

Abstract
Background:
Small molecule inhibitors of phosphatidylinositol-3 kinase (PI3K) have been developed as molecular therapy for cancer, but their efficacy in the clinic is modest, hampered by resistance mechanisms.
Methods:
We studied the effect of PI3K therapy in patient-derived tumor organotypic cultures (from five patient samples), three glioblastoma (GBM) tumor cell lines, and an intracranial model of glioblastoma in immunocompromised mice (n = 4–5 mice per group). Mechanisms of therapy-induced tumor reprogramming were investigated in a global metabolomics screening, analysis of mitochondrial bioenergetics and cell death, and modulation of protein phosphorylation. A high-throughput drug screening was used to identify novel preclinical combination therapies with PI3K inhibitors, and combination synergy experiments were performed. All statistical methods were two-sided.
Results:
PI3K therapy induces global metabolic reprogramming in tumors and promotes the recruitment of an active pool of the Ser/Thr kinase, Akt2 to mitochondria. In turn, mitochondrial Akt2 phosphorylates Ser31 in cyclophilin D (CypD), a regulator of organelle functions. Akt2-phosphorylated CypD supports mitochondrial bioenergetics and opposes tumor cell death, conferring resistance to PI3K therapy. The combination of a small-molecule antagonist of CypD protein folding currently in preclinical development, Gamitrinib, plus PI3K inhibitors (PI3Ki) reverses this adaptive response, produces synergistic anticancer activity by inducing mitochondrial apoptosis, and extends animal survival in a GBM model (vehicle: median survival = 28.5 days; Gamitrinib+PI3Ki: median survival = 40 days, P = .003), compared with single-agent treatment (PI3Ki: median survival = 32 days, P = .02; Gamitrinib: median survival = 35 days, P = .008 by two-sided unpaired t test).
Conclusions:
Small-molecule PI3K antagonists promote drug resistance by repurposing mitochondrial functions in bioenergetics and cell survival. Novel combination therapies that target mitochondrial adaptation can dramatically improve on the efficacy of PI3K therapy in the clinic.
The phosphatidylinositol-3 kinase (PI3K) pathway (1) is a universal signaling node that integrates environmental cues of cellular growth with downstream networks of cell proliferation, survival, and bioenergetics (2). Exploited in virtually every human cancer, in some cases through the acquisition of activating mutations (3), PI3K signaling and its effectors Akt and mammalian target of rapamycin (MTOR) (4) are validated therapeutic targets, and several small molecule antagonists of this pathway have entered clinical testing (5).
However, the response to PI3K therapy in the clinic has been inferior to expectations, with modest single-agent activity, statistically significant toxicity, and short-lived patient benefits (6). The basis for this treatment resistance is unknown (7), and strategies to guide patient selection or incorporate PI3K therapy in more effective combination regimens have remained elusive (8). In this context, there is evidence that small-molecule inhibitors of PI3K/Akt/MTOR activate a broad transcriptional and signaling program in tumors, culminating with a paradoxical (re)activation of Akt in treated patients (9–11). How (and whether) this process contributes to drug resistance has not been clearly elucidated, but it is possible that it provides a general adaptive response to “environmental stress” imposed by molecular therapy (12). In this context, mechanisms of adaptation are important drivers of tumor diversity and treatment failure (13), hinging on a tight control of the protein-folding environment (14) by molecular chaperones of the Heat Shock Protein-90 (Hsp90) family (15).
In this study, we hypothesized that clinical resistance to small molecule PI3K antagonists depends on reprogramming of metabolic and survival networks in tumor cells and that this adaptive response may be exploited for novel drug combination strategies in the clinic.
Methods
Patients
Fresh, patient-derived and treatment-naïve tissues obtained from surgical resections of colon adenocarcinoma (one case), infiltrating ductal breast adenocarcinoma (four cases), non–small cell lung adenocarcinoma (three cases), and grade IV glioblastoma (GBM) (five cases) were used in this study. Informed consent was obtained from all patients, and the study was approved by an Institutional Review Board at the Fondazione IRCCS Ca’ Granda hospital (Milan, Italy). The clinicopathological characteristics of the patient series used in this study are presented in Supplementary Table 1 (available online).
Organotypic cultures
Short-term organotypic cultures from primary patient samples were established as described (16). Cultures were supplemented with vehicle (DMSO, 2.5 µL), pan-PI3K inhibitor LY294002 (50 or 100 µM) or PX-886 (2.5, 5, or 10 µM), mitochondrial-targeted Hsp90 inhibitor, Gamitrinib (10 or 25 µM) (17), or the combination of PX-886 plus Gamitrinib (each used at 10 µM). At the end of the experiment, one tissue slice per condition was formalin fixed and paraffin embedded and was further processed for morphological and immunohistochemical analysis. An additional tissue slice was embedded in optimal cutting temperature, and snap-frozen for molecular or immunofluorescence studies.
Statistical Methods
Data were analyzed using the two-sided unpaired t tests using a GraphPad software package (Prism 4.0) for Windows. Data are expressed as mean ± SD of replicates from a representative experiment out of at least two independent determinations. A P value of less than or equal to .05 was considered as statistically significant.
All other methods are described in detail in the Supplementary Methods (available online).
Results
PI3K Therapy and Mitochondrial Metabolism
To study how PI3K therapy affects tumor behavior, we first profiled the metabolome (18) of GBM LN229 cells in response to PX-866, a small-molecule antagonist of all PI3K subunits, currently in clinical trials (6). PI3K inhibition induced extensive defects in tumor mitochondrial metabolism. These included impaired oxidative phosphorylation (19), with reduced levels of pyruvate, α-ketoglutarate, succinate, fumarate, and malate (Figure 1A; Supplementary Figure 1A and Supplementary Table 2, available online), and defective arginine metabolism, with decreased expression of polyamines, agmatine, spermidine, putrescine, and 5′-deoxy-5′-(methylthio)adenosine (MTA) (Figure 1B; Supplementary Figure 1B and Supplementary Table 2, available online). Conversely, PI3K therapy resulted in higher levels of carnitine conjugates required for mitochondrial fatty acid β-oxidation (Figure 1C; Supplementary Figure 1C and Supplementary Table 2, available online), and elevation of long-chain fatty acids (Figure 1C; Supplementary Figure 1D and Supplementary Table 2, available online). Consistent with these findings, LN229 or prostate adenocarcinoma PC3 cells exposed to PI3K inhibitors, including PX-866, AZD6482, or GDC0941 (6), exhibited defects in glycolysis, with reduced glucose utilization (Figure 1D) and lactate production (Figure 1E), and inhibition of oxygen consumption (Figure 1F), a marker of oxidative phosphorylation (19). As a result of these bioenergetics defects, PI3K therapy considerably reduced adenosine triphosphate (ATP) production in tumor cells (Figure 1G).
Figure 1.
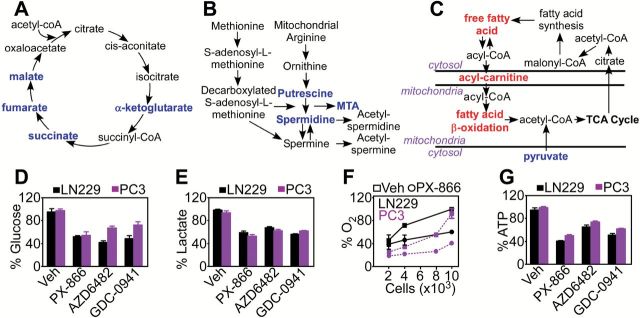
PI3K therapy and mitochondrial metabolic reprogramming. A-C) LN229 cells treated with PX-866 (10 μM for 48 hours) were analyzed in a global metabolomics screening (n = 5). Changes in expression levels of metabolites implicated in oxidative phosphorylation (A), polyamine metabolism (B), and fatty acid β-oxidation (C) are shown. Red, upregulation; blue, downregulation. D-G) LN229 or PC3 cells were treated with vehicle or PI3K inhibitors, PX-866 (10 μM), AZD6482 (10 μM), or GDC0942 (2 μM) and analyzed after 48 hours for changes in glucose utilization (D), lactate generation (E), oxygen consumption (F), or ATP production (G). Mean ± SD of at least two independent determinations. MTA = 5′-deoxy-5′-(methylthio)adenosine; Veh = vehicle.
Despite these bioenergetics defects, PI3K therapy did not appreciably kill tumor cells (Supplementary Figure 2A, available online) and only caused reduced cell proliferation (Supplementary Figure 2B, available online) because of G1 cell cycle arrest (Supplementary Figure 2, C and D, available online). These cytostatic effects were transient, as the long-term colony-forming ability of tumor cells treated with PI3K inhibitors was unaffected compared with control cultures (Supplementary Figure 2E, available online).
Akt Regulation During PI3K Therapy
We next searched for potential mediator(s) of resistance to PI3K therapy in cancer, and we focused on Akt, which becomes paradoxically reactivated under these conditions (9,10,20). A 48-hour exposure of organotypic cultures of GBM (16) (Figure 2A, Supplementary Figure 3A, available online) to PI3K inhibitors, PX-866, or LY294002 induced strong (re)phosphorylation of Akt (10,20) (vehicle vs PX-866 2.5 μM, P = .03; vehicle vs PX-866 5 μM, P = .005; vehicle vs PX-866 10 μM, P = .04 by two-sided unpaired t test) (Figure 2B) (vehicle vs LY294002 50 μM, P = .002; vehicle vs LY294002 100 μM, P ≤ .001 by two-sided unpaired t test) (Supplementary Figure 3B, available online) and higher levels of phosphorylated MTOR (vehicle vs LY294002 50 μM, P ≤ .001; vehicle vs LY294002 100 μM, P ≤ 0.001 by two-sided unpaired t test) (Supplementary Figure 3B, available online). PI3K inhibition also induced increased Ser473 phosphorylated Akt in organotypic cultures of breast adenocarcinoma (Supplementary Figure 3C, available online) (vehicle vs LY294002 50 μM, P ≤ .001; vehicle vs LY294002 100 μM, P = .007 by two-sided unpaired t test) (Supplementary Figure 3D, available online), or colon adenocarcinoma (Supplementary Figure 3E, available online) (vehicle vs LY294002 50 μM, P = .02, by two-sided unpaired t test) (Supplementary Figure 3F, available online). Other molecular therapies, including inhibition of cytosolic Hsp90 with 17-allylamino-demethoxygeldanamycin (17-AAG), had no effect on Akt or MTOR phosphorylation (Figure 2B).
Figure 2.
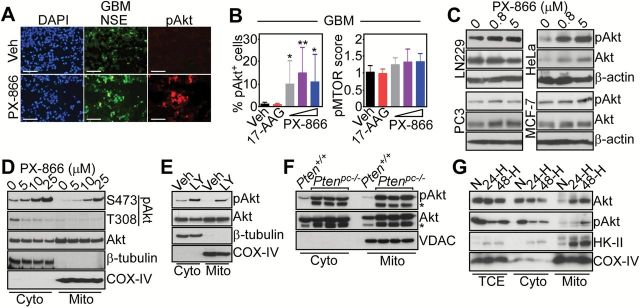
PI3K therapy and regulation of Akt signaling. A) Glioblastoma (GBM) organotypic cultures treated with vehicle or PX-866 (10 μM for 48 hours) were analyzed by immunofluorescence microscopy. DNA was counterstained with DAPI. Scale bar = 100 μm. B) The percentage of pAkt+ cells or a pMTOR immunohistochemical score was quantified in GBM organotypic cultures (PX-866, 2.5, 5, 10 μM, 17-AAG, 20 μM). None = untreated. Mean ± SD of at least three independent determinations. *P = .03-.04; **P = .005 by two-sided unpaired t test. C) The various tumor cell lines were treated with the indicated increasing concentrations of PX-866 and analyzed by western blotting. D) LN229 cells were treated with the indicated increasing concentrations of PX-866 and cytosol or mitochondrial fractions were analyzed by western blotting. E) LN229 cells were treated with vehicle or LY294002 (LY, 50 μM for 48 hours), fractionated in cytosol or mitochondrial extracts, and analyzed by western blotting. For (D) and (E), Cox-IV and β-tubulin were used as mitochondrial or cytosolic markers, respectively. F) Cytosol or mitochondrial fractions from prostate tissues of wild-type (Pten+/+) or Ptenpc-/- mice (three mice per condition) were analyzed by western blotting. * = nonspecific. VDAC was a mitochondrial marker. G) LN229 cells were incubated in normoxia or hypoxia (H, 0.5% O2) conditions for 24 to 48 hours, fractionated in cytosol or mitochondrial extracts and analyzed by western blotting. HK-II was used as a control for a hypoxia-regulated mitochondrial-associated protein. Cyto = cytosol; GBM = glioblastoma; Mito = mitochondrial; N = normoxia; NSE = neuron-specific enolase; pAkt = Ser473-phosphorylated Akt; TCE = total cell extracts; Veh = vehicle.
As an alternative experimental approach, we next silenced the expression of PI3K p110α subunit by small interfering RNA (siRNA), and looked at changes in signaling pathways. Similar to the results obtained with pharmacologic inhibition, PI3K knockdown in PC3 cells resulted in increased phosphorylation of Akt2 (see below), MTOR and its downstream effector, S6K (Supplementary Figure 3G, available online). This response was also associated with increased phosphorylation, ie, activation of ERK1/2 (Supplementary Figure 3G, available online), in agreement with previous observations (20). Consistent with these findings, PI3K therapy–induced Akt phosphorylation was observed in genetically heterogeneous tumor cell lines (Figure 2C), regardless of the presence of oncogenic “driver” mutation(s), for instance BRAF V600E melanoma cells (Supplementary Figure 3H, available online) and in response to structurally diverse PI3K antagonists currently in the clinic, including AZD6482, GDC0942, and BKM120 (Supplementary Figure 3I, available online). Both high (10 μM) and low (0.8 μM) concentrations of PX-866 induced Akt phosphorylation in tumor cells within 24 hours of treatment (Supplementary Figure 3J, available online).
In addition to Akt activation in cytosol (10,20), PI3K inhibitors induced the phosphorylation of a pool of Akt in mitochondria of tumor cells (Figure 2, D and E) (21). This involved the MTORC2 phosphorylation site on Akt (Ser473), whereas the PDK1 phosphorylation site (Thr308) was unaffected (Figure 2D). Mitochondrial Akt comprised predominantly the Akt2 isoform, whereas Akt1 was expressed at low levels in mitochondria (Supplementary Figure 4A, available online) (22). Accordingly, PI3K therapy with PX-866 resulted in robust and concentration-dependent isoform-specific phosphorylation of Akt2 on Ser474 in cytosol and mitochondrial extracts of treated tumor cells (Supplementary Figure 4B, available online), as well as primary GBM organotypic cultures (Supplementary Figure 4C, available online). In terms of submitochondrial localization, Akt2 predominantly accumulated in the organelle inner and outer membranes, and intermembrane space (Supplementary Figure 4D, available online), largely protected from proteinase K-dependent proteolysis of the outer membrane (Supplementary Figure 4E, available online). Akt is a known client protein for Hsp90 (15), and accordingly pretreatment of tumor cells with 17-AAG abolished the accumulation of phosphorylated Akt in cytosol, as well as mitochondria in response to PX-866 (Supplementary Figure 4F, available online). Mitochondrial Akt2 was broadly expressed in normal and tumor cell lines (Supplementary Figure 4G, available online) and in all normal mouse tissues examined (Supplementary Figure 4H, available online).
We next looked for other pathophysiological conditions that may activate mitochondrial Akt, independently of PI3K therapy. First, mice with prostate-specific deletion of Pten, an antagonist of the Akt pathway often deleted in tumors (23), showed constitutively high levels of Ser473-phosphorylated Akt in mitochondria (Figure 2F). Second, exposure of tumor cells to stress conditions, including hypoxia (Figure 2G) or glucose starvation induced by the nonmetabolizable analog, 2-deoxyglucose (2-DG), increased Akt recruitment to mitochondria and its phosphorylation on Ser473 (Figure 2G and Supplementary Figure 4I, available online). In contrast, ER (thapsigargin) or oxidative (H2O2) stress had no effect on Akt localization to mitochondria (Supplementary Figure 4I, available online).
Modulation of CypD Phosphorylation
We next searched public databases of mitochondria-localized proteins for the presence of potential Akt consensus phosphorylation site(s). We found that Cyclophilin D (CypD), a mitochondrial regulator of apoptosis (24) and bioenergetics (25), contained two potential Akt phosphorylation sites on Ser31 and Ser123 (Supplementary Figure 5A, available online). In immunoprecipitation (Figure 3A) and pull-down experiments (Figure 3B; Supplementary Figure 5B, available online), CypD formed a complex with Akt in tumor mitochondria. In addition, recombinant, active Akt1 (Supplementary Figure 5C, available online) or Akt2 (Supplementary Figure 5D) readily phosphorylated recombinant CypD, as well as its known substrate GSK3β in kinase assays. We next carried out mutagenesis studies to identify the Akt phosphorylation site(s) on CypD (Supplementary Figure 5A, available online). In kinase assays, recombinant Akt2 phosphorylated wild-type (WT) CypD or a CypD Ser123→Ala mutant (Figure 3C). In contrast, Akt2 phosphorylation of CypD Ser31→Ala mutant or a CypD Ser31/Ser123→Ala double mutant was abolished, and vehicle had no effect on CypD phosphorylation (Figure 3C).
Figure 3.
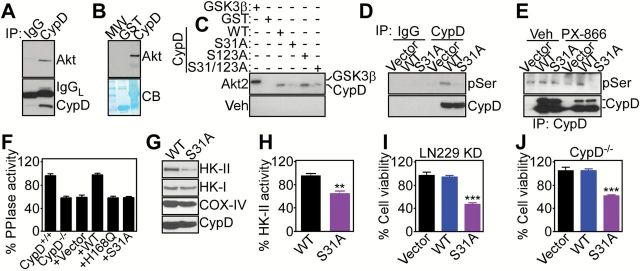
CypD phosphorylation. A) Mitochondrial extracts from LN229 cells were immunoprecipitated (IP) with IgG or an antibody to CypD, and pellets were analyzed by Western blotting. B) Recombinant GST-CypD or GST was incubated with mitochondrial extracts of LN229 cells, and bound proteins were analyzed by western blotting. C) The indicated recombinant proteins were incubated with recombinant active Akt2 or vehicle in a kinase assay, and radioactive proteins were visualized by autoradiography. GSK3β was a control Akt substrate. D) LN229 cells with stable CypD knockdown were reconstituted with the indicated CypD cDNAs, immunoprecipitated with IgG or an antibody to CypD, and pellets were analyzed with an antibody to phosphorylated Ser (pSer) by western blotting. E) LN229 cells with stable shRNA knockdown of CypD were transfected with the indicated FLAG-tagged CypD cDNAs, treated with vehicle or PX-866, and immunoprecipitated with anti-FLAG-M2 gel followed by western blotting with anti-pSer antibody. The position of full-length or mature CypD band is shown. F) CypD+/+ or CypD-/- mouse embryonic fibroblasts (MEFs) were transfected with wild-type or CypD mutant cDNAs and analyzed for peptidyl prolyl cis,trans isomerase (PPIase) activity. PPIase-defective CypD H168Q mutant was used as a control. Mean ± SD. G and H) LN229 cells with stable CypD knockdown were transfected with the indicated CypD cDNAs, and isolated mitochondrial extracts were analyzed by western blotting (G) or HK-II activity (H). Mean ± SD of replicates from a representative experiment out of at least two independent determinations.**P = .001 by two-sided unpaired t test. I and J) LN229 cells with stable shRNA knockdown (KD) of CypD (I) or CypD-/- MEFs (J) were transfected with the indicated CypD cDNAs and analyzed for cell viability by a 3-(4,5 dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay. Mean ± SD of replicates from a representative experiment out of at least two independent determinations. ***P < .001 by two-sided unpaired t test. CB = Coomassie blue staining; IgGL = Ig light chain; IP = immunoprecipitated; MW = molecular weight markers; PPIase = peptidyl prolyl cis,trans isomerase; Veh = vehicle; WT = wild-type.
To assess the status of CypD phosphorylation in vivo, we next reconstituted CypD-/- mouse embryonic fibroblasts (MEFs) (Supplementary Figure 5E, available online) or LN229 cells with stable knockdown of CypD with WT or mutant CypD cDNAs (Supplementary Figure 5F, available online). Treatment with PX-866 did not affect the levels of endogenous or overexpressed CypD in reconstituted cells (Supplementary Figure 5F, available online). Under these conditions, WT CypD immunoprecipitated from reconstituted LN229 cells reacted with an antibody to phosphorylated Ser (Figure 3D). In contrast, immune complexes containing CypD Ser31→Ala mutant did not react with phosphorylated Ser, and immune precipitates with nonbinding IgG were ineffective (Figure 3D). Similarly, WT CypD immunoprecipitated from reconstituted LN229 cells after treatment with PX-866 showed increased reactivity with an antibody to pSer, compared with control transfectants (Figure 3E). In contrast, pSer reactivity of CypD Ser31→Ala mutant was abolished in PX-866–treated cells, and exposure to vehicle did not affect CypD phosphorylation (Figure 3E).
Mitochondrial Tumor Reprogramming
We next studied how CypD phosphorylation by Akt affected mitochondrial functions. First, transfection of WT CypD in CypD-/- MEFs restored CypD peptidyl prolyl cis, trans isomerase (PPIase) activity to the levels of CypD+/+ MEFs (Figure 3F). In contrast, expression of a CypD Ser31→Ala mutant failed to restore PPIase activity in CypD-/- MEFs (Figure 3F). As control, reconstitution of CypD-/- MEFs with a PPIase-defective CypD His168→Gln mutant was also ineffective (Figure 3F). CypD PPIase activity is important for mitochondrial bioenergetics (25), as well as permeability transition-regulated apoptosis (24). Accordingly, expression of CypD Ser31→Ala mutant in CypD-depleted LN229 cells induced loss of the first enzyme of the glycolytic cascade, hexokinase-II (HK-II) (25), from the mitochondrial outer membrane (Figure 3G), resulting in decreased HK-II activity (WT CypD vs S31A CypD, P = .001 by two-sided unpaired t test) (Figure 3H), compared with WT CypD transfectants. The localization of HK-I was not affected (Figure 3G). Consistent with these data, tumor cells expressing CypD Ser31→Ala mutant exhibited defective mitochondrial bioenergetics, with reduced glucose utilization (WT CypD vs S31A CypD, P = .04 by two-sided unpaired t test) (Supplementary Figure 6A, available online), impaired oxygen consumption (WT CypD vs S31A CypD, P = .001 by two-sided unpaired t test) (Supplementary Figure 6B, available online), and decreased ATP production (WT CypD vs S31A CypD, P = .004 by two-sided unpaired t test) (Supplementary Figure 6C, available online), thus mimicking the bioenergetics defects induced by PI3K therapy (Figure 1).
When analyzed for markers of mitochondrial permeability transition (24), reconstitution of CypD-depleted LN229 cells with CypD Ser31→Ala mutant resulted in loss of organelle membrane potential (Supplementary Figure 6D, available online), reactivity for Annexin V (Supplementary Figure 6E, available online), and discharge of cytochrome c in the cytosol, compared with control transfectants (Supplementary Figure 6F, available online). As a result, expression of CypD Ser31→Ala mutant in CypD-depleted LN229 cells (WT CypD vs S31A CypD, P < .001 by two-sided unpaired t test) (Figure 3I) or CypD-/- MEFs (WT CypD vs S31A CypD, P < .001 by two-sided unpaired t test) (Figure 3J) reduced cell viability, whereas expression of WT CypD had no effect (Figure 3, I and J).
Mitochondrial Adaptation and PI3K Therapy
Many mitochondrial functions in tumors, including those mediated by CypD (18), depend on protein folding quality control maintained by mitochondrial-localized Hsp90 chaperones (26). Therefore, we asked whether this requirement provided new therapeutic opportunities, and we carried out a high-throughput drug screening of a small-molecule antagonist of mitochondrial Hsp90s, Gamitrinib (17), in combination with various molecular therapies (Figure 4A). In these experiments, Gamitrinib potently enhanced the anticancer activity of all structurally unrelated PI3K/Akt/MTOR pathway antagonists present in the screening (Figure 4, A and B). In contrast, other molecular therapies were not affected (Figure 4A). The combination of Gamitrinib plus PI3K inhibitor killed tumor cells via induction of mitochondrial apoptosis (24), with membrane depolarization (Figure 4C), increased Annexin V labeling (Figure 4D), and cleavage of effector caspase-3 and -7 and their substrate, poly-ADP ribose polymerase (PARP) (Supplementary Figure 7A, available online). Bliss independence analysis demonstrated that the combination of Gamitrinib plus PI3K inhibitor had synergistic anticancer activity (Supplementary Figure 7B, available online) and was associated with loss of antiapoptotic molecules implicated in tumor cell survival, including Bcl-2, XIAP, and survivin (Supplementary Figure 7C, available online).
Figure 4.

Mitochondrial adaptation and sensitivity to PI3K therapy. A) LN229 cells were incubated with the indicated small molecule targeted anticancer agents plus vehicle (DMSO) or a noncytotoxic concentration of Gamitrinib (Gam, 1 μM) and analyzed for cell viability after 18 hours. The heatmap represents compounds with statistically significant (P < .05) increased inhibitory effect in the presence of Gamitrinib. P = P value (Wilcoxon test); avg = average difference between inhibition shown in Gamitrinib vs DMSO across all concentration points. 2 = results from the combination of targeted compounds plus 2 μM Gamitrinib vs DMSO (only a subset of compounds were tested). B) LN229 cells were incubated with increasing concentrations of small molecule inhibitors of PI3K (GSK458, BKM120, BEZ235, GDC0941, AZD6482) or Akt (MK2206) in the presence of vehicle or Gamitrinib (Gam, 1 μM) and analyzed for inhibition of cell viability by MTT after 18 hours. Mean ± SD of two independent experiments. C and D) GBM U251 cells were treated with NVP-BEZ235 (BEZ, 0.5 μM) or LY294002 (LY, 50 μM), alone or in combination with Gamitrinib (0.5 μM or 5 μM, respectively), and analyzed for changes in mitochondrial membrane potential by JC1 staining (C) or Annexin V labeling (D) by multiparametric flow cytometry. The percentage of cells in each quadrant is indicated. Conc. = concentration; Veh = vehicle.
To validate these observations independently of pharmacologic inhibitors, we next transfected tumor cells with a PI3K Δ-p85 dominant negative mutant that interferes with PI3K signaling (Supplementary Figure 7D, available online). Expression of this mutant in GBM cell types reduced Akt phosphorylation on Ser473, but caused only modest activation of effector caspases (Supplementary Figure 7D, available online). In contrast, the combination of PI3K Δ-p85 mutant plus noncytotoxic concentrations of Gamitrinib increased caspase activation (Supplementary Figure 7D, available online), and enhanced tumor cell killing, compared with each treatment alone (Supplementary Figure 7E, available online). When analyzed in a preclinical model of intracranial GBM in mice, the combination of Gamitrinib plus a PI3K antagonist (NVP-BEZ235) inhibited tumor growth as determined by bioluminescence imaging (vehicle vs BEZ235, P = .08; vehicle vs Gamitrinib, P = .01; vehicle vs BEZ235+Gamitrinib, P = .001; Gamitrinib vs BEZ235+Gamitrinib, P = .04; BEZ235 vs BEZ235+Gamitrinib, P = .02, all by two-sided unpaired t test) (Figure 5, A and B; Supplementary Figure 8A, available online) and extended animal survival (vehicle: median survival = 28.5 days; Gamitrinib+BEZ235: median survival = 40 days, P = .003 by log-rank Mantel-Cox test), compared with single-agent treatment (BEZ235: median survival = 32 days, P = .02; Gamitrinib: median survival = 35 days, P = .008 by log-rank Mantel-Cox test) (Figure 5C). Histologic analysis of GBMs harvested from mice receiving the combination treatment showed extensive inhibition of cell proliferation (Supplementary Figure 8B, available online) and apoptosis (Supplementary Figure 8C, available online), compared with tumors in groups receiving each agent alone.
Figure 5.

Mitochondrial reprogramming and efficacy of PI3K therapy. A and B) Nude mice injected with U87-Luc glioblastoma (GBM) cells in the right cerebral striatum were treated as indicated, and tumor growth was assessed by bioluminescence imaging 28 days after injection (A) and quantified (B). **P = .001; *P = .01-.04 by two-sided unpaired t test. C) Overall survival of mice carrying intracranial GBMs in the various treatment groups. The statistical analysis by log-rank Mantel-Cox test among groups is as follows: Vehicle vs BEZ, P = .02 (χ2 = 4.82); Vehicle vs Gam, P = .008 (χ2 = 6.94); Vehicle vs Gam+BEZ, P = .003 (χ2 = 8.35); BEZ vs Gam+BEZ, P = .008 (χ2 = 7.01); Gam vs Gam+BEZ, P = .003 (χ2 = 4.29). See also Supplementary Table 3 (available online) for the numbers of mice at risk in each group at various time points. D and E) Breast adenocarcinoma organotypic cultures treated with PX-866 (10 μM) alone (left) or in combination with Gamitrinib (10 μM, right) were analyzed by immunohistochemistry and fluorescence microscopy (D), and pAkt- or pMTOR-expressing cells were quantified (E). Cytokeratin was an epithelial marker. DNA was counterstained with DAPI. Scale bar = 100 μm. Mean ± SD of individual replicates. *P = .01; **P = .007 by two-sided unpaired t test. F) Heatmap of proteins statistically significantly different in expression and/or phosphorylation in PC3 cells treated with vehicle (DMSO), PX-866 alone (PX) or PX-866 in combination with Gamitrinib (PX-Gam), as determined by Reverse Phase Protein Arrays. Fold indicates protein expression/phosphorylation signal in a sample vs average DMSO. A-C), internal replicates (n = 3). CK = cytokeratin; HE = hematoxylin & eosin staining; pAkt = Ser473-phosphorylated Akt; PX = PX-866 alone; Veh = vehicle.
Synergistic Enhancement of PI3K Therapy
We next analyzed the impact of this novel combination on therapy adaptive signaling induced by PI3K therapy (9–11). When combined with Gamitrinib, PX-866 no longer promoted the reactivation of Akt (Ser473) (PX 10 μM vs PX 10 μM+Gam, P = .01 by two-sided unpaired t test), or the phosphorylation of MTOR (PX 10 μM vs PX 10 μM+Gam, P = .007 by two-sided unpaired t test) in breast cancer organotypic cultures (Figure 5, D and E; Supplementary Figure 8D, available online). When analyzed in a Reverse Phase Protein Array (RPPA) screening in different tumor cell types, the addition of Gamitrinib reversed many of the adaptive transcriptional and signaling responses induced by PI3K therapy (9–11), affecting Akt/MTOR (4EBP1, S6, RICTOR) and growth factor receptor (EGFR, ErbB2, ErbB3, NRG) signaling, effectors of cell invasion (Snail, Tyro3, Src, PAI1), cell cycle control (CDKN1A, MAPK8, MAPK14), and endogenous tumor suppression (PRKAA1) (Figure 5F; Supplementary Figure 8E, available online).
Discussion
In this study, we have shown that PI3K therapy currently in the clinic is a powerful driver of tumor adaptation, reprogramming mitochondrial functions in bioenergetics and apoptosis to promote cell survival and treatment resistance. This pathway is centered on a pool of Akt2 recruited to mitochondria, and its phosphorylation of the mitochondrial regulator, CypD, on Ser31. Conversely, the combination of PI3K therapy with an antagonist of CypD protein folding currently in preclinical development, Gamitrinib, reverses this adaptive response and delivers potent, synergistic anticancer activity in vivo.
Despite their ability to target a fundamental cancer node (2), small-molecule inhibitors of PI3K/Akt/MTOR have shown modest efficacy in the clinic (6). The data presented here identify the paradoxical reactivation of Akt in response to PI3K therapy (9-11,20), as a pivotal effector of drug resistance to these regimens (8). Centered on the recruitment of Akt2 to mitochondria, this pathway differs from other mechanisms of drug resistance mediated by intratumor heterogeneity (27), acquisition of new mutations (28), or crosstalk within the tumor microenvironment (29).
Once in mitochondria, Akt2 associated with the organelle regulator CypD (30) and phosphorylated CypD on Ser31 to preserve its PPIase activity, maintain energy production, and antagonize apoptosis (24). There is prior evidence that post-translational modifications, for instance acetylation (31), affect CypD activity. Here, Ser31 is positioned at the NH2 terminus of the mature form of CypD and becomes readily phosphorylated by Akt in vitro and in vivo. However, the complete Akt consensus phosphorylation site for Ser31 extends into the mitochondrial-import sequence, and it is possible that a fraction of CypD is phosphorylated on Ser31 during mitochondrial trafficking. Akt plays a central role in tumor bioenergetics (32), influencing aerobic glycolysis (33), as well as oxidative phosphorylation (34). An antiapoptotic role of Akt2 phosphorylation of CypD is also consistent with a physical assembly of CypD in a mitochondrial permeability transition pore (24) that regulates stress-associated cell death (30).
The functions of CypD in bioenergetics (25) and apoptosis (24) require protein folding quality control maintained by mitochondrial-localized Hsp90s (26). Accordingly, the combination of a small-molecule inhibitor of mitochondrial-localized Hsp90s currently in preclinical development, Gamitrinib (17), converted a transient, cytostatic effect of PI3K antagonists into potent, synergistic anticancer activity in vivo. The idea of targeting mitochondrial integrity for cancer therapy (19) has recently gained attention (35), and regulators of Bcl-2 proteins (36), oxidative phosphorylation (37), and redox mechanisms (38) have progressed through (pre)clinical development. Gamitrinib is an attractive candidate for this approach given its ability to simultaneously disable multiple pathways of mitochondrial homeostasis in bioenergetics, gene expression, and redox balance selectively in tumors (18).
In addition, the combination with Gamitrinib reversed adaptive tumor reprogramming induced by PI3K therapy, with respect to Akt (re)activation, growth factor receptor signaling, cell proliferation, and endogenous tumor suppression. Small-molecule inhibitors of PI3K (10,20), Akt (11), or MTOR (9) have been shown to activate a broad gene expression program in tumor cells, potentially as a compensatory response via derepression of FOXO-dependent transcription. Our RPPA screening suggests that mitochondrial reprogramming maintained by organelle Hsp90s is important for this response, potentially via mitochondria-to-nuclei “retrograde” signaling (39). Accordingly, mitochondria-derived “retrograde” mediators that affect nuclear gene expression have been identified in model systems (40), and CypD contributes to retrograde signaling via activation of STAT3-dependent cell migration and invasion (41).
One limitation of our study is that the increased efficacy of PI3K therapy when combined with Gamitrinib was limited to mechanistic and preclinical readouts and not patient data. In addition, Gamitrinib—or other agents with comparable activity or specificity—is not yet available for clinical testing, as it is currently completing late stages for preclinical and safety evaluation.
In summary, Akt2-directed repurposing of mitochondrial functions provides a novel adaptive mechanism of tumor resistance to PI3K therapy. This pathway likely limits the activity of these agents in the clinic (8), but may confer a unique “addiction” of tumor cells to mitochondrial adaptation In this context, the combination of small-molecule PI3K antagonists plus inhibitors of mitochondrial homeostasis (17,26), like Gamitrinib, may eliminate mitochondrial adaptation and dramatically improve on the efficacy of PI3K therapy in the clinic.
Funding
This work was supported by National Institutes of Health grants P01 CA140043 (DCA, LRL), R01 CA78810 (DCA), F32 CA177018 (MCC), R01 CA089720 (LRL), K08 NS083732 (MDS), American Association for Cancer Research-National Brain Tumor Society Career Development Award for Translational Brain Tumor Research (MDS), American Brain Tumor Association Translational Grant (MDS), and the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No. W81XWH-13-1-0193 (DCA). Support for Core Facilities utilized in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Supplementary Material
The study sponsors had no role in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
We thank Dr. Herlyn (The Wistar Institute) for melanoma cell lines, and the MD Anderson Cancer Center Reverse Phase Protein Array (RPPA) Facility for high-throughput analysis.
The authors declare no competing financial interest RDM is an employee of Metabolon, Inc. Correspondence and requests for materials should be addressed to DCA (daltieri@wistar.org).
References
- 1. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. [DOI] [PubMed] [Google Scholar]
- 2. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. [DOI] [PubMed] [Google Scholar]
- 4. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. [DOI] [PubMed] [Google Scholar]
- 5. Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodon J, Dienstmann R, Serra V, et al. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–153. [DOI] [PubMed] [Google Scholar]
- 7. Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8(9):709–723. [DOI] [PubMed] [Google Scholar]
- 8. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chakrabarty A, Sanchez V, Kuba MG, et al. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2012;109(8):2718–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9(9):665–674. [DOI] [PubMed] [Google Scholar]
- 13. Fodale V, Pierobon M, Liotta L, et al. Mechanism of cell adaptation: when and how do cancer cells develop chemoresistance? Cancer J. 2011;17(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balch WE, Morimoto RI, Dillin A, et al. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. [DOI] [PubMed] [Google Scholar]
- 15. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. [DOI] [PubMed] [Google Scholar]
- 16. Vaira V, Fedele G, Pyne S, et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc Natl Acad Sci U S A. 2010;107(18):8352–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chae YC, Caino MC, Lisanti S, et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell. 2012;22(3):331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chae YC, Angelin A, Lisanti S, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun. 2013;4:2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Serra V, Scaltriti M, Prudkin L, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30(22):2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res. 2010;88(1):7–15. [DOI] [PubMed] [Google Scholar]
- 22. Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol. 2010;298(3):C580–C591. [DOI] [PubMed] [Google Scholar]
- 23. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. [DOI] [PubMed] [Google Scholar]
- 24. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. [DOI] [PubMed] [Google Scholar]
- 25. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang BH, Plescia J, Dohi T, et al. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131(2):257–270. [DOI] [PubMed] [Google Scholar]
- 27. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13(11):795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009. [DOI] [PubMed] [Google Scholar]
- 30. Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. [DOI] [PubMed] [Google Scholar]
- 31. Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J Cell Sci. 2010;123(Pt 6):894–902. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Garcia-Cao I, Song MS, Hobbs RM, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149(1):49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. [DOI] [PubMed] [Google Scholar]
- 34. Li C, Li Y, He L, et al. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med. 2013;60:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fulda S, Kroemer G. Mitochondria as therapeutic targets for the treatment of malignant disease. Antioxid Redox Signal. 2011;15(12):2937–2949. [DOI] [PubMed] [Google Scholar]
- 36. Czabotar PE, Lessene G, Strasser A, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. [DOI] [PubMed] [Google Scholar]
- 37. Kluckova K, Bezawork-Geleta A, Rohlena J, et al. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim Biophys Acta. 2013;1827(5):552–564. [DOI] [PubMed] [Google Scholar]
- 38. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. [DOI] [PubMed] [Google Scholar]
- 39. Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14(1):1–15. [DOI] [PubMed] [Google Scholar]
- 40. Haynes CM, Yang Y, Blais SP, et al. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37(4):529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tavecchio M, Lisanti S, Lam A, et al. Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. J Biol Chem. 2013;288(8):5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.