

Abstract
Background:
Phase I trials often include a dose expansion cohort (DEC), in which additional patients are treated at the estimated maximum tolerated dose (MTD) after dose escalation, with the goal of ensuring that data are available from more than six patients at a single dose level. However, protocols do not always detail how, or even if, the additional toxicity data will be used to reanalyze the MTD or whether observed toxicity in the DEC will warrant changing the assigned dose. A DEC strategy has not been statistically justified.
Methods:
We conducted a simulation study of two phase I designs: the “3+3” and the Continual Reassessment Method (CRM). We quantified how many patients are assigned the true MTD using a 10 to 20 patient DEC and how a sensible reanalysis using the DEC changes the probability of selecting the true MTD. We compared these results with those from an equivalently sized larger CRM that does not include a DEC.
Results:
With either the 3+3 or CRM, reanalysis with the DEC increased the probability of identifying the true MTD. However, a large CRM without a DEC was more likely to identify the true MTD while still treating 10 or 15 patients at this dose level.
Conclusions:
Where feasible, a CRM design with no explicit DEC is preferred to designs that fix a dose for all patients in a DEC.
In phase I clinical trial design, it has become common to extend the protocol beyond the standard dose escalation phase (eg, the cohorts-of-three “3+3” algorithm, accelerated titration, or continual reassessment method [CRM] [1,2]), which estimates a maximum tolerated dose (MTD). This protocol extension, called a dose expansion cohort (DEC), treats additional patients at the estimated MTD, sometimes stratifying enrollment of the DEC by multiple disease sites. The typical aim is to ensure that a sufficient number of patients has been treated at the estimated MTD by the trial’s completion to begin to characterize efficacy, tolerability, and pharmacokinetic or pharmacodynamic (PK/PD) endpoints (3–11). Recent discussions have highlighted the increasing size and complexity of DECs as well as the need to prospectively justify corresponding aims and analyses, which may be lacking (9–11). This paper provides a statistically based assessment of DECs and offers recommendations based on these results.
A systematic review of 611 single-agent phase I trials published from 2006 to 2011 found that 149 (24%) included a DEC, with the use of DECs increasing over time (9). Thirty-eight of these trials did not include any specific objectives for the DEC; among those that did, safety was most common (89 trials, 60%), followed by efficacy (50 trials, 34%). There is also heterogeneity in the size of DECs. Among the 149 trials identified by Manji, et al., the median DEC size was 17, and the range was two to 271 (9). In 10 case studies from Iasonos and O’Quigley, seven had a DEC of size less than 25, and the remaining three were 43, 97, and 100 (10). Dahlberg, et al. report a median DEC size of 27 from 60 phase I trials submitted at Harvard in 2011 (11). Reviewing all phase I trials at our institution (University of Michigan Comprehensive Cancer Center) currently enrolling patients (as of June, 2014), we found that 54 have a planned DEC in the trial protocol, with a median size of 19 and range of four to 105. Trials with a small DEC, eg, fewer than 25 patients, are of greatest relevance in a phase I context, where safety remains of primary importance and preliminary evidence of efficacy is secondary. Because these studies are not statistically powered to evaluate efficacy, they fall short of being true phase I/II trials. Recent papers have proposed dose-finding designs based on efficacy and toxicity (12–14). Although DECs may have additional endpoints beyond toxicity, a DEC strategy should first be rigorously evaluated with regard to patient safety, both to future patients, that is, finding the true MTD, as well as patients in the current trial, that is, treating many current patients at the true MTD (15–17). Thus, our focus here is on the role of DECs in traditional phase I designs based on toxicity considerations.
A principled dose escalation mechanism should operate for the duration of the trial to satisfy the safety metrics just described. We support this assertion with a simulation study of trial designs with a 10 to 20 patient DEC. The benefit conferred by a DEC may depend on the trial design, and we evaluate two common dose escalation designs: the 3+3 algorithm and the CRM (18). Because the final sample size of a 3+3 is not predetermined, adding a DEC increases the size of a trial that might otherwise be small. In contrast, the sample size of the CRM design may be predetermined, leading to a decision between inclusion of a DEC following completion of the CRM or a larger CRM in which the dose assignment mechanism operates throughout the entire trial. In investigator-initiated trials at our institution, the CRM design is used exclusively, making this decision crucial and recurrent. To the best of our knowledge, however, the strategy of appending a DEC to a CRM trial has not been statistically justified. Thus, we consider a small CRM trial paired with a DEC and an equivalently sized “large” CRM trial with no DEC.
Iasonos and O’Quigley provide guidance for safety monitoring during the DEC and present four strategies for reevaluating estimated dose-toxicity rates during and after the DEC based on safety or efficacy data (10). Also included was a simulation study of the 3+3 design to assess how frequently the final MTD estimate, after analyzing the DEC data, differs from the initial MTD estimate. Extending rule-based designs like the 3+3 with DECs is natural because such trials may otherwise terminate before enrolling enough patients to precisely evaluate secondary objectives such as efficacy or PK/PD (eg, 3–6). The authors found that the post-DEC MTD estimate from a complete analysis was more accurate than the pre-DEC estimate but did not report the frequency in which patients in the trial were assigned the true MTD.
A final important and distinguishing feature of this paper is that we differentiate between the final estimated, or selected, MTD and the true, unknown MTD, the latter being the dose level that induces dose-limiting toxicity (DLT) in x% of patients, where x may be selected based on characteristics of a particular therapy. Treating more than six patients at the estimated MTD is the motivation for using DECs and drives how precisely efficacy endpoints such as response rates may be estimated. However, although it is often implicitly assumed that the MTD determined from the dose-escalation phase is the true MTD, eg, “[t]he MTD was defined as the highest dose level with DLTs in <33% of the patients enrolled in the cohort” (p.700 [4]), this assumption is not true (19). Rather, this selected dose level is an estimate of the true MTD based on the limited available data. Given the small size of phase I studies, it frequently differs from the true MTD in a much larger population of patients. A DEC comprised of patients that are not enrolled at the true MTD will be less useful for characterizing later endpoints such as efficacy or PK/PD.
Methods
We conduct a simulation study to investigate variants of the 3+3 and CRM, with and without a DEC of size 10 to 20. These are listed as follows:
1) 3+3: A rule-based approach to dose assignments. Cohorts of three patients are enrolled at a dose level, and the highest dose level resulting in fewer than two DLTs out of six patients is the estimated MTD. The variant we used (20) allows for dose de-escalation.
2) 3+3+DEC: A 3+3 followed by a DEC treated at the estimated MTD, reestimating the MTD after the DEC.
3) CRMS: A model-based design with sample size equal to the average sample size from 3+3.
4) CRMS+DEC: CRMS followed by a DEC treated at the estimated MTD, reestimating the MTD after the DEC.
5) CRML: A single “large” CRM with sample size equal to the average sample size from 3+3+DEC and CRMS+DEC.
The Supplementary Methods (available online) contain details for implementing the 3+3 and CRM designs and the sensitivity of our results to modeling choices for the CRM. To the extent possible, we minimized all differences between designs not explicitly related to the DEC. For all designs, the first three patients were treated at dose level 2, reflecting a typical practice of including dose level one as a “fallback.”
We consider six scenarios for the true dose-toxicity relation over six dose levels (Table 1; Supplementary Figure 1, available online) and three additional scenarios with three dose levels. These vary with respect to the dose level corresponding to the true MTD, defined as the dose level with rate of DLT equal to 0.30, and the change in slope of the dose-toxicity curve. To establish equal average sample sizes within each dose-toxicity curve (DTC), we first simulated 2500 3+3 trials and determined the average number of patients enrolled (ˉn in Table 1), excluding trials that declare all dose levels too toxic. Then, 2500 trials of CRMS and CRML were run, enrolling ˉn and ˉn +15 patients, respectively, and stopping enrollment only when the estimated DLT rate at the lowest dose level exceeds 0.35, ie, 0.05 beyond the rate at the true MTD (Supplementary Results, available online). Thus, not counting trials that declare all dose levels too toxic, 3+3 and CRMS have equal average sample sizes, and the remaining designs, 3+3+DEC, CRMS+DEC, and CRML, have equal average sample sizes; this strategy was used in Ji and Wang (21). We record the number of patients treated at each dose level and the final estimated MTD, comparing all designs with respect to how frequently the estimated MTD is the true MTD and how many patients are treated at each dose level. We evaluate DECs of size 10, 15, and 20 and consider four additional scenarios in which the target rate of DLT for the CRM designs (and true MTD) is 0.25 rather than 0.30.
Table 1.
True probabilities of DLT for six DTCs evaluated in the simulation study and their corresponding sample sizes
| Probability of DLT | Dose Level | ˉn | ˉn+15 | |||||
|---|---|---|---|---|---|---|---|---|
| DTC | 1 | 2 | 3 | 4 | 5 | 6 | ||
| 1 | 0.02 | 0.07 | 0.12 | 0.18 | 0.24 | 0.30 | 17.6 | 32.6 |
| 2 | 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | 0.60 | 13.0 | 28.0 |
| 3 | 0.12 | 0.16 | 0.22 | 0.30 | 0.40 | 0.52 | 14.5 | 29.5 |
| 4 | 0.14 | 0.15 | 0.16 | 0.17 | 0.30 | 0.50 | 16.5 | 31.5 |
| 5 | 0.22 | 0.30 | 0.38 | 0.46 | 0.54 | 0.62 | 12.0 | 27.0 |
| 6 | 0.30 | 0.39 | 0.48 | 0.57 | 0.66 | 0.75 | 11.2 | 26.2 |
* ˉn is the average resulting sample size of the 3+3 design (see the Supplementary Methods, available online, for an explanation of how to account for fractional sample sizes). The DTCs are ordered by the probability of DLT at dose level one. The maximum tolerated dose is defined as the dose level with probability of DLT = 0.30. DLT = dose limiting toxicity; DTC = dose-toxicity curve.
Results
Probability of Selecting the True MTD
For each DTC, Figure 1 plots as circles the proportion of trials that selected each dose level. Within each column, the total area of the circles is the same, allowing for between-column comparisons. The shaded row corresponds to 0.30, ie, the true MTD, and values in these rows are annotated. A design performs well when the shaded circles are large. Supplementary Figures 2 and 3 (available online) give analogous results for DECs of size 10 and 20, respectively, Supplementary Figure 4 (available online) gives analogous results under three DTCs using three dose levels, and Supplementary Figure 5 (available online) gives analogous results under four DTCs with the true MTD defined as the dose level with probability of DLT equal to 0.25 rather than 0.30. The following relative comparisons remain almost identical with respect to these additional settings.
Figure 1.
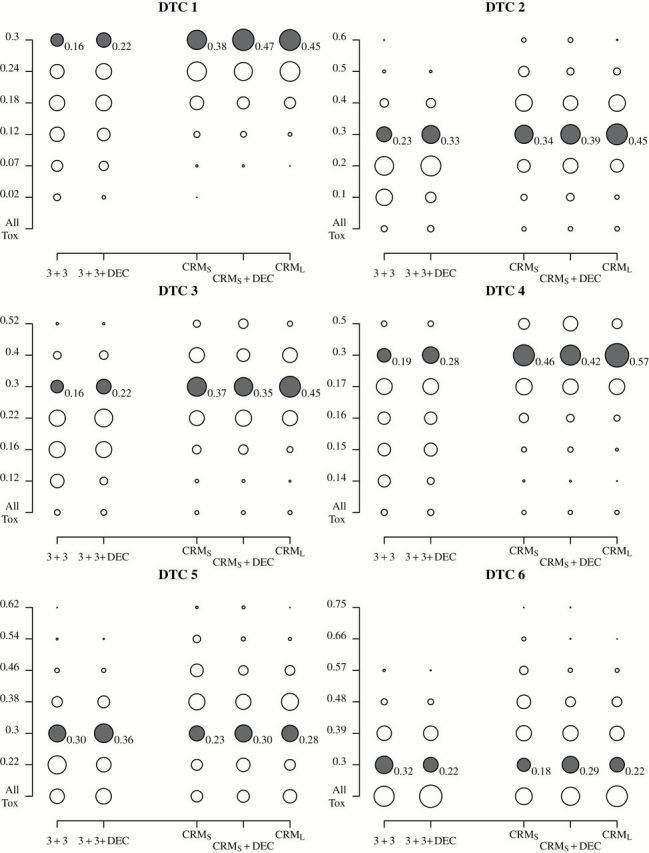
Probabilities of selecting each dose level for six DTCs. The columns correspond to designs, and the rows correspond to the true probabilities of toxicity (the bottom row denotes the probability of declaring all doses too toxic). Within a column, the area of the circle is proportional to the proportion of simulations that selected that dose as the MTD. The rows corresponding to the true MTD (closest to 0.30) are shaded gray and annotated with the probabilities of selecting that dose. 3+3 = `3+3’ trial; 3+3+DEC = `3+3’ trial followed by dose-expansion cohort; CRMS = small continual reassessment method trial; CRMS+DEC = small continual reassessment method trial followed by dose-expansion cohort; CRML = large continual reassessment method trial; DTC = dose-toxicity curve; MTD = maximum tolerated dose
Comparing 3+3 and 3+3+DEC in Figure 1 shows that reanalysis of the data after the DEC can increase the probability of selecting the true MTD, which confirms the findings of Iasonos and O’Quigley (10). This holds true for all DTCs considered except DTC 6, in which the lowest dose level is the true MTD. Comparing CRMS with CRMS+DEC, in DTCs 1, 2, 5, and 6, CRMS+DEC more frequently identifies the true MTD (respective differences of 0.09, 0.05, 0.07, and 0.11). In DTCs 3 and 4, the differences between CRMS and CRMS+DEC are small, less than 0.04, and within simulation variability. In four out of six DTCs, using an equivalent average number of patients, CRMS is more likely to select the true MTD than 3+3. Adding the DEC, CRMS+DEC more frequently selects the true MTD than does 3+3+DEC in all but DTC 5.
CRML more frequently selects the true MTD than CRMs+DEC: CRML is preferred in DTCs 2 through 4 (respective differences of 0.06, 0.10, and 0.15), and CRMs+DEC is preferred in DTC 6 (0.07). For DTCs 1 and 5, the differences are within simulation variability. Intuitively, when CRMS selects a dose that is close to the MTD, such that the subsequent DEC patients are treated at or within one dose level of the true MTD, the MTD is accurately reestimated at the trial’s completion. These results favoring CRML also hold when we use the same two-parameter model to reestimate the DLT rates at the end of the trial for both CRML and CRMs+DEC. (Supplementary Results, available online).
Number of Patients Treated at the Estimated and True MTD
Often, DECs are used to ensure that more than six patients have been treated at the estimated MTD by trial’s end. Table 2 presents statistics relevant to this metric: the probabilities of treating at least 10 or 15 patients at the estimated or true MTD. Also included is the probability of declaring all doses too toxic. For all six DTCs, 3+3+DEC most frequently declares all doses too toxic, followed by CRML and then CRMs+DEC. As we will discuss, however, this comparison does not take into account what may happen in reality when toxicity is observed during the DEC. Table 2 also presents the proportion of trials which, at completion, have treated at least 10 or 15 patients at the final estimated MTD, and no method is uniformly best with respect to this.
Table 2.
A summary of the simulation study*
| Metric | DTC 1 | DTC 2 | |||||
|---|---|---|---|---|---|---|---|
| 3+3+ DEC | CRM S +DEC | CRM L | 3+3+ DEC | CRM S +DEC | CRM L | ||
| Prob (declaring all doses toxic) | 0.00 | 0.00 | 0.00 | 0.04 | 0.02 | 0.02 | |
| Proportion of trials that treat ≥ k patients at estimated MTD | k = 10 | 0.55 | 0.71 | 0.83 | 0.55 | 0.52 | 0.68 |
| k = 15 | 0.55 | 0.69 | 0.69 | 0.55 | 0.51 | 0.36 | |
| Proportion of trials that select the true MTD and treat | |||||||
| ≥ k patients at true MTD | k = 10 | 0.14 | 0.31 | 0.36 | 0.17 | 0.22 | 0.31 |
| k = 15 | 0.14 | 0.31 | 0.31 | 0.17 | 0.22 | 0.16 | |
| DTC 3 | DTC 4 | ||||||
| 3+3+ DEC | CRM S +DEC | CRM L | 3+3+ DEC | CRM S +DEC | CRM L | ||
| Prob (declaring all doses toxic) | 0.03 | 0.02 | 0.02 | 0.04 | 0.02 | 0.02 | |
| Proportion of trials that treat ≥ k patients at estimated MTD | k = 10 | 0.52 | 0.57 | 0.74 | 0.50 | 0.54 | 0.76 |
| k = 15 | 0.52 | 0.56 | 0.48 | 0.50 | 0.52 | 0.57 | |
| Proportion of trials that select the true MTD and treat | |||||||
| ≥ k patients at true MTD | k = 10 | 0.11 | 0.21 | 0.35 | 0.15 | 0.28 | 0.48 |
| k = 15 | 0.11 | 0.21 | 0.23 | 0.15 | 0.27 | 0.39 | |
| DTC 5 | DTC 6 | ||||||
| 3+3+ DEC | CRM S +DEC | CRM L | 3+3+ DEC | CRM S +DEC | CRM L | ||
| Prob (declaring all doses toxic) | 0.25 | 0.15 | 0.19 | 0.52 | 0.34 | 0.45 | |
| Proportion of trials that treat ≥ k patients at estimated MTD | k = 10 | 0.52 | 0.40 | 0.54 | 0.37 | 0.25 | 0.33 |
| k = 15 | 0.52 | 0.40 | 0.27 | 0.37 | 0.25 | 0.14 | |
| Proportion of trials that select the true MTD and treat | |||||||
| ≥ k patients at true MTD | k = 10 | 0.21 | 0.16 | 0.22 | 0.22 | 0.13 | 0.10 |
| k = 15 | 0.21 | 0.16 | 0.11 | 0.22 | 0.13 | 0.03 | |
* Included are the proportion of trials declaring all doses too toxic, the proportion treating at least 10 or 15 patients at the estimated MTD, and the proportion treating at least 10 or 15 patients at the true MTD. 3+3+DEC = `3+3’ trial followed by dose-expansion cohort; CRMS+DEC = small continual reassessment method trial followed by dose-expansion cohort; CRML = large continual reassessment method trial; DTC = dose-toxicity curve; MTD = maximum tolerated dose
However, in the context of the dual safety objectives of a phase I trial, a more important metric is the joint probability of selecting the true, rather than estimated, MTD and treating at least 10 or 15 patients at this dose level, which is necessarily smaller than the previous metric. With respect to 10 patients, CRML is best in DTCs 1 through 5, reflecting its superiority at finding the true MTD. As before, the exception to this is DTC 6, in which 3+3+DEC is best. With respect to 15 patients, CRML is preferred in DTCs 1 through 4. By the nature of the 3+3+DEC design, either 2 through 6 or 21 patients are assigned the true MTD, depending on whether the initial 3+3 selects the true MTD. In summary, although reanalysis with the DEC data can increase the accuracy of estimating the MTD, a DEC approach still depends on the dose escalation scheme and is not necessarily ideal for ensuring many patients receive the MTD.
Discussion
Our results demonstrate that, when the dose-finding portion of a phase I trial enrolls approximately 12 to 18 patients, a 10 to 20 patient dose expansion cohort substantially increases the probability of selecting the true MTD, regardless of design. Even CRMS, which itself more frequently selects the true MTD than 3+3, benefits from the addition of the DEC. Based on this, the use of expansion cohorts to further characterize toxicity would seem to be supported by our results, provided all data are reanalyzed at the trial’s completion. However, a provision for how the DEC data will be analyzed is often not made (9–11). Furthermore, the probability that the DEC following the 3+3 was assigned the true MTD, which is equal to the probability that the 3+3 identifies the true MTD, is very small in the scenarios we considered (0.16–0.33) (Figure 1). Randomly choosing a dose level to be the estimated MTD would yield a similar probability, 1/6 or approximately 0.17, of identifying the true MTD. This is far from the implicit assumption that the 3+3 “defines” the MTD. Had we considered the simpler but more prevalent variant of the 3+3 that does not de-escalate dose assignments, its performance would likely have been worse. Although the small CRM design had more success in assigning DEC patients to the true MTD (0.18–0.46), its performance was also limited by small sample size. Twelve to 18 patients, the numbers typically enrolled by the 3+3 design, cannot accurately estimate the MTD. The additional patients from a DEC can increase the likelihood of identifying the MTD, but only when included in a reanalysis of toxicity data, and these patients may not necessarily receive the true MTD. A CRM with no explicit DEC but equal average total sample size confers this same benefit but is equipped to change dose assignments for all patients.
The reported results corresponding to the sixth DTC, in which the true MTD was the lowest dose level and 3+3+DEC evidently performed best, do not contradict those of the remaining DTCs, nor do they necessarily suggest that scenarios exist in which 3+3+DEC is preferred. Rather, the reality of how a DEC would proceed in this overly toxic scenario is difficult to simulate, because a pure approach that ignores the inevitable toxicity during the DEC would not be implemented (an exception being if enrollment was very fast during the DEC). In practice, if many toxicities are observed during a DEC, the investigator will lower the assigned dose, if possible, or stop the trial, which is an ad hoc dose-assignment scheme. The results of these overly toxic DTCs therefore highlight an additional challenge of a DEC strategy, namely prospectively defining how any observed toxicity in the DEC will be accommodated. In contrast, an equally sized (on average) CRM design with no explicit DEC, ie, CRML, automatically adjusts the dose assignment in the presence of toxicity, taking into account the entire history of the trial. This helps protect against the overdosing of patients without extemporaneous adjustments to the dose assignment. As an aside, these results reiterate the need for a safety dose on which to fallback in the presence of too much toxicity.
One limitation of our study is that we have focused exclusively on toxicity endpoints. While this is how most Phase I trials are currently conducted, we recognize that, when possible, it is preferable to involve efficacy assessments in dose-finding trial designs. An additional limitation of our study is the inability of our (and arguably any) prospectively defined algorithm to capture the way dosing decisions are made in real time by a data-safety monitoring committee. Such decisions are often subjective and depend on patient characteristics, lower-grade toxicities, and other subtleties not captured in our modeling of DLT data only. In anticipation of phase II study, investigators want to have experience at a single-dose level, perhaps stratifying enrollment of the DEC based on disease site or other factors. We acknowledge this role that DECs play; however, the belief that a DEC provides information to assess other endpoints, eg, efficacy, from many patients treated at the true MTD is contradicted by the results of Table 2. In light of this, together with our results that 12 to 18 patients are inadequate for accurately selecting the true MTD, our recommendation is to allow the dose escalation mechanism to operate throughout the DEC, moving away from the incorrect mindset that the MTD has been established upon completion of the dose escalation phase and need not be revisited. Specifically, a trial could enroll all comers for the first, say, 12 to 18 patients and thereafter stratify enrollment by disease site, while still allowing for the dose assignment to change according to a dose escalation mechanism. This recommendation does not preclude enrolling large DECs of many dozen patients at multiple disease sites and is, in fact, even more crucial in such cases where so many patients are at risk of being enrolled at inefficacious or toxic doses (11). Rather, our approach acknowledges that MTD estimates based on too few patients are highly variable (Figure 1). If the dose-assignment mechanism uses toxicity information from the entire history of the trial, as it should, dose assignments will change during the DEC only if the data warrant it. If the trial has indeed arrived at the true MTD within the first few patients, then many patients in the DEC will be assigned a single dose (Table 2). Not leveraging all patient data to estimate the MTD is a fundamental flaw of the 3+3 and a key reason why model-based approaches such as the CRM can more accurately identify the MTD (1,21–24). Our recommendation incorporates the DEC paradigm while preserving the essential safety imperatives of a phase I trial: selecting the true MTD and treating many patients at this dose level.
Funding
This work was supported by the National Institutes of Health (grant number P30 CA046592).
Supplementary Material
The study sponsor had no role in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
The code for the simulation study was written using SAS (9.3, UNIX) and R (3.0.2, Windows) software (25), and the continual reassessment method simulations were based on modified code from the package, (26). SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc., Cary, NC.
References
- 1. O’Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46(1):33–48. [PubMed] [Google Scholar]
- 2. Garrett-Mayer E. The continual reassessment method for dose-finding studies: a tutorial. Clin Trials. 2006;3(1):57–71. [DOI] [PubMed] [Google Scholar]
- 3. Srokowski TP, Liebmann JE, Modiano MR, et al. Pixantrone dimaleate in combination with fludarabine, dexamethasone, and rituximab in patients with relapsed or refractory indolent non‐Hodgkin lymphoma. Cancer. 2011;117(22):5067–5073. [DOI] [PubMed] [Google Scholar]
- 4. Gordon MS, Sweeney CJ, Mendelson DS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor–neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16(2):699–710. [DOI] [PubMed] [Google Scholar]
- 5. Isambert F, Freyer G, Zanetta S, et al. Phase I dose-escalation study of intravenous aflibercept in combination with docetaxel in patients with advanced solid tumors. Clin Cancer Res. 2012;18(6):1743–1750. [DOI] [PubMed] [Google Scholar]
- 6. Alsina M, Trudel S, Furman RR, et al. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin Cancer Res. 2012;18(17):4830–4840. [DOI] [PubMed] [Google Scholar]
- 7. Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemoth Pharm. 2010;65(4):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diamond JR, Bastos BR, Hansen RJ, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(4):849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manji A, Brana I, Amir E, et al. Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single-agent phase I cancer trials. J Clin Oncol. 2013;31(33):4260–4267. [DOI] [PubMed] [Google Scholar]
- 10. Iasonos A, O’Quigley J. Design considerations for dose-expansion cohorts in phase I trials. J Clin Oncol. 2013;31(33):4014–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dahlberg SE, Shapiro GI, Clark JW, Johnson BE. Evaluation of statistical designs in phase I expansion cohorts: The Dana-Farber/Harvard Cancer Center experience. J Natl Cancer Inst. 2014;106(7)dju163 10.1093/jnci/dju163. [DOI] [PubMed] [Google Scholar]
- 12. Thall PF, Cook JD. Dose-finding based on efficacy-toxicity trade-offs. Biometrics. 2004;60(3):684–693. [DOI] [PubMed] [Google Scholar]
- 13. Braun TM. The bivariate CRM: Extending the CRM to phase I trials of two competing outcomes. Control Clin Trials. 2002;23(3):240–256. [DOI] [PubMed] [Google Scholar]
- 14. Yuan Y, Yin G. Bayesian dose-finding by jointly modeling toxicity and efficacy as time-to-event outcomes. J R Stat Soc Ser C. 2009;58(5):719–736. [Google Scholar]
- 15. Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst. 2009;101(10):708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ratain MJ, Mick R, Schilsky Rl, Siegler M. Statistical and ethical issues in the design and conduct of phase I and II clinical trials of new anticancer agents. J Natl Cancer Inst. 1993;85(20):1637–1643. [DOI] [PubMed] [Google Scholar]
- 17. Joffe S, Miller FG. Rethinking risk-benefit assessment for phase I cancer trials. J Clin Oncol. 2006;24(19):2987–2990. [DOI] [PubMed] [Google Scholar]
- 18. Rogatko A, Schoeneck D, Jonas W, Tighiouart M, Khuri FR, Porter A. Translation of innovative designs into phase I trials. J Clin Oncol. 2007;25(31):4982–4986. [DOI] [PubMed] [Google Scholar]
- 19. Rosenberger WF, Haines LM. Competing designs for phase I clinical trials: a review. Stat Med. 2002;21(18):2757–2770. [DOI] [PubMed] [Google Scholar]
- 20. Korn EL, Midthune D, Chen TT, Rubinstein LV, Christian MC, Simon RM. A comparison of two phase I trial designs. Stat Med. 1994;13(18):1799–1806. [DOI] [PubMed] [Google Scholar]
- 21. Ji Y, Wang SJ. Modified toxicity probability interval design: A safer and more reliable method than the 3+3 design for practical phase I trials. J Clin Oncol. 2013;31(14):1785–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisenhauer EA, O’ Dwyer PJ, Christian M, Humphrey JS. Phase I clinical trial design in cancer drug development. J Clin Oncol. 2000;18(3):684–684. [DOI] [PubMed] [Google Scholar]
- 23. Ahn C. An evaluation of phase I cancer clinical trial designs. Stat Med. 1998;17(14):1537–1549. [DOI] [PubMed] [Google Scholar]
- 24. Doussau A, Asselain B, Le Deley MC, et al. Dose-finding designs in pediatric phase I clinical trials: Comparison by simulations in a realistic timeline framework. Contemp Clin Trials. 2012;33(4):657–665. [DOI] [PubMed] [Google Scholar]
- 25. R Core Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. Accessed July 7, 2014. [Google Scholar]
- 26. Cheung K: dfcrm: Dose-finding by the continual reassessment method. package version 0.2–2. http://CRAN.R-project.org/package=dfcrm. Accessed July 7, 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.