

Abstract
We developed two-photon scanning patterned illumination microscopy (2P-SPIM) for super-resolution two-photon imaging. Our approach used a traditional two-photon microscopy setup with temporally modulated excitation to create patterned illumination fields. Combing nine different illuminations and structured illumination reconstruction, super-resolution imaging was achieved in two-photon microscopy. Using 2P-SPIM we achieved a lateral resolution of 141 nm, which represents an improvement by a factor of 1.9 over the corresponding diffraction limit. We further demonstrated super-resolution cellular imaging by 2P-SPIM to image actin cytoskeleton in mammalian cells and three-dimensional imaging in highly scattering retinal tissue.
Keywords: two-photon microscopy, structured illumination, super-resolution imaging
I. INTRODUCTION
Super-resolution microscopy revolutionized conventional light microscopy by breaking Abbe’s diffraction limit and providing a unique platform to optically study nanostructures in biological imaging [1-12]. Inventions of stimulated emission depletion (STED) microscopy, photoactivated localization microscopy (PALM), and stochastic optical reconstruction microscopy (STORM) have reached ~10 nm resolution by manipulating the activation and depletion (on-off) of fluorescent tags. Other techniques have achieved more modest resolution enhancement by increasing the effective numerical aperture [5-7], confocal spatial phase modulation [10], anti-bunching properties of photons from single molecules [13], structure of the illuminated or emitted light [8, 11, 12, 14], or a combination of the aforementioned techniques [9, 15].
Structured illumination microscopy (SIM) is a nanoscopic imaging technology that does not rely on the “on-off” properties of particular fluorophores. SIM relies on the generation of moiré fringes caused by the structured excitation illumination field, Iex(r), where r is the central position of the illumination field on the imaging plane [4]. As a most common type of excitation used in wide-field SIM, the illumination field is described by , where ωr represents the spatial frequency of the illumination field in radians per unit length [16]. The illumination field is normally created using a grating or grid mask. The illumination pattern is shifted twice from the original incident angle by radians to extend the bandwidth isotropically in frequency domain or k-space. For each illumination field, a wide-field image is captured with an array detector for final reconstruction. Image reconstruction is typically conducted in the frequency domain, but can also be accomplished optically at the expense of higher resolutions allowed by higher harmonics [17-19]. SIM offers excellent flexibility on imaging various biological structures including mitochondria [20, 21], bacteria [22, 23], and other biological specimens [24-26]. Alterations of SIM have added advantages, such as saturated SIM [9, 27] and speckle pattern reconstruction [12]. Although SIM and it’s modifications potentially offer real-time in vivo super-resolution live cell imaging, they suffer from scattered and out of focus background radiation [28]. Consequently, the current excitation depth resolution in non-transparent samples is limited to a few tens of micrometers from the sample surface. Also, there is a significant loss of resolution at increasing imaging depths as a result of signal-to-noise ratio (SNR) degradation [29-32].
II. BACKGROUND
In the attempt to enhance the imaging depth in SIM, two-photon (2P) excitation was implemented using a similar wide-field structured illumination [18, 19, 33-35]. The longer wavelength excitation (usually in the NIR range) has much lower attenuation than visible-light excitation, which yields comparatively large penetration depth. Unfortunately, illumination areas are often limited due to the need for high photon density for 2P excitation. 2P type SIM cannot easily be realized using the grating or grid shaped optical masks (commonly used in wide-field SIM) due to power constrains. Using masks or gratings to create wide-field structured 2P excitation requires abnormally high average power outputs beyond what is commonly available from the fundamental output beam of pulsed lasers [28, 36]. 2P temporal focusing addressed some technical challenges of implementing masks and gratings for 2P type SIM, but still suffers from low fluences that can be a few orders of magnitude weaker than focused point beams used in laser scan microscopy (LSM) [33-35]. Later, laser scanning microscopy schemes were proposed for 2P type SIM fluorescence [18, 19], overcoming the power limitation problems from wide-field structured illumination. However, implemented techniques require either major modification to established 2P microscope setups or confocal pin-holes in tandem with micro-lens arrays that reduce useable signal [17-19]. More importantly, these methods employed optical domain image reconstruction to increase imaging speed, but limits the resolution far below what is theoretically possible in frequency domain reconstruction when utilizing higher frequency harmonics created during fluorophore saturation [9, 27].
III. THEORY
We developed a new 2P technique, referred to as 2P scanning patterned illumination microscopy (2P-SPIM), for nanoscopic imaging of fluorescent species. By using focused laser-scanning 2P excitation, we reduced laser scattering and confined fluorescence to the focal region. Our excitation scheme increased SNR and pushed lateral resolutions to 1.9-fold (141 nm) greater than the corresponding diffraction limit. Furthermore, 2P-SPIM can quickly be integrated with existing 2P microscopes by adding a few additional components and a scanning control.
Combining laser scanning 2P excitation with SIM requires mathematical and physical re-evaluation [9, 16]. Here we use a one dimensional case for simplicity. A similar theoretical analysis was previously reported by Lu et al [16]. For 2P scanning excitation, the intensity of the emission field, I2P em, can be described at any position of the central beam spot (r) during scanning. Depending on the temporal modulation frequency (ωt), which is determined by the electro-optical modulator (EOM), and scanning speed, the emission field is represented by . Here is the peak intensity of the 2P point spread function (PSF), h2P ex is the PSF of the 2P excitation wavelength, s(r) is the concentration of fluorophores, and t represents both time and position of the laser spot given a constant beam scanning speed as previously used [9, 16]. At constant beam scanning speed, the central beam position can be described by . For each beam position r, the excitation volume is determined by h2P ex. During a single frame scan, the signal is collected by the imaging objective and projected onto an integrating imaging detector in detector space (x) to form the final image. In single photon scanning SIM, we have
where represents the convoluted product of the of the peak intensity of the scan spot and excitation wavelength PSF (hex) in detector space. For continuously integrated laser scanning (i.e. the wide-field detector integrates signal over a single complete scan) is equivalent to wide-field illumination with the spatially modulated light pattern implemented in wide-field SIM. However, for 2P scanning, the quadratic relation changes the final picture to the form
The optical transfer function (OTF), derived from the Fourier transform of the 2P effective PSF determines the cutoff frequency , from which the first harmonic falls beyond the frequency bandwidth range. Care must be taken to keep ωt within the maximum 2P optical transfer function (OTF) or the first harmonic will be outside the frequency bandwidth range for reconstruction.
IV. METHOD
For imaging, we used three modulated directional laser scans of the sample at angles θ of 50°, 170°, and 290° with respect to normal incidence. For each angle, the modulation was retarded with three equally spaced phase shifts φ. We took nine images from each sample region to form an image stack for reconstruction, (Fig. 1a). To reconstruct a super-resolution image, we first recovered the un-attenuated frequency spectrum by Weiner filtering each individual image. The parameters of the Weiner filter, including the system PSF and SNR, were predetermined based on image analysis of a pre-patterned scanned liquid fluorescein sample (Fig. 1a). SNR parameters were readjusted for cell samples based on the signal intensity of the respective contrast agent. We then performed a two-dimensional Fourier transform to convert each image into k-space. After a 4-fold up-sampling, we gained precise control of the frequency shift (up to 1/4 of the pixel resolution). The precise modulation angle and frequency were decoded by analyzing the location of the first-order harmonic peaks as they appeared in the two-dimensional spectrum image. Once we determined the exact modulation frequency and angle, the phase retardation was estimated by shifting and matching the theoretical cosine pattern against the original image in the spatial domain.
Figure 1.

(a) Scanned patterned illumination acquired from a fluorescein solution phantom. The phase (φ) was shifted in equal increment three times along a single direction before the incident angle (θ) was shifted by radians. This process was repeated to acquire nine images. (b) The phase and angle movement information corresponding to the Fourier components were calculated from each incident angle (colored circles) and the sequence was reconstructed to achieve super-resolution. (c) Schematic of 2P-SPIM experimental setup. (d) Imaged 110 nm fluorescent nanosphere by direct 2P microscopy (top) and 2P-SPIM (middle) images used to extract the corresponding PSFs (bottom).
The corresponding baseband spectrum and modulated high frequency components were recovered using matrix-based image algebra. We classified the nine images into three groups based on the modulation angles. For each group, the corresponding base and modulated frequency components, X0,X−, and X+, can be solved using equation
where φ1, φ2, and φ3, are the estimated modulation phase retardation; Yφ1, Yφ2, and Yφ3, are the corresponding vectorized expressions of the modulated image in the frequency domain.
The recovered frequency components were then reshaped into a 2D matrix and shifted back to their true positions for re-assembly (Fig. 1b). For the overlapping areas between the frequency components, we calculated the total energy for each component and normalized the higher order terms before collective summation. Finally, discontinuities in the frequency domain were removed using a low pass filter during reconstruction to reduce reconstruction artifacts.
V. EXPERIMENTAL SETUP AND RESULTS
The 2P-SPIM system was built upon a commercial inverted microscope platform (IX81, Olympus) as shown in Fig. 1c. The 795 nm output of a 80 MHz Ti:Sapphire laser with 100-fs pulse width (Solstice, Newport) was routed through an electro-optic modulator (EOM) to generate a sinusoidal output modulation beam. Next, the beam was conditioned through a telescopic beam condenser containing a confocal pinhole to improve the Gaussian profile of the incident light. The beam was directed onto X-Y scanning mirrors (QS-7 OPD, Nutfield) and passed through a beam expander before entering the microscope body. The NIR light entering the microscope was reflected off a dichroic beam splitter (69218, Edmund Optics) and then projected onto the sample through a 1.4-NA 100x oil immersion objective (UPlanSApo, Olympus). A custom Labview program synchronized the timing of the X-Y scan with the EOM sinusoidal modulation frequency to produce the desired patterns of incident NIR light onto the imaging plane. 2P excited fluorescent emissions were then collected by the objective and passed through the dichroic beam splitter. A NIR filter (49822, Edmond Optics) was placed before an imaging EM-CCD (C9100-13, Hamamatsu) to remove stray laser light. The detector integration time, EOM and scanning mirrors were synchronized to achieve a single image for each incident angle and phase. We selected an EM-CCD detector for lower dark current and decreased picture acquisition time, however any sensitive array detector can be used if it provides sufficient SNR. Image reconstruction was done using a homemade Matlab program.
Two types of phantom samples composed of fluorescent nanospheres were fabricated to (1) measure the experimental resolution of our 2P and 2P-SPIM systems and (2) optimize parameters for cellular imaging. For resolution quantification, 110 nm diameter fluorescent nanospheres (Fluoresbrite Plain YG 0.1 Micron Microspheres, Polyscience Inc.) were deposited onto a treated hydroscopic glass substrate via spin coating. The fluorescent nanospheres were chosen for their high resistance to photobleaching and for their size below the maximum possible resolution of the first harmonic 2P-SPIM. For an ideal imaging demonstration, a second phantom was created by depositing 200 nm fluorescent nanospheres (Fluoresbrite Plain YG 0.2 Micron Microspheres, Polyscience Inc.) using the same method previously described. For both phantoms, the spin speed in spin coating was carefully adjusted to give a monolayer of spheres, which was confirmed by SEM prior to imaging. For biological imaging, HeLa cells were cultured and affixed on glass coverslips in PFA mounting media. The cytoskeleton of the cell was stained with a fluorescent contrast agent (Alexafluor 546 phalloidin, Lifetechnologies) before the slides were sealed against a second glass slide for imaging. Three-dimensional volumetric imaging was performed on GFP infused ganglion axonal structures in retinal tissue [37]. Retinal slices were fixed in mounting media and sealed between two glass slides.
We quantified the resolution of 2P-SPIM using 110 nm fluorescent nanospheres. The theoretical maximum resolution using the first harmonic should be approximately 135 nm. In Fig. 1d, the full-width at half-max (FWHM) of the PSF was measured from single nanospheres of direct 2P and 2P-SPIM reconstructed images. As shown in Fig. 1d, experimental results indicated a resolution of 270 nm for direct 2P microscopy and 141 nm for 2P-SPIM, respectively, which demonstrated a 1.9-fold improvement in lateral resolution.
To confirm the resolving power of our system, we imaged both congested and sparse self-assemblies of 200 nm nanospheres under glycerol, respectively shown in Figs. 2 (a-c) and Figs. 2 (d-e). SEM comparison from a similar sample was used to verify the single layer self-assembly of the nanospheres and image small assemblies for comparison [Fig. 2 (f)]. Sphere structures can clearly be resolved using 2P-SPIM, whereas the diffraction-limited 2P microscope failed to show the triple-nanosphere assembly. These results confirmed the improved spatial resolution of 2P-SPIM and allowed us to optimize it for biological samples.
Figure 2.
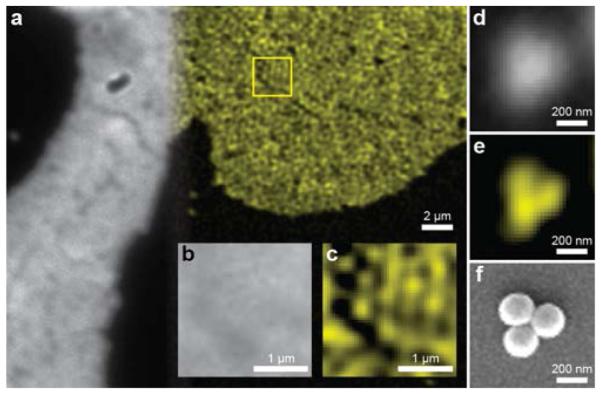
(a) Comparison between direct 2P (gray-scale) and 2P-SPIM (pseudo-color) images of same fluorescence nanosphere phantom. A region within the sample as highlighted by the yellow box was magnified to compare (b) direct 2P microscopy and (c) 2P-SPIM. The monolayer of the 200 nm fluorescent spheres can be resolved using 2P-SPIM. Smaller clusters of spheres were also imaged using (d) direct 2P microscopy and (e) 2P-SPIM to compare structures visualized by (f) SEM microscopy.
An image of the actin cytoskeleton of a HeLa cell using 2P-SPIM is shown in Fig. 3. Laser fluence was decreased (< 13KW/cm2) in cellular studies to combat photobleaching. As expected, direct 2P microscopy of the cytoskeleton yielded diffraction-limited resolutions. Comparatively, the 2P-SPIM image increased in both contrast and resolution. Fine features and spacing, which were not observable in direct 2P microscopy, were clearly visible in 2P-SPIM images as shown in Fig. 3 (b) and Fig. 3 (c). Line-outs of two nearby microtubules from the cellular images suggest that the experimental resolution was approximately 180 nm [Fig. 3 (d)]. The decreased resolution compared with fluorescence nanosphere imaging can be explained by the red-shifted emission wavelength and lower SNR from the fluorescent contrast agent.
Figure 3.
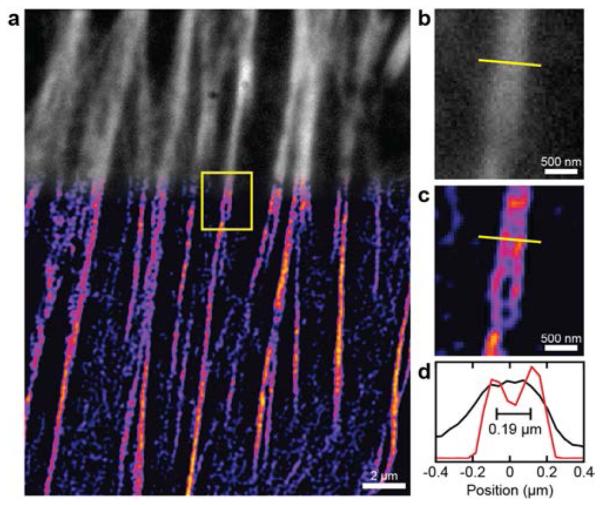
(a) Direct 2P microscopy (grayscale) and 2P-SPIM (pseudo-color) images of the actin cytoskeleton in the same HeLa cell stained with Alexafluor 546 phalloidin. (b) Magnified direct 2P microscopy image of the region within the yellow box. (c) Magnified 2P-SPIM microscopy image of the region within the yellow box. (d) One-dimensional profiles of imaged microtubules at the location highlighted by the yellow lines. Resolution is 180 nm in 2P-SPIM image.
In vitro volumetric imaging of retinal ganglion cell dendrites is shown in Fig. 4. A modified rabies virus was used to produce strong expression of GFP in a retinal ganglion cell [37-39]. The retina was excised and fixed, and the GFP signal was further intensified by applying an anti-GFP primary antibody followed by an Alexafluor488-linked secondary antibody. The labeled ganglion cell was excited using a 780 nm incident beam. Intense fluorescence allowed for lower laser fluence (< 10 KW/cm2), shorter imaging times and higher resolutions as compared to HeLa cell samples. At an imaging depth greater than 100 μm, a lateral resolution of ~145 nm can still be realized.
Figure 4.
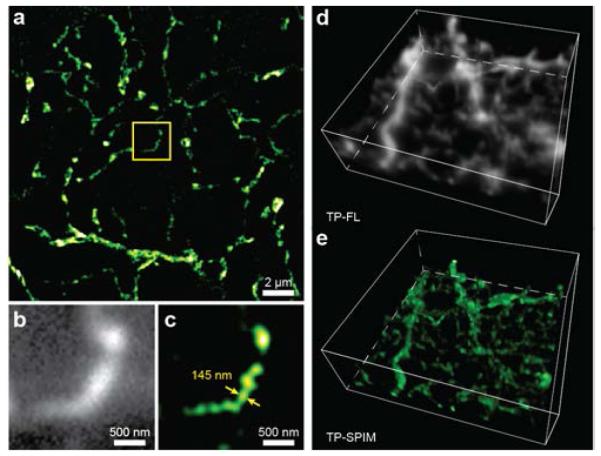
Volumetric imaging using 2P-SPIM. (a) Two-dimensional 2P-SPIM reconstructed image of ganglion cell dendrites in ground squirrel retina at a depth of 100 μm from the vitreal surface. A magnified comparison (within the yellow box) of (b) direct 2P microscopy (grayscale) and (c) 2P-SPIM (pseudo-colored) images. The 2P-SPIM reconstruction can resolve the object with a size of ~145 nm, which is still much beyond its diffraction limit, at the depth of 100 μm. Three-dimensional image stacks at depths from 94 μm to 110 μm in the retinal tissue were created using both (d) direct 2P microscopy (grayscale) and (e) 2P-SPIM (pseudo-colored). A total of 16 slices acquired at 1 μm depth interval were stacked to form the volumetric images. The cell soma, at top, is removed. Z-axis resolution is limited by the 2P point spread function.
VI. DISCUSSION AND CONCLUSION
There are some limitations in 2P-SPIM that should be considered. The experimentally achievable resolution of the 2P-SPIM is limited by the modulation depth and temporal modulation frequency of the projected incident light grating pattern. Next, the signal necessary for 2P-SPIM reconstruction is significantly higher than standard 2P microscopy due to the reliance on ballistic photons. The EOM in our current setup limited the modulation depth peak values to around 30% for the chosen incident beam fluence and separation frequency of the projected grating pattern. Such modulation value was sufficient, although not ideal, for super resolution image reconstruction. Increasing the temporal modulation period increased modulation depth, but resulted in lower resolution due to overlapping grating edges of the scanned pattern. Increasing beam fluence also increased SNR; however it negatively affected modulation depth due to enlarged volumetric excitation. Therefore, beam fluence and temporal period must be adjusted to achieve sufficient modulation depth and SNR for super resolution reconstruction.
2P absorption efficiency, quantum yield, and photobleaching properties of contrast agents need careful consideration in 2P-SPIM. Ideally, 2P fluorescent agents should exhibit high resistance to photobleaching, efficient 2P-absorption and quantum yield (QY). Poor emission intensity requires longer detector integration times, increasing background noise and image acquisition time, while poor 2P-absorption and QY force the use of higher laser fluence which can potentially damage biological tissues. In practice, the majority of fluorescent agents have more serious photobleaching under 2P excitation, causing catastrophic issues with image reconstruction. Due to the patterned illumination, sufficiently high beam fluence in 2P-SPIM can induce grid shaped patterns in the reconstructed image due to photobleaching along the original modulated angles. Thus, to avoid reconstruction artifacts, the optimal spatial frequency separation and laser fluence parameters must be individually predetermined for each contrast agent to achieve an optimal balance between SNR and minimal photobleaching.
To summarize, we created a laser scanning patterned illumination technique to realize 2P super-resolution microscopy with a resolution of 141 nm. Experimental results indicate an improvement by a factor of 1.9 in lateral resolution. Compared to other reported methods, our technique requires little modification to existing 2P microscopy systems, making quick implementation possible for numerous groups. Moreover, frequency domain reconstruction is used to allow higher theoretical resolution. Imaging speed of 2P-SPIM was nine times slower than direct 2P microscopy. By using an optimized scanning system, video-rate image acquisition is technically achievable with sufficiently bright contrast agents. With penetration depths significantly deeper than single photon SIM, wide beam line tunability, and a multiplex of contrast agents, 2P-SPIM is ideal for tissue studies where larger penetration depths and resolutions greater than the diffraction limit are necessary.
Acknowledgements
We thank Christina Chan from our lab for her help with cell sample preparation and Dr. Kevin Jia and Mrs. Julie Ives from Olympus for experimental assistance. We also acknowledge the generous financial support from the National Institutes of Health (R01EY019951, R24EY022883, R01EY019484 and R01EY018204) and National Science Foundation (DBI-1353952, CBET-1055379, and CBET-1066776). Ji Yi is also supported by a postdoctoral fellowship from Juvenile Diabetes Research Foundation International.
References
- [1].Rust MJ, Bates M, Zhuang X. Nature methods. 2006;3:793. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Henriques R, Griffiths C, Hesper Rego E, Mhlanga MM. Biopolymers. 2011;95:322. doi: 10.1002/bip.21586. [DOI] [PubMed] [Google Scholar]
- [3].Heller I, Sitters G, Broekmans OD, Farge G, Menges C, Wende W, Hell SW, Peterman EJ, Wuite GJ. Nature methods. 2013;10:910. doi: 10.1038/nmeth.2599. [DOI] [PubMed] [Google Scholar]
- [4].Gustafsson MG. Journal of microscopy. 2000;198:82. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- [5].Hell S, Stelzer EH. Optics Communications. 1992;93:277. [Google Scholar]
- [6].Gugel H, Bewersdorf J, Jakobs S, Engelhardt J, Storz R, Hell SW. Biophysical journal. 2004;87:4146. doi: 10.1529/biophysj.104.045815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gustafsson M, Agard D, Sedat J. Journal of microscopy. 1999;195:10. doi: 10.1046/j.1365-2818.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- [8].Müller CB, Enderlein J. Physical review letters. 2010;104:198101. doi: 10.1103/PhysRevLett.104.198101. [DOI] [PubMed] [Google Scholar]
- [9].Laporte GP, Stasio N, Sheppard CJ, Psaltis D. Optica. 2014;1:455. [Google Scholar]
- [10].Gong W, Si K, Chen N, Sheppard CJ. Optics letters. 2009;34:3508. doi: 10.1364/OL.34.003508. [DOI] [PubMed] [Google Scholar]
- [11].Lim D, Chu KK, Mertz J. Optics letters. 2008;33:1819. doi: 10.1364/ol.33.001819. [DOI] [PubMed] [Google Scholar]
- [12].Mudry E, Belkebir K, Girard J, Savatier J, Le Moal E, Nicoletti C, Allain M, Sentenac A. Nature Photonics. 2012;6:312. [Google Scholar]
- [13].Monticone DG, et al. Physical review letters. 2014;113:143602. doi: 10.1103/PhysRevLett.113.143602. [DOI] [PubMed] [Google Scholar]
- [14].Lim D, Ford TN, Chu KK, Mertz J. Journal of biomedical optics. 2011;16:016014. doi: 10.1117/1.3528656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heintzmann R, Hanley Q, Arndt-Jovin D, Jovin T. Journal of microscopy. 2001;204:119. doi: 10.1046/j.1365-2818.2001.00945.x. [DOI] [PubMed] [Google Scholar]
- [16].Lu J, Min W, Conchello J-A, Xie XS, Lichtman JW. Nano letters. 2009;9:3883. doi: 10.1021/nl902087d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].York AG, Chandris P, Dalle Nogare D, Head J, Wawrzusin P, Fischer RS, Chitnis A, Shroff H. Nature methods. 2013;10:1122. doi: 10.1038/nmeth.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ingaramo M, York AG, Wawrzusin P, Milberg O, Hong A, Weigert R, Shroff H, Patterson GH. Proceedings of the National Academy of Sciences. 2014;111:5254. doi: 10.1073/pnas.1314447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Winter PW, York AG, Nogare DD, Ingaramo M, Christensen R, Chitnis A, Patterson GH, Shroff H. Optica. 2014;1:181. doi: 10.1364/OPTICA.1.000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tauber J, Dlasková A, Šantorová J, Smolková K, Alán L, Špaček T, Plecitá-Hlavatá L, Jabůrek M, Ježek P. The international journal of biochemistry & cell biology. 2013;45:593. doi: 10.1016/j.biocel.2012.11.019. [DOI] [PubMed] [Google Scholar]
- [21].Swayne TC, et al. Current Biology. 2011;21:1994. doi: 10.1016/j.cub.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wheeler R, Mesnage S, Boneca IG, Hobbs JK, Foster SJ. Molecular microbiology. 2011;82:1096. doi: 10.1111/j.1365-2958.2011.07871.x. [DOI] [PubMed] [Google Scholar]
- [23].Olshausen P. v., Defeu Soufo HJ, Wicker K, Heintzmann R, Graumann PL, Rohrbach A. Biophysical journal. 2013;105:1171. doi: 10.1016/j.bpj.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA. Biology open. 2012 doi: 10.1242/bio.20122337. BIO20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Markaki Y, Smeets D, Fiedler S, Schmid VJ, Schermelleh L, Cremer T, Cremer M. Bioessays. 2012;34:412. doi: 10.1002/bies.201100176. [DOI] [PubMed] [Google Scholar]
- [26].Fitzgibbon J, Bell K, King E, Oparka K. Plant physiology. 2010;153:1453. doi: 10.1104/pp.110.157941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gustafsson MG. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13081. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Helmchen F, Denk W. Nature methods. 2005;2:932. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- [29].Richter V, Bruns T, Weber P, Strauss WS, Wittig R, Schneckenburger H. European Conferences on Biomedical Optics. :80890D. (International Society for Optics and Photonics, 2011) [Google Scholar]
- [30].York AG, Parekh SH, Dalle Nogare D, Fischer RS, Temprine K, Mione M, Chitnis AB, Combs CA, Shroff H. Nature methods. 2012;9:749. doi: 10.1038/nmeth.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carlton PM, et al. Proceedings of the National Academy of Sciences. 2010;107:16016. [Google Scholar]
- [32].Fischer RS, Wu Y, Kanchanawong P, Shroff H, Waterman CM. Trends in cell biology. 2011;21:682. doi: 10.1016/j.tcb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Isobe K, et al. Biomedical optics express. 2013;4:2396. doi: 10.1364/BOE.4.002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Choi H, Yew E, Hallacoglu B, Fantini S, Sheppard CJ, So PT. Biomedical optics express. 2013;4:995. doi: 10.1364/BOE.4.000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Therrien O, Aubé B, Pagès S, Koninck PD, Côté D. Biomedical optics express. 2011;2:696. doi: 10.1364/BOE.2.000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].So PT, Dong CY, Masters BR, Berland KM. Annual review of biomedical engineering. 2000;2:399. doi: 10.1146/annurev.bioeng.2.1.399. [DOI] [PubMed] [Google Scholar]
- [37].Zhu Y, Xu J, Hauswirth WW, DeVries SH. The Journal of Neuroscience. 2014;34:7845. doi: 10.1523/JNEUROSCI.2960-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wickersham IR, Finke S, Conzelmann K-K, Callaway EM. Nature methods. 2006;4:47. doi: 10.1038/NMETH999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wickersham IR, Lyon DC, Barnard RJ, Mori T, Finke S, Conzelmann K-K, Young JA, Callaway EM. Neuron. 2007;53:639. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]