar et al. 2009; Bartz et al. 2006). These microtubule-targeted agents have been shown to be more biologically potent with Aurora A inhibition due to the activation of the spindle assembly checkpoint and prolonged mitotic arrest thereby enhancing apoptosis (Green et al. 2011; Mazumdar et al. 2009). While we observed limited synergy between MK-5108 and cisplatin, most of our study focused on the combination with docetaxel. Shimomura et al. showed in cervical and ovarian cancer cells that MK-5108 sensitized tumor cells to docetaxel and enhanced apoptosis by concurrent treatment and sequential administration of docetaxel first (Shimomura et al. 2010). We similarly showed that concurrent treatment of MK-5108 and docetaxel synergistically enhanced cell growth inhibition and apoptosis in NSCLC cells. In sequential drug experiments, MK-5108 also improved the antitumor activity of docetaxel when docetaxel was administered first in NSCLC cells; however, concurrent treatment still exhibited greater sustained growth inhibition. Investigating the molecular mechanisms driving the synergistic interaction between MK-5108 and docetaxel would help to explain the advantage of concurrent treatment over sequential treatment. While dosing and scheduling drug studies are challenging to model in cell lines, we did not note any antagonistic effects attributable to the cell cycle effects of the individual agents that would require pharmacodynamic separation of agents (Davies et al. 2006).
While there was heterogeneity in cell line response, the selective Aurora A kinase inhibitor MK-5108 can, in NSCLC cell lines, effectively inhibit proliferation, induce mitotic accumulation and polyploidy, and enhance the antitumor activity of docetaxel. The in vitro results reported prompt the need to evaluate the anticancer activity of MK-5108 in xenograft studies, which may provide a more substantial basis for clinical application. Clinically relevant dose concentrations of MK-5108 were explored, yet further work will be required to determine how best to integrate Aurora inhibitors into current treatment regimens in use for lung cancer. The determination of specific biomarkers predictive of clinical benefit for NSCLC would be extremely useful to optimize the efficacy of Aurora A inhibitors whether as monotherapy or in combination with other drugs.
Supplementary Material
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00432-014-1675-6) contains supplementary material, which is available to authorized users.
Conflict of interest Research was directed solely by the PI and staff with no direct involvement from Merck, Incorporated (Whitehouse Station, NJ, USA), who sponsored the study.
Ethical standards The manuscript does not contain clinical studies or patient data.
Contributor Information
Danielle C. Chinn, Division of Hematology/Oncology, UC Davis Cancer Center, 4501 X Street, Suite 3016, Sacramento, CA 95817, USA
William S. Holland, Division of Hematology/Oncology, UC Davis Cancer Center, 4501 X Street, Suite 3016, Sacramento, CA 95817, USA
Philip C. Mack, Email: pcmack@ucdavis.edu, Division of Hematology/Oncology, UC Davis Cancer Center, 4501 X Street, Suite 3016, Sacramento, CA 95817, USA.
References
- Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120(Pt 17):2987–2996. doi: 10.1242/jcs.013136. [DOI] [PubMed] [Google Scholar]
- Bartz SR, Zhang Z, Burchard J, Imakura M, Martin M, Palmieri A, Needham R, Guo J, Gordon M, Chung N, Warrener P, Jackson AL, Carleton M, Oatley M, Locco L, Santini F, Smith T, Kunapuli P, Ferrer M, Strulovici B, Friend SH, Linsley PS. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol Cell Biol. 2006;26(24):9377–9386. doi: 10.1128/MCB.01229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebbington D, Binch H, Charrier JD, Everitt S, Fraysse D, Golec J, Kay D, Knegtel R, Mak C, Mazzei F, Miller A, Mortimore M, O’Donnell M, Patel S, Pierard F, Pinder J, Pollard J, Ramaya S, Robinson D, Rutherford A, Studley J, Westcott J. The discovery of the potent aurora inhibitor MK-0457 (VX-680) Bioorg Med Chem Lett. 2009;19(13):3586–3592. doi: 10.1016/j.bmcl.2009.04.136. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007;6(1):70–74. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006;12(23):6869–6875. doi: 10.1158/1078-0432.CCR-06-1405. [DOI] [PubMed] [Google Scholar]
- Chen YP, Lin HJ, Chen JS, Tsai MY, Hsieh HP, Chang JY, Chen NF, Chang KC, Huang WT, Su WC, Yang ST, Chang WC, Hung LY, Chen TY. CDKN1A-mediated responsiveness of MLL-AF4-positive acute lymphoblastic leukemia to Aurora kinase-A inhibitors. Int J Cancer. 2013 doi: 10.1002/ijc.28708. [DOI] [PubMed] [Google Scholar]
- Chinn DC, Holland WS, Yoon JM, Zwerdling T, Mack PC. Anti-tumor activity of the HSP90 inhibitor SNX-2112 in pediatric cancer cell lines. Pediatr Blood Cancer. 2012;58(6):885–890. doi: 10.1002/pbc.23270. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Chowdhury A, Chowdhury S, Tsai MY. A novel Aurora kinase A inhibitor MK-8745 predicts TPX2 as a therapeutic biomarker in non-Hodgkin lymphoma cell lines. Leukemia Lymphoma. 2012;53(3):462–471. doi: 10.3109/10428194.2011.619018. [DOI] [PubMed] [Google Scholar]
- Davies AM, Ho C, Lara PN, Jr, Mack P, Gumerlock PH, Gandara DR. Pharmacodynamic separation of epidermal growth factor receptor tyrosine kinase inhibitors and chemotherapy in non-small-cell lung cancer. Clin Lung Cancer. 2006;7(6):385–388. doi: 10.3816/CLC.2006.n.021. [DOI] [PubMed] [Google Scholar]
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14(6):1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, Santo L, Tai YT, Nahar S, Zheng M, Bandi M, Carrasco RD, Raje N, Munshi N, Richardson P, Anderson KC. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Yasui Y, Nigg EA, Inagaki M. Aurora-B phosphorylates Histone H3 at serine28 with regard to the mitotic chromosome condensation. Genes Cells. 2002;7(1):11–17. doi: 10.1046/j.1356-9597.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- Green MR, Woolery JE, Mahadevan D. Update on Aurora Kinase targeted therapeutics in oncology. Expert Opin Drug Discov. 2011;6(3):291–307. doi: 10.1517/17460441.2011.555395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Gong Y, Huang M, Lu C, Spitz MR, Wu X. Polymorphisms of STK15 (Aurora-A) gene and lung cancer risk in Caucasians. Carcinogenesis. 2007;28(2):350–355. doi: 10.1093/carcin/bgl149. [DOI] [PubMed] [Google Scholar]
- Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, Marumoto T, Saya H, Horii A. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005;65(7):2899–2905. doi: 10.1158/0008-5472.CAN-04-3981. [DOI] [PubMed] [Google Scholar]
- Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, Yuan J, Ye J, Ozeck M, Shi ST, Zheng X, Rejto PA, Kan JL, Christensen JG, Pavlicek A. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther. 2012;11(3):710–719. doi: 10.1158/1535-7163.MCT-11-0184. [DOI] [PubMed] [Google Scholar]
- Jung CK, Jung JH, Park GS, Lee A, Kang CS, Lee KY. Expression of transforming acidic coiled-coil containing protein 3 is a novel independent prognostic marker in non-small cell lung cancer. Pathol Int. 2006;56(9):503–509. doi: 10.1111/j.1440-1827.2006.01998.x. [DOI] [PubMed] [Google Scholar]
- Kanda A, Kawai H, Suto S, Kitajima S, Sato S, Takata T, Tatsuka M. Aurora-B/AIM-1 kinase activity is involved in Rasmediated cell transformation. Oncogene. 2005;24(49):7266–7272. doi: 10.1038/sj.onc.1208884. [DOI] [PubMed] [Google Scholar]
- Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, Jove R, Forman SJ, Yen Y, Kirschbaum MH. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011;71(11):3912–3920. doi: 10.1158/0008-5472.CAN-10-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy PJ, Hunter JJ, Hoar KM, Burke KE, Shinde V, Ruan J, Bowman D, Galvin K, Ecsedy JA. Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser558 by Aurora A: a novel pharmacodynamic method for measuring Aurora A activity. Cancer Res. 2007;67(11):5362–5370. doi: 10.1158/0008-5472.CAN-07-0122. [DOI] [PubMed] [Google Scholar]
- Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci USA. 2002;99(24):15440–15445. doi: 10.1073/pnas.202606599202606599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Iacono M, Monica V, Saviozzi S, Ceppi P, Bracco E, Papotti M, Scagliotti GV. Aurora Kinase A expression is associated with lung cancer histological-subtypes and with tumor de-differentiation. J Transl Med. 2011;9:100. doi: 10.1186/1479-5876-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok W, Klein RQ, Saif MW. Aurora kinase inhibitors as anticancer therapy. Anticancer Drugs. 2010;21(4):339–350. doi: 10.1097/CAD.0b013e3283350dd1. [DOI] [PubMed] [Google Scholar]
- Lu LY, Yu X. The balance of Polo-like kinase 1 in tumorigenesis. Cell Div. 2009;4:4. doi: 10.1186/1747-1028-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455(7209):119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, Wysong DR, Zhang M, Bolen JB, Claiborne CF. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA. 2007;104(10):4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Zhang D, Saya H. Aurora-A—a guardian of poles. Nat Rev Cancer. 2005;5(1):42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL. Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck. 2009;31(5):625–634. doi: 10.1002/hed.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton SE, LoRusso P, Lockhart AC, et al. A phase I study of MK-5108, an oral aurora A kinase inhibitor, in both monotherapy and in combination with docetaxel in patients with advanced solid tumors. J Clin Oncol. 2010 doi: 10.1007/s10637-015-0306-7. (15_suppl (May 20 Supplement)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle. 2012;11(4):807–817. doi: 10.4161/cc.11.4.19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai M, Albaugh D. Millennium initiates pivotal phase 3 trial of MLN8237 in patients with relapsed or refractory peripheral T-cell lymphoma. 2012 http://investor.millennium.com/phoenix.zhtml?c=80159&p=irol-newsArticle&ID=1669234&highlight.
- Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem. 2009;52(9):2629–2651. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- Sardon T, Pache RA, Stein A, Molina H, Vernos I, Aloy P. Uncovering new substrates for Aurora A kinase. EMBO Rep. 2010;11(12):977–984. doi: 10.1038/embor.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc Natl Acad Sci USA. 2004;101(23):8625–8630. doi: 10.1073/pnas.04029661010402966101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan W, Akinfenwa PY, Savannah KB, Kolomeyevskaya N, Laucirica R, Thomas DG, Odunsi K, Creighton CJ, Lev DC, Anderson ML. A small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcoma. Clin Cancer Res. 2012;18(12):3352–3365. doi: 10.1158/1078-0432.CCR-11-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, Takahashi I, Miki S, Kawanishi N, Ohkubo M, Kotani H, Iwasawa Y. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 2010;9(1):157–166. doi: 10.1158/1535-7163.MCT-09-0609. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Hashimoto Y, Ito T, Kondo K, Higashiyama M, Tsunoda S, Ortiz C, Sakai Y, Inazawa J, Shimada Y. The suppression of aurora-A/STK15/BTAK expression enhances chemosensitivity to docetaxel in human esophageal squamous cell carcinoma. Clin Cancer Res. 2007;13(4):1331–1340. doi: 10.1158/1078-0432.CCR-06-1192. [DOI] [PubMed] [Google Scholar]
- Vidarsdottir L, Steingrimsdottir G, Bodvarsdottir SK, Ogmundsdottir HM, Eyfjord JE. Sensitivity of BRCA2 mutated human cell lines to Aurora kinase inhibition. Invest New Drugs. 2012;30(2):425–434. doi: 10.1007/s10637-010-9566-4. [DOI] [PubMed] [Google Scholar]
- Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rubsamen-Waigmann H, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14(5):543–549. doi: 10.1038/sj.onc.1200862. [DOI] [PubMed] [Google Scholar]
- Yao R, Natsume Y, Saiki Y, Shioya H, Takeuchi K, Yamori T, Toki H, Aoki I, Saga T, Noda T. Disruption of Tacc3 function leads to in vivo tumor regression. Oncogene. 2012;31(2):135–148. doi: 10.1038/onc.2011.235onc2011235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.