

Abstract
Vacuolar protein sorting-35 (VPS35) is a retromer component for endosomal trafficking. Mutations of VPS35 have been linked to familial Parkinson’s disease (PD). Here we showed that specific deletion of the VPS35 gene in DA neurons resulted in PD-like deficits including loss of DA neurons and accumulation of α-synuclein. Intriguingly, mitochondria became fragmented and mal-functional in VPS35-deficient DA neurons, phenotypes that could be restored by expressing VPS35 wild type, but not PD-linked mutant. Concomitantly, VPS35 deficiency or mutation increased mitochondrial E3 ubiquitin ligase-1 (MUL1) and thus led to mitofusin-2 (MFN2) degradation and mitochondrial fragmentation. Suppression of MUL1 expression ameliorated MFN2-reduction and DA neuron-loss, but not α-synuclein-accumulation. These results provide a cellular mechanism for VPS35-dysfunction in mitochondrial impairment and PD pathogenesis.
Keywords: VPS35, retromer, MFN2, MUL1, mitochondrial fusion/fission, dopamine neurons
Graphical Abstract
INTRODUCTION
VPS35 (vacuolar protein sorting-35, also called Park17) is a key component of the cargo-selective retromer complex that consists of VPS35, VPS29 and VPS26. This retromer complex sorts cargos into tubules for retrieval to the Golgi apparatus or to the cell surface (Seaman, 2012). Prominent retromer cargos include: the cation independent mannose 6-phosphate receptor (CI-MPR) (Seaman, 2004), the iron transporter DMT1-II (Tabuchi et al., 2010), the Wnt transport protein Wntless (Eaton, 2008), and SorLA (sortilin-related receptor) (Nielsen et al., 2007). Dysfunction of VPS35/retromer is a risk factor for both Alzheimer’s disease (AD) and Parkinson’s disease (PD) pathogenesis (Muhammad et al., 2008; Small et al., 2005; Small and Petsko, 2015). VPS35 is reduced in the hippocampus of late-onset AD patients (Small et al., 2005). VPps35-haploinsufficiency enhances AD-like neuropathology in the Tg2576 mouse model of AD (Wen et al., 2011). Suppressing VPS35 expression in neonatal hippocampal neurons results in “degenerative-like” morphology including abnormal dendritic spines and swollen axons (Wang et al., 2012). Intriguingly, VPS35 mutations have been identified in autosomal dominant PD patients (Vilarino-Guell et al., 2011; Zimprich et al., 2011), and VPS35 is also reduced in the substantia nigra of PD patients (MacLeod et al., 2013). However, it remains unclear whether and how VPS35-deficiency or mutation contributes to PD pathogenesis.
Dysfunctional mitochondrial fusion/fission dynamics are believed to be a culprit for neurodegenerative disorders, including AD and PD (Knott et al., 2008). Interestingly, several genes identified in PD patients encode proteins important for mitochondrial fusion/fission dynamics, which include mutations in α-synuclein (also called Park1) and leucine rich repeat kinase 2 (Lrrk2) (Park8) that cause autosomal dominant PD, and mutations in parkin (Park2), DJ-1 (Park7), Pink1 (Park6), and ATP13A2 that have been linked to autosomal recessive PD (Gasser, 2007; Lee et al., 2012b; Martin et al., 2011; Savitt et al., 2006; von Coelln et al., 2006). Whereas these studies have pointed to the impaired mitochondrial fusion/fission dynamics as a pathophysiological mechanism for both familial and sporadic PD, the underlying molecular mechanisms of mitochondrial fusion/fission, and the relationship between dynfunctional mitochondrial fusion/fission and PD pathogenesis remain to be further explored.
The fusion/fission dynamics of mitochondria are critical not only for controlling mitochondrial shape, size, and number, but also for regulating mitochondrial functions (such as respiration and cell death/survival), clearance (e.g., mitophage), and distribution. Fission facilitates the distribution of mitochondria in response to local demand for ATP, whereas fusion helps to replace damaged mitochondrial DNA (mtDNA) (Chan, 2006b; Karbowski and Youle, 2003). The fusion/fission dynamics are controlled by opposing actions of different dynamin-family members such as dynamin-related protein 1 (Drp1) and mitofusins (Mfns) (Chan, 2006a; Itoh et al., 2013; Okamoto and Shaw, 2005). Fission requires Drp1 and its targeting to mitochondria via outer membrane binding partners including mitochondrial fission factor (Mff) (Loson et al., 2013). The activity of Drp1 is regulated by post-translational modifications including phosphorylation, SUMOylation, and ubiquitination (Westermann, 2010). On the other hand, fusion requires mitochondrial outer membrane proteins Mfns, including Mfn1 and MFN2. Both Mfns are subjected to ubiquitination and proteasomal degradation, and MFN2 is highly expressed in the brain and implicated in Charcot-Marie-Tooth Type 2A, an axonal degenerative disorder (Zuchner et al., 2004).
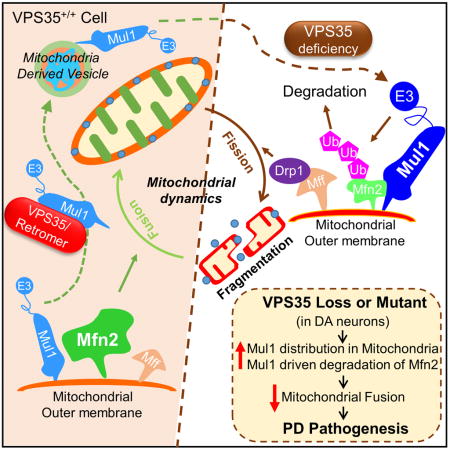
In this paper, we provide evidence linking VPS35-deficiency and mutation to mitochondrial impairments and DA neuronal loss. VPS35-deficiency or mutation impairs mitochondrial fusion and function in cultured DA neurons, neuroblastoma cell lines (SH-SY5Y and NLT), and in the ventral midbrain. Additionally, we show that VPS35 regulates mitochondrial fusion by promoting MUL1 (a mitochondrial associated E3 ubiquitin ligase) degradation and increasing MFN2 protein stability. Suppression of MUL1 expression in VPS35-deficient DA neurons in culture and in the ventral midbrain restored the PD-relevant deficits, including MFN2-decrease, mitochondrial fragmentation, and DA neuronal loss, but not the increase of α-synuclein. Taken together, our results suggest that VPS35 is a critical regulator for MUL1 trafficking and degradation, thus increasing MFN2 and promoting MFN2-mediated mitochondrial fusion and DA neuron survival. Dysregulation of VPS35/retromer (due to deficiency or VPS35 mutation) impairs MUL1 degradation and mitochondrial fusion, which may underlie PD pathogenesis.
RESULTS
PD-relevant pathology in VPS35+/− mice and in mice with specific depletion of VPS35 in DA neurons
Null allele VPS35 mutant mice (VPS35−/−) die early during embryonic development (Wen et al., 2011). To address whether VPS35-deficiency contributes to PD pathogenesis, we used two other mouse models, VPS35 heterozygotes (VPS35+/−) and a newly generated mouse model with specific deletion of VPS35 gene in DA neurons (VPS35DAT-Cre). The VPS35+/− mice survived to adult age, without a defect in life span (Wen et al., 2011). Before age of 6 months (M) old, the PD-relevant deficits, such as loss of DA neurons and increase of α-synuclein, were not obviously detected in VPS35+/− mice (Tang et al., 2015). However, at age of 12-M old, ~20% of DA neuron loss was detected in the SNpc of VPS35+/− brain, compared with that of VPS35+/+ controls, and VPS35+/− mice showed an increase in α-synuclein levels in the VM too (Tang et al., 2015). These results demonstrate that VPS35+/− mice display an age-dependent DA neuron-loss and accumulation of α-synuclein, both pathological features of PD, suggesting that VPS35-haploinsufficiency promotes PD-pathogenesis.
VPS35 is expressed in multiple cell types in the brain, including DA neurons (MacLeod et al., 2013) and glial cells (data not shown). To address if VPS35 expression in DA neurons is essential for prevention of PD-like deficits, we generated VPS35DAT-Cre mice by crossing floxed VPS35 (VPS35f/f) with DAT-Cre mice (Figs. S1A–S1C), where Cre recombinase is under the control of the promoter of DA transporter gene (DAT) and specifically expressed in DA neurons (Fig. S1C) (Zhuang et al., 2005). Thus, the VPS35 protein was specifically depleted in SNpc-DA neurons (Fig. S1D). The VPS35DAT-Cre mice survived to adult age (up to 6-M of age examined) with a slight reduction in body weight and size (Fig. S1E). Remarkably, at young adult age (2–3-M old), VPS35DAT-Cre mice showed a marked reduction in TH+ DA neurons at the SNpc, and a decrease in TH+ fibers in STR, compared with that of littermate controls (VPS35f/f) (Figs. 1A–1D). The neuronal loss in SNpc of VPS35DAT-Cre mice was confirmed by NeuN staining analysis (Figs. 1A–1B). The reduction of TH proteins was further verified by Western blot analysis (Figs. 1E–1F). In addition, increased α-synuclein levels were also detected in the VM/SNpc-DA neurons of VPS35DAT-Cre mice (Figs. 1E–1H). Furthermore, we examined if VPS35DAT-Cre mice show PD-relevant motor deficit. In the automatic gait test, VPS35DAT-Cre mice displayed significant unsteady and weaker gaits (Fig. 1I). VPS35DAT-Cre mice also exhibited a decline in hindlimb stepping (rearing behavior) in the cylinder observational test (Fig. 1J), and spent more time to descend the pole by the Vertical pole test (Fig. 1K). In the open- eld test, VPS35DAT-Cre mice showed a signi cant reduction in the overall locomotor activity, including shorter total distance travelled and increased immobile time (Figs. 1L). These results indicate that VPS35DAT-Cre mice exhibite reduced locomotor activity.
Figure 1. PD-relevant neuropathology in VPS35DAT-Cre mice.

(A–B) Loss of TH+-DA neurons in 2~3 M old VPS35DAT-Cre SNpc. Shown in (A) were representative images of midbrain coronal sections of VPS35DAT-Cre and f/f littermates. Scale bars: 200 μm. B, Data were quantified by stereological analysis total numbers of TH+ or NeuN+ cells in SNpc and shown as mean ± SEM (n = 4; *p < 0.05). (C–D) Immunostaining analysis showing reduced TH+ neuronal processes in 2~3-M old VPS35DAT-Cre STR. C, Representative images of the whole STR; Scale bars: 400 μm. Inset: close up views of neuronal processes; Scale bars: 10 μm. D, Quantification of TH optical density (mean ± SEM; n = 4; *p < 0.05). (E–F) Western blot analysis of TH and α-synuclein (α-syn) levels from ventral midbrain (VM) of 2~3-M old VPS35DAT-Cre and f/f mice. Data were quantified and presented in F as mean ± SEM. n = 4. *p < 0.05. (G–H) Accumulation of α-synuclein in VPS35DAT-Cre SNpc-DA neurons. Midbrain coronal sections of indicated mice were immunostained with α-synuclein (red) and TH (Green) antibodies. Representative images were shown in G. Scale bar, 10 μm. Quantitative data of α-synuclein somatic accumulation in TH+ neurons (%, over total TH+ neurons) in SNpc were presented in H (mean ± SEM; n = 4. *p < 0.01). (I~L) Behavioral tests on VPS35DAT-Cre mice and control littermates were as follow: (I) Grip strength. Forelimb grip strength was determined by a digital weight gauge; (J) Cylinder observational test. Rearing frequency were record; (K) Pole test. Each mouse was placed head upwards on a round vertical pole and performance were determined by the latency(s) of the mouse to turn downwards and completely descend the pole. (L) Open-field tests. Total distance travelled and immobile time in open-field test were quantified in E. For I-L, n=4~6, Data were presented as mean ± SEM. *, p < 0.05.
Taken together, these results demonstrate that VPS35DAT-Cre mice exhibit earlier onset of PD-relevant pathological features than that of VPS35+/− mice, revealing the necessity of VPS35 expression in DA neurons for prevention of PD pathogenesis.
Reduced mitochondrial MFN2 in VPS35-deficient DA neurons
To understand how VPS35 in DA neurons prevents PD pathogenesis, we examined whether VPS35-deficiency altered levels of proteins critical for endosomes (EEA1), Golgi (GM130), lysosomes (LAMP1), mitochondrial fission/fusion (Drp1 and MFN2), mitochondria (Tom20), and proteins implicated in PD pathogenesis such as Lrrk2, Pink1, and Parkin. Homogenates prepared from various brain regions including VM, hippocampus (HIP), and STR of control (VPS35f/f) and VPS35DAT-Cre mice were subjected to Western blotting analyses. As shown in Figs. 2A–2B, no obvious change was observed for EEA1, GM130, LAMP1, Lrrk2, Pink1, Parkin, Tom20, and Drp1. However, MFN2 was significantly reduced in the VM, but not HIP or STR region, of VPS35DAT-Cre mice (Figs. 2A–2B). MFN2 is a GTPase embedded in the outer membrane of the mitochondria (Chen et al., 2003). We thus examined if mitochondrial MFN2 is reduced in VPS35-deficient brain. Mitochondrial fractions were purified from the VM region of control and VPS35+/− mice at different ages and subjected to Western blotting. In consistent, MFN2 was reduced in VPS35+/− VM mitochondria, whereas no obvious change was observed for Lrrk2, Pink1, Parkin, ATP5a, Tom20, and Drp1, even in aged mutant mice (Figs. 2C–2D). We next examined if mitochondrial MFN2 is reduced in VPS35-deficient neurons in culture. Indeed, a marked MFN2 reduction in Mito-RFP labeled mitochondria was detected in neurons expressing miRNA-VPS35, which reduced VPS35 by >80% (Wang et al., 2012) (Figs. 2E–2F). MFN2-reduction was also confirmed by Western blot analysis of lysates of cultured DA-neurons from VPS35DAT-Cre mice (Figs. 2G–2H). A similar MFN2-reduction was detected in shRNA-VPS35 expressing SH-SY5Y and NLT cells by both Western blot and immunostaining analyses (Fig. S2). Taken together, these results suggest that VPS35-deficiency in DA neurons as well as neuronal cell lines reduced mitochondrial MFN2, identifying a potential molecular target.
Figure 2. Reduced mitochondrial MFN2 in Vps35-deficient DA neurons.

(A–B) Reduced MFN2 in Vps35DAT-Cre VM (ventral midbrain), but not HIP (hippocampus) or STR (striatum). Soluble extracts from HIP, VM and STR of indicated mice were subjected to Western bot analysis. A, Representative blots; B, Quantification of protein levels (compared with Vps35f/f controls). Data presented were mean ± SEM (n = 3 mice/genotype); *p < 0.05. (C–D) Reduced mitochondrial MFN2 in aged Vps35+/− VM. Mitochondrial fractions of indicted mice were subjected to Western bot analysis. C, Representative blots; D, Quantification of protein levels (compared with Vps35+/+ controls); Data presented were mean ± SEM (n = 3 mice/genotype); *p < 0.05. (E–F) Reduced mitochondrial MFN2 in miRNA-Vps35 expressing neurons. E, Representative images of transfected neurons; F, Quantitative data (normalized by control miR) (mean ± SEM; n = 5 neurons; *p < 0.05). (G–H) Reduced MFN2 levels in Vps35DAT-Cre primary DA neurons. G, Representative blots; H, Quantification of protein levels (normalized with Vps35f/f controls). Data presented were mean ± SEM (n = 3 mice/genotype); *p < 0.05.
Mitochondrial fragmentation in VPS35DAT-Cre DA neurons and neurons expressing PD-linked VPS35 mutant
MFN2 is essential for mitochondrial fusion (Chen et al., 2003), which is frequently altered by PD-linked genetic mutations (Haelterman et al., 2014). We thus examined mitochondrial morphology in VPS35-deficient DA neurons. Overall, VPS35DAT-Cre DA neurons (TH+) in culture showed no obvious defect in neuronal processes (revealed by TH and Tuj1 staining) (Fig. 3A and data not shown). However, mitochondria of VPS35-depleted DA neurons (revealed by Tom20, a marker for mitochondrial outer membrane) were shorter and rounder than those in control neurons (Figs. 3A–3B), suggesting mitochondrial fragmentation. Importantly, the mitochondrial fragmentation in VPS35-depleted neurons was mitigated by expressing GFP-fused VPS35-WT (wild type), but not VPS35-D620N, the PD-linked VPS35 mutant (Figs. 3C–3D). These results thus reveal a cell-autonomous function of VPS35 in preventing mitochondrial fragmentation in neurons, and provide a possible pathophysiological mechanism of the PD-linked VPS35-D620N mutation.
Figure 3. Mitochondrial fragmentation in VPS35DAT-Cre DA neurons.

(A–B) Increased mitochondrial fragmentation in primary DA/TH+ neurons (DIV7) from VPS35DAT-Cre midbrain, compared to VPS35f/f controls. Midbrain neurons of E18.5 mice were cultured for 7 days (DIV7). Mitochondria were labeled by Tom20 staining. A, Representative images and close up views of mitochondria in neurites; A′, Close up views of mitochondria in neuron soma region; Scale bars: 10 μm. B, Quantifications of mitochondrial length and aspect ratio (mitochondrial major axis over minor axis). Shown were grouped column scatter (left, with mean indicated by the line) and as aspect ratio (right). n=400 mitochondria from 20 different neurons of each group. *p < 0.05. (C–D) Reduced mitochondrial fragmentation in VPS35DAT-Cre DA neurons expressing VPS35-WT, but not VPS35-D620N. Primary VPS35DAT-Cre DA neurons were transfected with VPS35-WT-GFP or VPS35-D620N-GFP (GFP was fused with the VPS35-C-terminus) at DIV6 and analyzed for mitochondrial morphology at DIV10. C, Representative images; Scale bars: 10 μm. D, Quantitation of mitochondrial. n = 200 mitochondria from 10 different neurons; *p < 0.01.
Notice that mitochondrial fragmentation was also observed in NLT and SH-SY5Y neuronal cell lines that were transfected with miRNA-VPS35 (Figs. S3A–D) or shRNA-VPS35 (Figs. S3E–G). These mitochondrial fragmentation was restored by expressing shRNA-resistant, but not shRNA-sensitive, VPS35 in shRNA-VPS35 expressing NLT cells (Figs. S3E–G). These results demonstrate that VPS35 deficiency or mutation results in mitochondrial fragmentation in DA neurons as well as neuronal cell lines.
Impaired mitochondrial function in VPS35-deficient DA neurons
Mitochondrial fusion morphology is believed to be critical for maintenance of mitochondrial respiratory capacity in mammalian cells (Westermann, 2012). De cits in mitochondrial respiration are critically associated with DA neuronal dysfunction in PD (Itoh et al., 2013). We thus examined mitochondrial respiratory capacity in VPS35-deficient DA neurons by use of the Seahorse platform to measure real-time oxygen (O2) consumption rates (OCR). The basal oxygen consumption rate (BR) was slightly higher in DA neurons from VPS35DAT-Cre mice compared with controls, whereas no difference in oxygen consumption was observed following treatment of neurons with oligomycin (an inhibitor of ATP synthase), which serves as a negative control (Figs. 4A–4B). However, the maximal respiratory rates (MR) induced by mitochondrial un-coupler (Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP)) treatment was markedly reduced in VPS35-depleted DA neurons (Figs. 4A–4B), which resulted in a de ciency in spare respiratory capacity (SR) (Fig. 4C). A similar mitochondrial MR deficit was also detected in VPS35+/− DA neurons (data not shown). These results thus indicate dysfunctional mitochondria in cultured VPS35-deficient DA neurons.
Figure 4. Impaired mitochonchrial function in VPS35DAT-Cre DA neurons and VPS35-deficient NLT cells.

(A–B) Real-time O2 consumption rate (OCR) was measured in DIV7 primary DA neurons by a Seahorse XF96 analyzer. For validation of the measurement, we used the ATP synthase inhibitor oligomycin (2 uM) after recording basal line, followed by the treatments of the pharmaceutical uncoupler FCCP (3 uM) and the Complex I inhibitor rotenone (1uM). Representative trace of OCR are indicated in A, each time point representing the mean ± SEM, and averaged OCR profile from at least 4 animals are showed in B. *p < 0.05. (C) The spare respiratory capacity changes (compared with BR) are showed in C with bar graph. *p < 0.01. (D–F) Decreased mitochondrial membrane potential in miRNA-VPS35 expressing NLT cells. D, Representative TMRM time-lapse images of transfected NLT cells, without or with 20 uM CCCP. Scale bar, 10 μm. E, Quantitative data. Arrow indicates CCCP treatment. F, ΔΨm in NLT cells expressing control or miR-VPS35 (mean ± SEM; n = 50 cells; *p < 0.01).
Mitochondrial membrane potential (Ψm) is another indicator of mitochondrial health and function (Chen, 1988). We then asked if VPS35-deficiency alter mitochondrial membrane potential by using the potentiometric fluorescent dye, tetramethylrhodamine methylester (TMRM), as its fluorescent intensity reflects the mitochondrial membrane potential. NLT, a GnRH neuronblastoma cell line, was used, because VPS35 is highly expressed in NLT cells (Wen et al., 2011) and the spread cell morphology in NLT cells provides a technical advantage for this assay. As shown in Figs. 4D–4E, TMRM accumulated in control NLT cells and the fluorescence was decreased by treatment with CCCP (carbonyl cyanide m-chlorophenyl hydrazine), a proton ionophore that depolarizes mitochondria, indicating the specificity of TMRM labeling. However, TMRM signal was reduced in miRNA-VPS35 expressing cells which agrees with the increased respiratory rate in OCR test (Figs. 4D–4F). Taken together, these results suggest that mitochondrial dysfunction occurs in VPS35-deficient DA neurons as well as NLT cells.
In vivo mitochondrial deficit in VPS35-deficient midbrain
To determine whether mitochondrial structures were altered in VPS35-deficient brain, we carried out transmission electron microscopy (TEM) analysis. As shown in Figs. S4A–S4D, mitochondria were smaller in VPS35+/− SNpcs in 4-M old mice. The deficit was un-detectable in STR of the mutant mice, but became prominent in 12-M old mutant mice (Figs. S4B, S4C, and S4E). The STR mitochondria in 12-M old mutant mice were condensed and damaged (Fig. S4F). These results thus indicate that mitochondria appear to be fragmented and abnormal in morphology in aged VPS35+/− midbrains and STRs, which may underlie PD-relevant neuropathology.
To access if mitochondria in VPS35+/− brain are mal-functional, we measured mitochondrial DNAs (mtDNAs), which are largely dependent on fusion and have been used as an indicator of mitochondrial function (Chan, 2006a). The ratio of mtDNAs over nuclear DNAs (ncDNAs) in VM (ventral midbrain) and STR (striatum) was similar between VPS35+/+ and +/− mice at 3-M of age (Fig. S4G). However, the ratio was markedly decreased in 12-M old VPS35+/− VM and STR, compared with that of WT controls (Fig. S4G, bottom panel). This effect was not observed in the hippocampus, indicating regional specificity. These results provide additional evidence for age-dependent and brain region-specific mitochondrial mal-function.
MFN2 protein reduction associated with increased ubiquitination
We then investigated the underlying molecular mechanisms for MFN2 reduction in VPS35-deficient brain/neurons. The MFN2 reduction was not caused by a problem in transcription, because a comparable level of MFN2 mRNA was detected between control and mutant mice (Fig. S2E). MFN2 can be degraded by the ubiquitin-proteasome-system (UPS) (Tanaka et al., 2010). We thus speculated that the MFN2-reduction might be due to MFN2 degradation by the UPS. This notion was supported by the observation that MG132, a proteasome inhibitor, restored MFN2 levels in VPS35-deficient DA neurons (Figs. S5A–5B) or SH-SY5Y cells (Figs. S5C–S5D). Consistently, MFN2-ubiquitination was increased in VPS35-deficient SH-SY5Y cells or cells expressing VPS35 mutant (D620N) (Figs. S5E–S5F). Interestingly, expression of shRNA-resistant VPS35-WT, but not VPS35-D620N, also restored MFN2 levels in VPS35-deficient SH-SY5Y cells (Figs. S5C–S5D). These results suggest that VPS35-deficiency or mutation (D620N) reduces MFN2 protein likely by UPS-mediated MFN2-degradation.
Increase of mitochondrial E3 ubiquitin ligase MUL1
A previous proteomic study suggests that MUL1 (also known as MAPL for mitochondrial-anchored protein ligase) interacts with VPS35/retromer (Braschi et al., 2010). Recently, MUL1, a mitochondrial outer membrane protein, was found to be a critical E3 ubiquitin ligase of MFN2 (Lokireddy et al., 2012). To determine if MUL1 was involved in VPS35 regulation of MFN2, we examined MUL1 protein levels in mitochondrial fractions of different brain regions of control and mutant mice (at 4-M old of age). MUL1 levels were selectively elevated in mitochondrial fractions of VPS35+/− VMs and STRs, but not hippocampus, corroborating with MFN2-reduction (Figs. 5A–5B). In VPS35-deficient SH-SY5Y cells, MUL1 increase was also detectable, which was associated with a decrease of MFN2 (Figs. 5C–5D), revealing a reciprocal relationship between MUL1 and MFN2 that suggests MFN2 as a MUL1 substrate. Additionally, the MUL1 increase was mitigated by co-expressing shRNA-resistant VPS35-WT, but not VPS35-D620N (Figs. 5C–5D). Finally, expression of this and other PD-associated mutants (P316S or R524W) was sufficient to increase MUL1 and reduce MFN2 (Figs. 5E–5F). These results suggest that VPS35 regulates MUL1 protein level, which may be involved in VPS35-deficiency-induced MFN2-reduction.
Figure 5. Increased MUL1 in VPS35+/− midbrains and in SH-SY5Y cells expressing shRNA-VPS35 or PD-linked VPS35 mutants.

(A–B) Increased mitochondrial-MUL1 protein in 4-M old VPS35+/− VM and STR, but not Hip. Mitochondrial fractions were subjected to Western blotting with indicated antibodies. A, Representative blots; B, Quantification of data (normalized by VPS35+/+ control). (C–D) Association of increased MUL1 with decreased MFN2 in shRNA-VPS35-expressing SH-SY5Y cells, which was restored by expressing shRNA-resistant VPS35-WT, but not VPS35-D620N. C, Representative blots; D, Quantification of MUL1 and MFN2 protein (normalized by Mock control). (E–F) Association of increased MUL1 with decreased MFN2 in SH-SY5Y cells expressing PD-linked VPS35 mutants. E, Representative blots; F, Quantification of MUL1 and MFN2 protein levels (normalized by VPS35-WT control). Data in B, D, and F are presented as mean ± SEM (n = 3; *p < 0.05).
To understand how VPS35 regulates MUL1 protein level, we measured the half-life of MUL1 in control and shRNA-VPS35 expressing SH-SY5Y cells. The half-life of MUL1 in control cells was about 4 hours (Figs. 6A–6B). However, it dramatically increased in shRNA-VPS35-expressing cells, with no detectable reduction within 8 hours of experiments (Figs. 6A–6B). These results indicate that VPS35 is necessary for MUL1-degradation. To determine if MUL1 distribution in mitochondria was altered in VPS35-deficient cells, we stained for MUL1 in miRNA-VPS35 expressing NLT cells and VPS35+/− neurons. As shown in Figs. 6C–6E, exogenous MUL1 (GFP-MUL1) and endogenous MUL1 in mitochondria were markedly increased in VPS35 deficient NLT and neurons, compared to those in control cells. The increase of mitochondrial-MUL1 in VPS35+/− neurons was also ameliorated by expressing VPS35-WT, but not VPS35-D620N (Figs. 6F–6G). These results were in agreement with increased MUL1 in mitochondrial fractions of VPS35+/− VMs (Figs. 5A–5B). These results led us to propose that VPS35 promotes the degradation of MUL1 to reduce MUL1 level in mitochondria, which may be critical for maintaining MFN2 protein level for mitochondrial fusion.
Figure 6. Impaired MUL1-degradation and increased MUL1-distribution in mitochondria in VPS35-deficient cells.

(A–B) MUL1 protein degradation was inhibited by shRNA-VPS35. SH-SY5Y cells were transfected with control and shRNA-VPS35 (at ~80% efficiency). Transfected cells were incubated with CHX (100 mg/ml) for indicated times. Cell lysates were subjected to Western blotting using indicated antibodies. A, Representative blots; B, Quantification of MUL1 levels (normalized with time 0). (C–E) MUL1 distribution in mitochondria was increased in miRNA-VPS35 expressing NLT cells. GFP-MUL1 (C) and endogenous MUL1 (D) were increased in mito-RFP-labeled mitochondria of miRNA-VPS35 expressing cells. C and D, Representative images. Scale bar: 5 μm. MUL1 colocalization with mitochondria (Mito-RFP) was presented in E as mean ± SEM, n = 3, each with 20 cells; *p < 0.05. (F–G) MUL1 distribution in mitochondria was increased in VPS35+/− neurons, which was reduced by GFP-VPS35-WT, but not GFP-VPS35-D620N. F, Representative images. Scale bar: 10 μm. G, Quantification of MUL1 co-localization with mitochondria (mean ± SEM; n = 3, each with 20 cells; *p < 0.05).
Requirement of MUL1 for MFN2-reduction, mitochondrial fragmentation, and DA neuron loss in VPS35-deficient cells or midbrain
To investigate if MUL1 is involved in MFN2-reduction in VPS35-deficient cells, we generated shRNA-MUL1 (shR-MUL1) which specifically and efficiently suppressed MUL1 expression (Figs. S6A–S6C). As shown in Figs. S6A–S6B, co-expressing shR-MUL1 prevented miRNA-VPS35 from reducing MFN2 in SH-SY5Y cells. Moreover, shR-MUL1 also prevented VPS35 deficiency from causing mitochondrial fragmentation in either miR-VPS35-expressing NLT cells (Figs. S6D–S6E) or neurons (Figs. S6F–S6G). These results demonstrate that MFN2-reduction and mitochondrial fragmentation by VPS35-deficiency require MUL1, providing a molecular mechanism by which VPS35-deficiency causes MFN2 degradation and mitochondrial fragmentation.
We further tested this view by determining if MUL1 causes MFN2-reduction in VPS35-deificent DA neurons in vivo. To this end, adenovirus (serotype 5)-encoding shRNA against mouse MUL1 (AAV-shR-MUL1-GFP) was generated, which was injected into VPS35+/+ and +/− SNpc (at 6-M old of age) (Fig. 7A). 2-M post virus injection, GFP expression throughout the injected SNpc was observed (Fig. 7B), and >80% of TH-positive neurons expressed GFP (Fig. 7B insets). As DA neurons of the SNpc project to the STR to create the nigral-striatal pathway, the GFP signal in the ipsilateral, but not contralateral, STR was also detected, indicating the specificity of the injection (Fig. 7B). Western blot analysis confirmed that AAV-shR-MUL1-GFP infection efficiently suppressed MUL1 expression in both WT and VPS35+/− VMs (Figs. 7C–7D). Remarkably, the MFN2 level was restored to normal in VPS35+/− VMs, and increased in WT VMs by suppressing MUL1 expression (Figs. 7C–7D), indicating the necessity of MUL1 for the MFN2-reduction in vivo.
Figure 7. Restoration of MFN2-reduction and DA neuron loss by suppressing MUL1 expression in VPS35+/− SNpc.

(A) Schematic of AAV injection strategy. AAV5-shR-MUL1-GFP were stereotactically injected into VPS35+/+ or Vps+/− mice brains at 6-M old of age. Two months post injection, coronal brain sections, ventral midbrain or STR homogenates were collected and subjected to immunohistochemical staining, Western blot and HPLC analysis. (B) AVV5 infect the DA neurons in coronal brain sections. Contralateral (CON) and ipsilateral (shR-MUL1) substantia nigra (SNpc) are indicated (Scale bar = 1mm). Right insets show magnified images of the marked squares (Scale bar = 50μm). (C) Representative Western blots of homogenates of ventral midbrain derived from the virus ipsilateral (shR-MUL1) and contralateral (CON) hemispheres. (D) Quantification of data from C. Relative MUL1 and MFN2 protein levels were normalized by un-incetion side of VPS35+/+. Means ± SEM (n = 3) were shown. *p < 0.05. (E–F) Stereological analysis of TH-positive dopaminergic neurons in the SNpc. E, Representative images of TH immunostaining analysis in the virus injected SNpc (Scale bar = 200 μm). Insets show magnified images (Scale bar = 10 μm) and arrows indicate axon swelling. Quantification analysis of TH+ or NeurN+ cells in each hemisphere (mean ± SEM, n = 3) were presented in F. *p < 0.05 by one-way ANOVA with post hoc analysis. (G–H) Distribution of TH immunoreactive fibers in the STR. G, Representative images of TH staining analysis in adjacent sections of the injected STR (Scale bar = 400 μm). Insets show magnified images (Scale bar = 10 μm). G, Quantification of TH-immunoreactive intensity in the STR. Data are expressed as the percentage of the TH intensity of VPS35+/+ CON side. Mean ± SEM, n = 3. *p < 0.05 by one-way ANOVA with post hoc analysis. (I) Dopamine levels in the VPS35+/+ and VPS35+/− STR were measured by HPLC analysis. Isoproterenol was added as internal control, n=3, *p < 0.05 by one-way ANOVA with post hoc analysis.
Interestingly, upon AAV-shR-MUL1-GFP injection, TH+ neuron counts in VPS35+/− SNpc were restored to a nearly normal range (Figs. 7E–7F). The TH+ neurons in the contralateral (CON) side of VPS35+/− SNpc remained lower than that of VPS35+/+ controls (Figs. 7E–7F), indicating the specificity. Consistently, TH protein was reduced in VPS35+/− VMs, which was restored by infection with the virus of AAV-shR-MUL1-GFP (Fig. 7C). TH+ dopaminergic nerve terminals were also decreased in VPS35+/− STR, compared with that of WT controls, which was also rescued by suppressing MUL1 expression (Figs. 7G–7H). We further measured DA neurotransmitter level by HPLC (high-performance liquid chromatography) in VPS35+/+ and +/− STR injected with or without the shR-MUL1-GFP virus. Whereas DA was markedly reduced in VPS35+/− STR (compared with that of WT controls), suppressing MUL1 expression prevented this DA reduction (Fig. 7I). It is noteworthy that the increase of α-synuclein in VPS35+/− VM was un-changed by suppression of MUL1 expression (Figs. 7C). Collectively, these findings suggest that VPS35-deficiency reduces MFN2 and promotes DA neuronal loss, which can be alleviated by suppression of MUL1 expression.
We next determined if MUL1 also causes MFN2-reduction in DA neurons expressing PD-linked VPS35 mutant (D620N) in vivo. To this end, AAV5-encoding GFP-T2A-VPS35-D620N was generated, which was injected or co-injected with shR-MUL1 virus into WT-SNpc (at 3-M old of age) (Fig. S7A). Western blot and immunostaining analysis confirmed the VPS35-D620N expression and the MUL1 suppression by infecting WT VM with of both viruses (Figs. S7B–S7D). Indeed, MFN2 was reduced in VMs expressing VPS35-D620N, which was restored to normal in VMs co-expressing shRNA-MUL1 (Figs. S7B–S7C). Accordantly, the TH+ neurons were lower in VPS35-D620N expressing SNpc, which was attenuated by co-expression of shR-MUL1 (Figs. S7G–S7H). Interestingly, in VPS35-D620N expressing DA neurons, mitochondria were shorter and round (indicators of mitochondrial fragmentation), compared with that of uninfected or AAV-GFP infected control neurons (Figs. S7E–S7F). Moreover, co-injection with shR-MUL1 virus prevented mitochondrial fragmentation in these VPS35-D620N expressing DA neurons (Figs. S7E–S7F). Taken together, these results suggest that VPS35-mutation (D620N) acts as VPS35-deficiency, promoting DA neuronal loss likely by increasing MUL1-mediated MFN2 degradation.
DISCUSSION
This study has the following major findings. First, VPS35 expression in DA neurons is essential for prevention of PD-relevant deficits. Second, VPS35 expression in DA neurons is necessary for mitochondrial fusion and function, an event critical for DA neuron survival. Third, VPS35 regulates the trafficking and degradation of the E3 ubiquitin ligase MUL1, thus suppressing MUL1-mediated MFN2 degradation and promotes mitochondrial fusion. Fourth, elevated MUL1 in VPS35-deficient DA neurons contributes to MFN2-reduction, mitochondrial fragmentation, and DA neuron loss, but not to the accumulation of α-synuclein. Our work thus provides evidence for VPS35/retromer’s unrecognized function in regulating mitochondrial fusion dynamics, and suggests a working model, in which dys-regulation of VPS35/retromer’s function results in an increase of mitochondrial MUL1 E3 ubiquitin ligase, decrease in MFN2, and impairment of mitochondrial fusion and function, which may underlie VPS35-deficiency/mutation-associated PD pathogenesis.
The dysfunction of VPS35/retromer is believed to be a risk factor for the pathogenesis of both AD and PD. Whereas VPS35-deficienty enhances AD neuropathology in the Tg2576 mouse model of AD (Wen et al., 2011), it remains unclear if VPS35-deficiency contributes to PD pathogenesis. Here we provide evidence for VPS35-deficiency or mutation to promote PD-pathogenesis. First, VPS35+/− mice exhibit age-dependent PD-like deficit, including TH+ DA neuron loss and accumulation of α-synuclein (Tang et al., 2015). Second, young adult VPS35DAT-Cre mice, which specifically delete VPS35 gene in DA neurons (Fig. S1), show an earlier onset PD-like deficit than that of VPS35+/− mice (Fig. 1), revealing a genetic dose-dependency and the importance of VPS35 expression in DA neurons. Third, DA neurons expressing VPS35-D620N (a PD-linked mutation) (via AAV virus) also exhibited a reduction TH+ neurons (Fig. S7).
How does VPS35-deficiency or mutation in DA neurons promote PD pathogenesis? The underlying mechanisms may be a complex, as VPS35/retromer regulates the function and trafficking of numerous cargos (Small and Petsko, 2015). Here we propose that dysfunction in mitochondrial fusion dynamics may be a critical mechanism underlying VPS35-deficiency or mutation associated PD pathogenesis for the following reasons. First, the MFN2 protein, a GTPase essential for mitochondrial fusion, was decreased in both VPS35+/− and VPS35DAT-Cre ventral midbrain and in VPS35-deficient primary DA neurons (Fig. 2), which correlates well with the mitochondrial deficit. Second, expression of VPS35-WT, but not the PD-linked mutant D620N, in VPS35-deficient cells was capable of restoring the phenotypes (such as MFN2-reduction and mitochondrial fragmentation) (Figs. 3 and 5). Third, DA neuron-selective MFN2 knock-out mice (MFN2−/− or MFN2DAT-Cre) show mitochondrial and PD-relevant deficits (Lee et al., 2012a; Pham et al., 2012). Fourth, MFN2 is mutated in human axonal degenerative disorder Charcot-Marie-Tooth Type 2A (Zuchner et al., 2004), and MFN2-loss is implicated in the pathogenesis of not only Charcot-Marie-Tooth type 2A, but also PD. Finally, the dysregulation of mictochondrial function has been linked to the pathogenesis of multiple neurodegenerative disorders, and the evidence in PD is particularly strong (Haelterman et al., 2014). Mutations in several genes identified in PD patients encode proteins critical for mitochondrial dynamics as well as quality control. Among these genes, α-synuclein, Pink1, Parkin, and DJ-1’s functions on mitochondrial dynamics have been extensively investigated. Although their functions in regulating mitochondrial fusion or fission dynamics remain controversial, studies from Drosophila, mouse, and human cell lines support the view for the critical roles that these genes play in maintaining mitochondrial morphology, function, and mitophage mediated clearance of damaged mitochondria in PD (Lee et al., 2012b; Pickrell and Youle, 2015).
VPS35 is a well-recognized key component of the retromer essential for endosome-to-Golgi retrieval of transmembrane proteins/cargos. This study suggests a crucial role for VPS35 in regulating mitochondrial fusion dynamics, raising a question of how VPS35/retromer, a critical endosomal pathway regulator, regulates MFN2 protein degradation and mitochondrial protein trafficking. Results in our cellular and in vivo mouse studies lead to a working model, in which VPS35 promotes mitochondrial fusion dynamics by increasing MUL1 E3 ubiquitin ligase degradation and suppressing MUL1 mitochondrial targeting, thus preventing MUL1-mediated MFN2-ubiquitination and degradation. This VPS35-MUL1-MFN2 pathway is in line with the reports that MUL1/MAPL promotes ubiquitination and degradation of MFN2 and enhances mitochondrial fission (Braschi et al., 2009; Lokireddy et al., 2012). It is also in agreement with the observations that VPS35 interacts with MUL1/MAPL and is required for the formation of MUL1/MAPL associated MDVs (mitochondrial derived vesicles) or MUL1/MAPL containing peroxisomes in the MCF7 tumor cell line (Braschi et al., 2010). While this model is supported by both in vitro and in vivo evidence, it is intriguing and raises additional questions. How does VPS35 regulate MUL1/MAPL degradation? Are the sorting nexins and other retromer complex proteins (e.g., Vps26 and 29) involved in this event? Does MUL1/MAPL-mediated Drp1-sumoylation also contribute to the mitochondrial deficit in VPS35 deficient cells/brain? Addressing these questions in future studies may reveal further insights into VPS35/retromer regulation of mitochondrial fusion/fission dynamics and PD pathogenesis.
Notice that altered mitochondrial morphology or fusion dynamics were not only detected in VPS35-deficient DA neurons, but also in VPS35-depleted neuroblastoma cell lines. These results suggest that VPS35 may be a general regulator for mitochondrial fusion dynamics in vitro. However, in vivo/in VPS35+/− brain, mal-functional mitochondria appeared to be brain-region selective and age-dependent, mainly detected in PD-vulnerable brain regions, such as SNpc and STR (Fig. S4). Consistently, the MUL1-increase and MFN2-reduction in VPS35 deficient mice were also brain-region selective, having been detected in SNpc and STR, but not hippocampus (Figs. 2 and 5). These observations suggest a brain region-specific factor that may also regulate MUL1 degradation and mitochondrial function. This factor may be higher in the hippocampus, but lower in the SNpc and STR. Alternatively, additional factor(s) that occur in VPS35 deficient midbrain lower the threshold to tolerate VPS35/retromer dysfunction in DA neurons as compared to that in other types of neurons, which may contribute to the different results between in vitro and in vivo experiments.
In aggregate, this study provides evidence that loss of VPS35 function in DA neurons results in impaired MUL1 trafficking and degradation and increased MUL1, thus promoting MFN2 degradation, mitochondrial fusion and function impairments, and DA neuronal loss.
MATERIALS AND METHODS
Animals
Two VPS35-deficient mice were generated and used in this study. First, VPS35+/− mice were described previously (Wen et al., 2011). Second, VPS35DAT-Cre mice were generated by crossing floxed allele of VPS35 (VPS35f/f) with DAT-Cre mice. The VPS35f/f mice were generated as described in Supplemental Experimental Procedures. All the mice were backcrossed with C57BL/6 mice for more than 6–10 generations. All the mice were housed in a room with a 12 hours light/dark cycle with water and standard rodent chow diet. All the experimental procedures were approved by the IACUC (Institutional Animal Care and Use Committee) at Georgia Regents University.
Stereological analysis
To quantify the number of DA (TH+) or NeuN+ neurons in SNpc, the unbiased stereological estimation of the total number of the TH+/NeuN+ cells in VM (ventral midbrain) was employed as previously described (Yin et al., 2013). In brief, every fifth section from the bregma −2.92 to −4.16 mm coronal sections across the midbrain were stained with TH/NeuN antibody. The total number of TH+/NeuN+ neurons in each section of SNpc was mathematically calculated using the Stereo Investigator program.
Primary culture and transfection
Dopaminergic (DA) neuronal culture was prepared as previously described (Lin et al., 2012). Briefly, individual ventral midbrain of E18.5 VPS35f/f or VPS35+/m mice was dissected and placed in cold HBSS. The brain pieces were then digested in papain (5U/ml; Worthington Biochemicals) for 20 min at 37°C. Cells were dissociated using increasingly smaller pipette tips, washed twice, resuspended in the plating medium (80% Neurobasal-A and 20% fetal bovine serum), and plated onto Poly-D-Lysine-coated coverslips at a density of 130K cells/well (24 well plate). The growth media supplemented with 1X B27, 2mM GlutaMAX-1 and 1% penicillin/streptomycin were replaced 4-hrs later. At DIV 6, neurons were subjected to transient transfection by using the calcium phosphate-mediated gene transfer method as previously described (Zhu et al., 2007).
Measurement of oxygen consumption rate
Oxygen consumption rate (OCR) was analyzed in an X96 Extracellular Flux Analyzer with XF Cell Mito Stress Test Kit (Seahorse Biosciences) at 37°C. DA neurons were plated on Poly-D-Lysine coated XF96 cell culture plate at 40K per well and cultured for 7 days with 80ul neuronal culture medium/well. Four wells without neuron seeding from each plate were set as temperature and background control. For measurement, neurons were gently rinsed with 100ul/well assay medium (XF Base medium (Seahorse Biosciences) with 2mM Glutamine and 10mM glucose), put into 175ul/well fresh assay medium and assayed. Three baseline recordings were made, followed by sequential injection of the injection of the ATP synthase inhibitor oligomycin, the mitochondrial uncoupler Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) and the Complex I inhibitor rotenone. Two minutes OCR measurement were performed at 3 minute intervals with mixing and each condition was measured in independent well.
Measurement of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was measured with a cationic fluorescent dye, tetramethylrhodamine methyl ester (TMRM+) (Molecular probes, Carlsbad, CA). Briefly, NLT cells were transfected with miR- scramble or miR-VPS35 on poly-D-lysine coated cover slips, and 48 hours post transfection, cells were washed and incubated with TMRM+ at 10 nM for 30 minutes in a Dulbecco’s Phosphate Buffered Saline (D-PBS, Hyclone). After treatment, cells were imaged at an excitation wavelength of 555 nm and emission wavelength of 580 nm by the fluorescence microscope (Olympus IX70) and captured every 60 sec with a 60X objective (Olympus, USA) and further analysis of TMRM fluorescence was performed using Image-Pro Plus software.
AAV generation and injection
Adenovirus 5 carrying shRNA against mouse-MUL1 (AAV-shR-MUL1) was generated at the Emory virus core facility. The shR-MUL1 (5′-GAGCTGTGCGGTCTGTTAA -3′) was synthetized and subcloned into a pAAV-U6-GFP expression vector (Cell Biolabs). The eGFP cDNA was used as a control after sub-cloning into pAAV-MCS vector (Cell Biolabs). The full length of mouse VPS35-D620N cDNA was sub-cloned into the pAAV -EGFP-T2A-V5 vector (Vector Biolabs). Infected 293 cells from 25 to 50 15-cm tissue culture plates were collected and centrifuged. Then the cells were lysed by three freeze–thaw cycles. The AAV virus was purified using the iodixanol gradient centrifugation procedure and the virus titers were measured by real-time PCR. For stereotaxic injection, 1.5 μl viral solution (1 × 1012 vg/ml) was injected into the right ventral midbrain (anterior, −2.8 mm; lateral, −1.3mm; dorsal, −4.2 mm relative to bregma) with a 30-gauge microsyringe at a rate of 0.5 μl/min.
Statistical analysis
Data were analyzed using Prism 5.0 software (GraphPad). Student’s t test and one-way ANOVA with Bonferroni post hoc test were used as indicated. Significance was defined as p < 0.05.
Supplementary Material
Figure S1. Generation of DA neuron-specific VPS35 knock-out (VPS35DAT-Cre) mice, related to Figure 1.
(A) Schematic of generation of DA neuron-specific VPS35 knock-out (VPS35DAT-Cre) mice. First panel, genomic structure of the VPS35; second panel, VPS35 allele after homologous recombination in ES cells; third panel, targeted VPS35 allele without the LacZ-Neo cassette; forth panel, targeted VPS35 allele without exon 6 (VPS35DA cKO). Frt, Flp recognition target; neo, Neomycin resistance cassette. (B) Genotyping of VPS35DAT-Cre mice and control littermates. Genomic DNA was isolated from mouse tail for PCR with indicated primers. Cre primers generate 750-bp products. VPS35-Flox primers generate 180-bp products in wild type, or 240-bp products in floxed allele. The genotype DAT-Cre; VPS35-loxP/loxP was considered as the conditional knockout (VPS35DAT-cre). (C) DAT-Cre recombinase expressed in DA neurons. Rosa::LSL::tdTomato mice were crossed with or without DAT-Cre mice. Immunofluorescence for TH (Green) and tdTomato (Red) in coronal sections from Rosa::LSL::tdTomato and Rosa::LSL::tdTomato;DAT-Cre shows in C. Scale bars: 20 μm. (D) Decrease of VPS35 expression in DA neurons in VPS35DAT-Cre mice. Dual immuno uorescence for VPS35 (Green) and TH (Red) in coronal sections. Scale bars: 20 μm. (E) Slight reduction of body weight in VPS35DAT-Cre mice. Body weight was measured in 2-M old and 3-M old VPS35DAT-Cre mice (n = 6) and control littermates (n = 8). Data are presented as mean ± SEM. * p < 0.05.
Figure S2. Decreased Mfn2 in VPS35-deficient neuroblastomal cells and normal Mfn2 transcription in aged VPS35+/− brain areas, related to Figure 2.
(A–B) Immunoblot analysis of Mfn2 levels in SH-SY5Y cells transfected with indicated plasmids. A, Representative blots; B, Quantification of Mfn2 levels (mean ± SEM, n = 3; *p < 0.05). (C–D) Mitochondrial Mfn2 was reduced in NLT cells expressing miR-VPS35. Transfected NLT cells were fixed and subjected to immunostaining analysis using indicated antibodies. Mitochondria were labeled by anti-ATP5a antibody. C, Representative images; Scale bars: 5 μm. D, Quantification of mitochondrial-Mfn2 (mean ± SEM; n = 20 cells; *p < 0.05). (E) No difference in Mfn2 mRNA between VPS35+/+ and +/− mice in different brain regions. The mRNA of 12-M old mice was analyzed by real time PCR analysis.
Figure S3. Mitochondrial fragmentation in VPS35-deficient neuroblastomal cells and rescue of shR-VPS35-induced mitochondrial fragmentation by shRNA-resistant VPS35, related to Figure 3.
(A–D) NLT and SH-SY5Y cells were transfected with miRNA-VPS35 or scramble control (marked by eBFP) and examined for mitochondrial morphology. A and C, Representative images; B and D, Quantitative data. *p < 0.05; Scale bars: 5 μm. (E) Restoration of VPS35 protein by co-expressing shRNA-resistant, but not shRNA-sensitive, VPS35, in SH-SY5Y cells. SH-SY5Y cells were transfected with indicated plasmids. Cell lysates were subjected to Western blot analysis. (F–G) Rescue of shRNA-VPS35-induced mitochondrial fragmentation in NLT cells by shRNA-resistant VPS35. F, Representative images. Areas in squares are enlarged in bottom panels. Bar, 5 μm. G, Quantification of mitochondrial lengths as column scatter (n = 3 independent experiments, with > 200 mitochondria per experiment; *p < 0.05).
Figure S4. Abnormal and dysfunctional mitochondria in VPS35+/− midbrains, related to Figure 4.
(A–E) Reduced mitochondrial length in VPS35+/− SNpc and STR by transmission electron microscopic (TEM) analysis. A and B, Representative TEM images. Scale bars, 0.5 μm. C, 3D scatter plot graphs of the distribution of area/width/length of individual mitochondria. D and E, Quantitative analysis of TEM data. Data are presented as mean ± SEM; n > 200 mitochondria; p values are indicated in the figure. (F) The proportion of normal, damaged or condensed/aggregated mitochondria in 4 or 12-M old VPS35+/− STR. Data are presented as mean ± SEM; n = 3 mice/genotype; *p < 0.05. (G) Decreased mtDNA/nuDNA ratio in aged, but not young, entorhinal cortex (Ec-ctx), STR, and VM. Relative copy numbers of mtDNA and ncDNA were calculated from the DCt by qPCR. Data are presented as mean ± SEM; n = 3; *p < 0.05.
Figure S5. Increased Mfn2 ubiquitination and degradation in VPS35-deficient cells, related to Figure 5.
(A–B) Restoration of Mfn2 levels in VPS35+/− neurons by MG132. Neurons were treated with 30 mM MG132 for 6 hrs and blotted for different proteins (A). Mfn2 level is quantified in B (mean ± SEM; n = 5; *p < 0.05). (C–D) Restoration of Mfn2 levels in VPS35-deficeint SH-SY5Y cells by MG132 and expressing VPS35-WT, but not VPS35-D620N. C, Representative blots; D, Quantitative data (normalized by b-actin loading and Mock control) (mean ± SEM; n = 3; *p < 0.05). (E–F) Increased Mfn2-ubiquitination in VPS35-deficient (miR-VPS35) and VPS35 mutation (VPS35-DN) SH-SY5Y cells. Mfn2 was immunoprecipitated from lysates of transfected SH-SY5Y cells, and the resulting complex was subjected to immunoblotting analyses using indicated antibodies. E, Representative blots; F, Quantification of ubiquitinated Mfn2 (Ub-Mfn2). Level in control cells was set as 1. Data are shown as mean ± SEM (n = 3; *p <0.05).
Figure S6. Restoration of Mfn2 and mitochondrial deficits by suppressing Mul1 expression in VPS35-deficient NLT and neurons, related to Figure 7.
(A–B) Immunoblot analysis of Mul1 and Mfn2 levels in SH-SY5Y cells transfected with indicated plasmids. A, Representative blots; B, Quantification of Mul1 and Mfn2 levels (normalized by untransfected cells). (C) Endogenous Mul1 level was suppressed by shR-Mul1-1 in transfected NLT cells. Transfected cells are marked by white dash lines. Bar, 5 μm. (D–E) Mitochondrial fragmentation in miRNA-VPS35-expressing NLT cells was mitigated by co-expressing shRNA-Mul1. D, Representative confocal images. Scale bar, 5 μm. E, Quantitative data of mitochondrial length are shown as grouped column scatter and aspect ratio. Data are presented as mean ± SEM (n = 200 mitochondria from 10 different cells; *p < 0.05). (F–G) Mitochondrial fragmentation in VPS35-deficient neurons was ameliorated by expressing shRNA-Mul1. F, Representative confocal images. Higher magnification images are shown in bottom panels. Scale bar, 10 μm. Quantitative data of mitochondrial length is shown as grouped column scatter and aspect ratio. Data are presented as mean ± SEM (n = 200 mitochondria from 10 different cells; *p < 0.05).
Figure S7. Restoration of mitochondrial fragmentation and DA neuron loss by suppressing Mul1 expression in VPS35-D620N expressing SNpc, related to Figure 7.
(A) Schematic of AAV injection strategy. AAV5 coated GFP-2A-VPS35-D620N and GFP-shR-Mul1 were stereotactically injected into the SNpc of 3-M old WT mouse. Three months post-injection, coronal brain sections and ventral midbrain homogenates were collected and subjected to immunohistochemical staining and Western blot analyses, respectively. (B) Representative Western blots of homogenates of ventral midbrain derived from the ipsilateral (VPS35-DN or VPS35-DN+shR Mul1) and contralateral (CON) hemispheres. (C) Quantification of data from B. Relative VPS35, Mul1 and Mfn2 protein levels were normalized by CON side. Means ± SEM (n = 4) are shown. (D–F) Mitochondrial fragmentation in VPS35-D620N overexpression DA neurons (indicated by TH or GFP) were mitigated by co-expressing shRNA-Mul1. Representative images of Mul1 (D) or Tom20 (E) immunostaining in the ipsilateral (VPS35-DN or VPS35-DN+shR Mul1) and contralateral (CON) SNpc (Scale bar = 10 μm). Quantification of percentage of DA neurons with mitochondrial fragmentation is shown in F, (G–H) Stereological analysis of TH-positive dopaminergic neurons in SNpc. G, Representative images of TH immunostaining in the SNpc (Scale bar = 200 μm). Quantification analysis (mean ± SEM, n = 4 animals/group) is presented in H. *p < 0.05.
Acknowledgments
We thank Drs. Xiaoxi Zhuang (Univ. Chicago, USA), Philip Wang (Georgia Regents University), and Gia Voeltz (Univ. Colorado, USA) for reagents. We also thank Ms. Shan Xiong for VPS35 mutant mouse maintenance and genotyping, and members of the Xiong and Mei laboratories for helpful discussions. This study was supported in part by grants from National Institutes of Health (AG045781 to WCX) and Department of Veterans Affairs (BX00838 to WCX).
Footnotes
AUTHOR CONTRIBUTIONS
F-L.T. conducted all experiments and data analysis. W.L. performed immunostaining analysis. J.E. performed behavior test. J-X.H. prepared AAV vectors for viral injection. J.Y., L.M. and W-C.X. is the senior author who designed the project. F-L.T. and W-C.X. wrote the manuscript.
The authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Current biology : CB. 2010;20:1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Dissecting mitochondrial fusion. Developmental cell. 2006a;11:592–594. doi: 10.1016/j.devcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006b;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annual review of cell biology. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Eaton S. Retromer retrieves wntless. Developmental cell. 2008;14:4–6. doi: 10.1016/j.devcel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007;22(Suppl 17):S343–350. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]
- Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ. A mitocentric view of Parkinson’s disease. Annual review of neuroscience. 2014;37:137–159. doi: 10.1146/annurev-neuro-071013-014317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nature reviews Neuroscience. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Human molecular genetics. 2012a;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson’s disease: vertebrate genetics. Cold Spring Harb Perspect Med. 2012b:2. doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, et al. Conditional expression of Parkinson’s disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. doi: 10.1016/j.cmet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annual review of genomics and human genetics. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Gustafsen C, Madsen P, Nyengaard JR, Hermey G, Bakke O, Mari M, Schu P, Pohlmann R, Dennes A, et al. Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Molecular and cellular biology. 2007;27:6842–6851. doi: 10.1128/MCB.00815-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Pham AH, Meng SX, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Human molecular genetics. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. The Journal of cell biology. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. The retromer complex - endosomal protein recycling and beyond. Journal of cell science. 2012;125:4693–4702. doi: 10.1242/jcs.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nature reviews Neuroscience. 2015;16:126–132. doi: 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Yanatori I, Kawai Y, Kishi F. Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. Journal of cell science. 2010;123:756–766. doi: 10.1242/jcs.060574. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. The Journal of cell biology. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, Tang B, Mei L, Xiong WC. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J Neurosci. 2015;35:10613–10628. doi: 10.1523/JNEUROSCI.0042-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. VPS35 Mutations in Parkinson Disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, et al. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open. 2012;1:1248–1257. doi: 10.1242/bio.20122451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. The Journal of cell biology. 2011;195:765–779. doi: 10.1083/jcb.201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nature reviews Molecular cell biology. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochimica et biophysica acta. 2012;1817:1833–1838. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Yin DM, Chen YJ, Lu YS, Bean JC, Sathyamurthy A, Shen C, Liu X, Lin TW, Smith CA, Xiong WC, et al. Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron. 2013;78:644–657. doi: 10.1016/j.neuron.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XJ, Wang CZ, Dai PG, Xie Y, Song NN, Liu Y, Du QS, Mei L, Ding YQ, Xiong WC. Myosin X regulates netrin receptors and functions in axonal path-finding. Nat Cell Biol. 2007;9:184–192. doi: 10.1038/ncb1535. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. Journal of neuroscience methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation of DA neuron-specific VPS35 knock-out (VPS35DAT-Cre) mice, related to Figure 1.
(A) Schematic of generation of DA neuron-specific VPS35 knock-out (VPS35DAT-Cre) mice. First panel, genomic structure of the VPS35; second panel, VPS35 allele after homologous recombination in ES cells; third panel, targeted VPS35 allele without the LacZ-Neo cassette; forth panel, targeted VPS35 allele without exon 6 (VPS35DA cKO). Frt, Flp recognition target; neo, Neomycin resistance cassette. (B) Genotyping of VPS35DAT-Cre mice and control littermates. Genomic DNA was isolated from mouse tail for PCR with indicated primers. Cre primers generate 750-bp products. VPS35-Flox primers generate 180-bp products in wild type, or 240-bp products in floxed allele. The genotype DAT-Cre; VPS35-loxP/loxP was considered as the conditional knockout (VPS35DAT-cre). (C) DAT-Cre recombinase expressed in DA neurons. Rosa::LSL::tdTomato mice were crossed with or without DAT-Cre mice. Immunofluorescence for TH (Green) and tdTomato (Red) in coronal sections from Rosa::LSL::tdTomato and Rosa::LSL::tdTomato;DAT-Cre shows in C. Scale bars: 20 μm. (D) Decrease of VPS35 expression in DA neurons in VPS35DAT-Cre mice. Dual immuno uorescence for VPS35 (Green) and TH (Red) in coronal sections. Scale bars: 20 μm. (E) Slight reduction of body weight in VPS35DAT-Cre mice. Body weight was measured in 2-M old and 3-M old VPS35DAT-Cre mice (n = 6) and control littermates (n = 8). Data are presented as mean ± SEM. * p < 0.05.
Figure S2. Decreased Mfn2 in VPS35-deficient neuroblastomal cells and normal Mfn2 transcription in aged VPS35+/− brain areas, related to Figure 2.
(A–B) Immunoblot analysis of Mfn2 levels in SH-SY5Y cells transfected with indicated plasmids. A, Representative blots; B, Quantification of Mfn2 levels (mean ± SEM, n = 3; *p < 0.05). (C–D) Mitochondrial Mfn2 was reduced in NLT cells expressing miR-VPS35. Transfected NLT cells were fixed and subjected to immunostaining analysis using indicated antibodies. Mitochondria were labeled by anti-ATP5a antibody. C, Representative images; Scale bars: 5 μm. D, Quantification of mitochondrial-Mfn2 (mean ± SEM; n = 20 cells; *p < 0.05). (E) No difference in Mfn2 mRNA between VPS35+/+ and +/− mice in different brain regions. The mRNA of 12-M old mice was analyzed by real time PCR analysis.
Figure S3. Mitochondrial fragmentation in VPS35-deficient neuroblastomal cells and rescue of shR-VPS35-induced mitochondrial fragmentation by shRNA-resistant VPS35, related to Figure 3.
(A–D) NLT and SH-SY5Y cells were transfected with miRNA-VPS35 or scramble control (marked by eBFP) and examined for mitochondrial morphology. A and C, Representative images; B and D, Quantitative data. *p < 0.05; Scale bars: 5 μm. (E) Restoration of VPS35 protein by co-expressing shRNA-resistant, but not shRNA-sensitive, VPS35, in SH-SY5Y cells. SH-SY5Y cells were transfected with indicated plasmids. Cell lysates were subjected to Western blot analysis. (F–G) Rescue of shRNA-VPS35-induced mitochondrial fragmentation in NLT cells by shRNA-resistant VPS35. F, Representative images. Areas in squares are enlarged in bottom panels. Bar, 5 μm. G, Quantification of mitochondrial lengths as column scatter (n = 3 independent experiments, with > 200 mitochondria per experiment; *p < 0.05).
Figure S4. Abnormal and dysfunctional mitochondria in VPS35+/− midbrains, related to Figure 4.
(A–E) Reduced mitochondrial length in VPS35+/− SNpc and STR by transmission electron microscopic (TEM) analysis. A and B, Representative TEM images. Scale bars, 0.5 μm. C, 3D scatter plot graphs of the distribution of area/width/length of individual mitochondria. D and E, Quantitative analysis of TEM data. Data are presented as mean ± SEM; n > 200 mitochondria; p values are indicated in the figure. (F) The proportion of normal, damaged or condensed/aggregated mitochondria in 4 or 12-M old VPS35+/− STR. Data are presented as mean ± SEM; n = 3 mice/genotype; *p < 0.05. (G) Decreased mtDNA/nuDNA ratio in aged, but not young, entorhinal cortex (Ec-ctx), STR, and VM. Relative copy numbers of mtDNA and ncDNA were calculated from the DCt by qPCR. Data are presented as mean ± SEM; n = 3; *p < 0.05.
Figure S5. Increased Mfn2 ubiquitination and degradation in VPS35-deficient cells, related to Figure 5.
(A–B) Restoration of Mfn2 levels in VPS35+/− neurons by MG132. Neurons were treated with 30 mM MG132 for 6 hrs and blotted for different proteins (A). Mfn2 level is quantified in B (mean ± SEM; n = 5; *p < 0.05). (C–D) Restoration of Mfn2 levels in VPS35-deficeint SH-SY5Y cells by MG132 and expressing VPS35-WT, but not VPS35-D620N. C, Representative blots; D, Quantitative data (normalized by b-actin loading and Mock control) (mean ± SEM; n = 3; *p < 0.05). (E–F) Increased Mfn2-ubiquitination in VPS35-deficient (miR-VPS35) and VPS35 mutation (VPS35-DN) SH-SY5Y cells. Mfn2 was immunoprecipitated from lysates of transfected SH-SY5Y cells, and the resulting complex was subjected to immunoblotting analyses using indicated antibodies. E, Representative blots; F, Quantification of ubiquitinated Mfn2 (Ub-Mfn2). Level in control cells was set as 1. Data are shown as mean ± SEM (n = 3; *p <0.05).
Figure S6. Restoration of Mfn2 and mitochondrial deficits by suppressing Mul1 expression in VPS35-deficient NLT and neurons, related to Figure 7.
(A–B) Immunoblot analysis of Mul1 and Mfn2 levels in SH-SY5Y cells transfected with indicated plasmids. A, Representative blots; B, Quantification of Mul1 and Mfn2 levels (normalized by untransfected cells). (C) Endogenous Mul1 level was suppressed by shR-Mul1-1 in transfected NLT cells. Transfected cells are marked by white dash lines. Bar, 5 μm. (D–E) Mitochondrial fragmentation in miRNA-VPS35-expressing NLT cells was mitigated by co-expressing shRNA-Mul1. D, Representative confocal images. Scale bar, 5 μm. E, Quantitative data of mitochondrial length are shown as grouped column scatter and aspect ratio. Data are presented as mean ± SEM (n = 200 mitochondria from 10 different cells; *p < 0.05). (F–G) Mitochondrial fragmentation in VPS35-deficient neurons was ameliorated by expressing shRNA-Mul1. F, Representative confocal images. Higher magnification images are shown in bottom panels. Scale bar, 10 μm. Quantitative data of mitochondrial length is shown as grouped column scatter and aspect ratio. Data are presented as mean ± SEM (n = 200 mitochondria from 10 different cells; *p < 0.05).
Figure S7. Restoration of mitochondrial fragmentation and DA neuron loss by suppressing Mul1 expression in VPS35-D620N expressing SNpc, related to Figure 7.
(A) Schematic of AAV injection strategy. AAV5 coated GFP-2A-VPS35-D620N and GFP-shR-Mul1 were stereotactically injected into the SNpc of 3-M old WT mouse. Three months post-injection, coronal brain sections and ventral midbrain homogenates were collected and subjected to immunohistochemical staining and Western blot analyses, respectively. (B) Representative Western blots of homogenates of ventral midbrain derived from the ipsilateral (VPS35-DN or VPS35-DN+shR Mul1) and contralateral (CON) hemispheres. (C) Quantification of data from B. Relative VPS35, Mul1 and Mfn2 protein levels were normalized by CON side. Means ± SEM (n = 4) are shown. (D–F) Mitochondrial fragmentation in VPS35-D620N overexpression DA neurons (indicated by TH or GFP) were mitigated by co-expressing shRNA-Mul1. Representative images of Mul1 (D) or Tom20 (E) immunostaining in the ipsilateral (VPS35-DN or VPS35-DN+shR Mul1) and contralateral (CON) SNpc (Scale bar = 10 μm). Quantification of percentage of DA neurons with mitochondrial fragmentation is shown in F, (G–H) Stereological analysis of TH-positive dopaminergic neurons in SNpc. G, Representative images of TH immunostaining in the SNpc (Scale bar = 200 μm). Quantification analysis (mean ± SEM, n = 4 animals/group) is presented in H. *p < 0.05.