

Abstract
Thiocyanate (SCN) is used by the innate immune system, but less is known about its impact on inflammation and oxidative stress. Granulocytes oxidize SCN to evolve the bactericidal hypothiocyanous acid, which we previously demonstrated is metabolized by mammalian, but not bacterial, thioredoxin reductase (TrxR). There is also evidence that SCN is dysregulated in cystic fibrosis (CF), a disease marked by chronic infection and airway inflammation. To investigate antiinflammatory effects of SCN, we administered nebulized SCN or saline to β epithelial sodium channel (βENaC) mice, a phenotypic CF model. SCN significantly decreased airway neutrophil infiltrate and restored the redox ratio of glutathione in lung tissue and airway epithelial lining fluid to levels comparable to wild type. Furthermore, in Pseudomonas aeruginosa–infected βENaC and wild-type mice, SCN decreased inflammation, proinflammatory cytokines, and bacterial load. SCN also decreased airway neutrophil chemokine keratinocyte chemoattractant (also known as C-X-C motif chemokine ligand 1) and glutathione sulfonamide, a biomarker of granulocyte oxidative activity, in uninfected βENaC mice. Lung tissue TrxR activity and expression increased in inflamed lung tissue, providing in vivo evidence for the link between hypothiocyanous acid metabolism by TrxR and the promotion of selective biocide of pathogens. SCN treatment both suppressed inflammation and improved host defense, suggesting that nebulized SCN may have important therapeutic utility in diseases of both chronic airway inflammation and persistent bacterial infection, such as CF.
Keywords: hypothiocyanous acid, innate immunity, antioxidant, glutathione sulfonamide, nebulization
Clinical Relevance
Thiocyanate is an endogenous pseudohalide that plays an important role in lung host defense. These studies show that thiocyanate may also have novel antiinflammatory effects when given by inhalation in the lungs of the β epithelial sodium channel mouse with and without Pseudomonas aeruginosa infection.
The halogenation cycle of chordate peroxidases (e.g., lactoperoxidase, myeloperoxidase [MPO]) is a primary component of the innate immune system, catalytically evolving hypohalous acids (HOXs) from a halide and H2O2 (1). Hypochlorous acid (HOCl; i.e., chlorine bleach) is seemingly the most toxic and produced from chloride oxidation by MPO compound I. HOCl is a highly reactive oxidant associated with neutrophilic inflammation (2–4). Thiocyanate (SCN) is a ubiquitous pseudohalide in mammalian secretions, and is also oxidized by chordate peroxidases to yield hypothiocyanous acid (HOSCN) (5, 6), a milder, thiol-selective oxidant that is better tolerated by mammalian cells (7–10). SCN is the preferred two-electron substrate of chordate peroxidases (11, 12), replacing injurious HOX species with HOSCN in the inflammatory milieu (depicted in Figure 1). This reaction is primarily limited by the SCN concentration in extracellular fluids during inflammation (11). Similarly, SCN rapidly reduces HOX species, including HOCl (pseudo–first order rate constant, k = 2.34 × 107 M−1 s−1) and its chloramine metabolites, to form HOSCN (13, 14). Thus, HOSCN is the major end product of the mammalian halogenation cycle when SCN is abundant.
Figure 1.

Thiocyanate (SCN) influences mammalian biochemistry and inflammation. Scheme depicting the parallel paths of SCN to remove hypohalous acid (HOX) and their metabolites (e.g., monohaloamines, RNHX) from biological systems, avoiding irreversible damage and toxicity, while maintaining host defense. SCN is catalytically oxidized by peroxidases and can also directly and rapidly reduce HOX, producing hypothiocyanous acid (HOSCN) in each case. Thioredoxin reductase (TrxR) catalytically reduces HOSCN, which increases host tolerance of this species, while sustaining a host defense response. HOX is capable of irreversible macromolecular damage and may cause toxicity, in addition to its host defense function. The scheme is driven by inflammation, which provides a significant source of H2O2 and peroxidases. Modified with permission from Ref. 5. EPO, eosinophil peroxidase; LPO, lactoperoxidase; MPO, myeloperoxidase; NADPH, reduced nicotinamide adenine dinucleotide phosphate.
We previously determined that mammalian cells metabolize HOSCN through catalytic reduction by thioredoxin reductase (TrxR) (15) (Figure 1). Mammalian TrxR1 (cytosolic) and TrxR2 (mitochondrial) are selenoenzymes that rapidly reduce HOSCN in vitro, in cell lysates and in whole epithelia, to SCN and H2O. Bacteria encode a phylogenetically divergent family of TrxR genes unable to metabolize HOSCN and potently inhibited by it, contributing to bacterial growth arrest (15). This duality supports the function of HOSCN as selective biocide against pathogens, and highlights an important yet largely unappreciated role for mammalian TrxR in inflammation and innate immunity.
Cystic fibrosis (CF) lung disease results from one or more dysfunctional copies of the CF transmembrane conductance regulator (CFTR) gene (16) that cause decreased expression, maturation, and/or function of CFTR. CFTR is an apical ion channel that actively transports chloride, SCN, and other anions (17–20), and regulates the activity of other ion channels (e.g., the epithelial sodium channel [ENaC], which absorbs Na+ and H2O) (21). Although airway chloride is not affected in patients with CF, studies have reported deficient SCN secretion in human CF cells (22, 23), CFTR-knockout (KO) mice (9), and human patients (24). Furthermore, decreased SCN secretion by primary CF epithelia enhances bacterial survival (22, 23), SCN in nasal lining fluid correlates with better lung function (25), and hypersensitivity of CF epithelia to HOCl was ablated with replacement of CFTR, which restored SCN efflux (9). Although clearly not absent in CF secretions, dysregulation of SCN may contribute to chronic inflammation and dysfunctional bacterial clearance in the CF airway (5), providing a rationale for testing SCN as a therapeutic intervention in CF and related diseases.
In the present work, we used transgenic, airway-targeted ENaC subunit β (βENaC)-overexpressing mice as a constitutively inflamed model of the CF airway (26). Although CFTR KO mice share some of the antioxidant-deficient characteristics noted in CF (9), their airway physiology, cytokines, and cellularity resemble those of a healthy mouse (27), perhaps due to differential expression of CFTR and other anion transporters between mice and humans. βENaC mice more closely model spontaneous CF airway pathologies, such as increased mucus, impaired mucociliary clearance, and chronic neutrophilia (26), compared with CFTR KO mice. βENaC mice have not been investigated regarding the low basal epithelial lining fluid (ELF) SCN reported in CFTR KO mice (9); however, these studies were intended to assess the effects of supraphysiologic SCN in mice predisposed to inflammation rather than to compare the effects of different baseline values.
We nebulized βENaC mice and their wild-type (WT) littermates with SCN or saline, with or without Pseudomonas aeruginosa lung infection. SCN decreased airway neutrophilia, morbidity, lung bacteria, and cytokines in infected WT and βENaC mice, and decreased constitutive airway neutrophilia and neutrophil chemokine keratinocyte chemoattractant (KC; also known as C-X-C motif chemokine ligand 1) in uninfected βENaC mice. SCN also inhibited the accumulation of oxidized glutathione (GSSG) and glutathione sulfonamide (GSA), a HOCl-specific metabolite of reduced glutathione (GSH) (28), in uninfected βENaC mice. In addition, lung TrxR expression increased concurrent with inflammation, supporting this enzyme as a regulator of inflammation outcomes and HOSCN metabolism.
Materials and Methods
Additional details are available in the Materials and Methods in the online supplement.
Chemicals
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO), with the exceptions of bovine fibrinogen (MP-Biomedical, Santa Ana, CA) and human thrombin (GenTrac, Bristol, TN).
Tissue Culture
J774A.1 murine macrophage-like cells (TIB-67; ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagles medium with 10% FBS and antibiotics. Cells were plated in six-well plates at 2 × 105 cells ml−1 and used the following day. Cells were washed with prewarmed PBS containing 1.26 mM CaCl2 and 812 μM MgCl2, then treated with the indicated concentrations of HOCl and/or SCN in PBS for 15 minutes at 37°C. Afterward, buffer was replaced with media for 6 hours, and then cells were harvested by gentle scraping. Typical yield per well was approximately 106 cells. Cell culture experiments were repeated twice.
Flow Cytometry
Annexin V and propidium iodide (PI) were used to assess cell death, according to the manufacturer’s instructions (BD Biosciences, San Jose, CA). Cells were washed once with cold PBS, and then resuspended in 1 ml annexin V binding buffer (10 mM HEPES-NaOH, pH 7.4, with 140 mM NaCl and 2.5 mM CaCl2). Cells (105) were mixed with annexin V and PI for 15 minutes, then diluted to 300 μl with binding buffer and analyzed by FACSCalibur (BD Biosciences). Data was analyzed using FlowJo software version 9 (TreeStar Inc., Ashland, OR).
Animals
Airway-targeted βENaC–overexpressing mice (006438; Jackson Laboratories, Bar Harbor, ME) were bred in-house and maintained with nontransgenic littermates on C57BL/6J background. To initially characterize inflammation in the βENaC mice, six animals were killed per group. To study the effects SCN with or without infection, six mice were used per group with eight groups, for a total of 48 mice after completion of all experiments, which were repeated once. Males and females, aged 8–14 weeks, were distributed evenly to all groups according to both age and sex. Body weights were measured at 7:00 a.m. each day. Mice were killed 96 hours after infection (uninfected mice followed the same treatment schedule as infected mice), and samples were collected as previously described (10). For bronchoalveolar lavage (BAL), two sequential 0.75-ml aliquots of 310 mOsm L−1 sodium phosphate buffer (pH 6.8) were instilled once each and collected. Mean fluid recovery was 1.06 (±0.02) ml, or 70.6 (±1.5)% instilled. Urea dilution factor was measured as previously described (29), and the mean dilution in all study data was 63.1 (±3.9). Animal studies received prior approval by the National Jewish Health Animal Care and Use Committee, and were maintained in compliance with their guidelines.
Infection
Mucoid P. aeruginosa was clinically isolated from the sputum of a subject with CF by culturing on lysogeny broth agar, and verified as P. aeruginosa with cetrimide selection agar. Bacteria were administered to mice intratracheally as previously described (10). A total of 3.0 × 108 CFU of P. aeruginosa were instilled. SCN intervention began 24 hours after infection. Lung CFUs were determined by homogenizing whole left lung tissue in sterile saline and plating neat and diluted (101–103) homogenate on LB agar. Colonies were assessed after 24 hours at 37°C.
SCN Formulation and Administration
Normal 0.9% (wt/vol) saline and normal saline with SCN (0.5% NaSCN and 0.54% NaCl) were prepared, filter sterilized, and administered as previously described (10). Pharmacokinetics of this therapy in ELF and plasma of C57BL/6J mice were characterized previously (10). Mice were treated every 12 hours starting 24 hours after infection (uninfected mice followed the same treatment schedule) and ending 12 hours before being killed at 96 hours, for a total of six treatments at 24, 36, 48, 60, 72, and 84 hours after infection.
Cytology of BAL Cells
Leukocyte counts were determined as previously described (10). Cells were isolated from BAL fluid (BALF) by centrifugation (2,000 × g for 8 min at 4°C) and erythrocytes were removed with lysis buffer. Cells were resuspended in Isoton fluid (Beckman Coulter, Brea, CA) and a 100-μl volume containing 15,000 cells was loaded on Shandon Cytospin 3 cassettes with poly-L-lysine slides (Thermo Fisher, Waltham, MA) (600 rpm × 3 min). Cells were fixed and stained for identification using the Hema 3 kit (Fisher, Hampton, NH). A total of 100 cells was counted per field over four fields, and the relative proportion of leukocytes was normalized to the total leukocyte count.
Cytokines
Proinflammatory cytokines were assessed from BALF using the Pro-inflammatory Panel 1 (mouse) V-Plex Kit (Meso Scale Discovery, Rockville, MD). Analytes that were not above the lower limit of detection in three or more individuals per group were considered not detectable. Values greater than the upper limit of detection are noted in the legend of Figure 5.
Figure 5.

Cytokine panel from infected mice. Cytokines were assessed in BALF from the same mice described in Figure 4 using the Meso Scale Discovery 10-cytokine panel ELISA-like assay. Cytokines included (A) KC (βENaC, P < 0.001; SCN, P < 0.01; interaction, P > 0.05), (B) IL-1β (βENaC, P < 0.001; SCN, P < 0.01; interaction, P > 0.05), (C) TNF-α (βENaC, P < 0.01; SCN, P < 0.01; interaction, P > 0.05), (D) IL-6 (βENaC, P < 0.05; SCN, P > 0.05; interaction, P > 0.05) (one value in the WT+Pa column, two values in the ENaC+Pa column, and one value in the ENaC+Pa+SCN column are above the upper limit of detection for the IL-6 assay), (E) IFN-γ (βENaC, P > 0.05; SCN, P > 0.05; interaction, P > 0.05), (F) IL-12 p70 (βENaC, P > 0.05; SCN, P < 0.05; interaction, P > 0.05), (G) IL-10 (βENaC, P > 0.05; SCN, P < 0.01; interaction, P > 0.05), (H) IL-4 (βENaC, P > 0.05; SCN, P > 0.05; interaction, P < 0.01), (I) IL-2 (βENaC, P > 0.05; SCN, P > 0.05; interaction, P > 0.05), and (J) IL-5 (βENaC, P > 0.05; SCN, P > 0.05; interaction, P > 0.05). One-way ANOVA: *P < 0.05, **P < 0.01 compared with WT + Pa; #P < 0.05, ##P < 0.01, ###P < 0.001 comparing SCN and vehicle within a genotype. Data are expressed as mean (±SEM).
Statistical Analysis
Data are expressed as mean (±SEM). Prism 6 (GraphPad Software, La Jolla, CA) was used to perform and evaluate one-way ANOVA with Holm-Sidak multiple comparison test after log transformation to minimize variance or parametric t test with the F test for equal variances. Two-way ANOVA was also performed for all nested experimental designs (e.g., comparison of WT or βENaC treated with either vehicle or SCN), and the results for each factor and their interaction are given in the figure legends. Significance level for all tests was P less than 0.05. For one-way ANOVA results, asterisks denote significant difference of a variable group against vehicle-treated WT (animal studies) or of a variable group against control (cell culture), whereas pound symbols denote significant difference between vehicle and SCN within a genotype (animal studies) or HOCl and HOCl + SCN (cell culture). Where infected and uninfected animal data occur together, the uninfected, vehicle-treated WT serves as the primary control. For t test results, symbols are shown over a line connecting the two groups tested.
Results
SCN Protects Macrophage (J774A.1) Cells from HOCl-Mediated Necrosis
Macrophage-derived J774A.1 cells were exposed to 100 μM HOCl for 15 minutes with or without coadministered SCN. The dose of 400 μM SCN was chosen to model a pharmacologically achievable value previously established to convert a near-totality of HOX and/or peroxidase compound I to HOSCN in vitro (8, 15). After the exposure time, cells were submerged in fresh media for 6 hours and then harvested for annexin V–FITC and PI staining and flow cytometry (Figure 2A; see representative dot plots in Figure 2B). HOCl induced significant necrosis in J774A.1 cells (∼70% of gated cells) and coadministered SCN ablated HOCl-mediated necrosis. SCN alone had no effect.
Figure 2.

SCN protects J774A.1 cells from hypochlorous acid (HOCl)–mediated necrosis. Cells were exposed to 100 μM HOCl and/or 400 μM SCN in PBS for 15 minutes and given fresh media for 6 hours before harvest and staining for flow cytometry. (A) Quantification of annexin V and propidium iodide (PI) positive-gated cells by percentage of the total gated cell population (annexin V: HOCl P < 0.001, SCN P < 0.001, interaction P < 0.001; PI: HOCl P < 0.001, SCN P < 0.001, interaction P < 0.001) (results shown in parentheses are from two-way ANOVA). (B) Representative images of gated J774 cells stained with annexin V and PI. ***P < 0.001 compared with control; ###P < 0.001 compared with HOCl. Data are expressed as mean (±SEM).
Nebulized SCN Ablates Inflammation and Oxidative Stress in βENaC Mice
Naive βENaC mice were lavaged and leukocytes assessed to characterize airway inflammation. Neutrophils made up a significant portion of BAL cells in βENaC mice (see Figure E1A in the online supplement). Lung tissue from βENaC mice did not yield colonies in bacterial culture. Because neutrophilia is a potential source of oxidative stress, we hypothesized the GSH:GSSG redox ratio may be compromised in βENaC mice. The GSH:GSSG ratio was significantly decreased in the lung tissue of βENaC mice compared with WT, and a similar trend was observed in ELF (Figure E1B), suggesting that chronic inflammation causes significant oxidative stress in the airways of these mice.
To investigate the effect of SCN on unchallenged βENaC mice, nebulized SCN or saline vehicle was administered every 12 hours for 3 days, and mice were killed 12 hours after the final treatment. Airway leukocytes, proinflammatory cytokines, and GSH and GSSG in lung tissue and ELF were assessed. The 12-hour dosing regimen of 0.5% SCN was chosen based on previous observation of a 12-hour clearance time after a single dose (10); no significant differences in drug delivery were observed between βENaC mice and WT littermates (data not shown). No significant changes in body mass occurred over the course of the study (data not shown). Airway neutrophils were significantly increased in βENaC mice and decreased 68% by SCN treatment compared with vehicle (Figure 3A). Lymphocytes and macrophages obtained from BAL were significantly elevated in βENaC mice compared with WT, but SCN treatment had no effect (although lymphocytes in βENaC mice declined 48% with SCN treatment; Figure 3A). The neutrophil chemokine, KC (analog to human IL-8), was increased 15-fold in βENaC mice compared with WT, and decreased 40% with SCN treatment in βENaC mice (Figure 3B). All other detectable cytokines from the 10-cytokine panel were not affected by SCN treatment, though IL-1β and TNF-α were increased in βENaC mice (Figures 3C–3F). SCN treatment lowered steady-state levels of GSSG in lung tissue of βENaC mice, resulting in a GSH:GSSG redox ratio similar to WT; a similar trend was observed in ELF (Figures 3G and 3H). Steady-state GSH levels were similar for all groups. Nebulized SCN also increased ELF SCN 66 and 106% compared with vehicle control in WT and βENaC mice, respectively, whereas, in lung tissue, WT SCN levels were unchanged, but βENaC mice SCN levels increased 53% (Figure E2). In addition, mean ELF SCN was decreased 60% in vehicle-treated βENaC mice compared with WT mice (Figure E2B).
Figure 3.

Nebulized SCN rescues airway inflammation and oxidative stress in β-epithelial sodium channel (βENaC) mice. Wild-type (WT) and βENaC mice were nebulized with vehicle or SCN twice daily for 3 days. Two-way ANOVA results are given in parentheses here. (A) Bronchoalveolar lavage (BAL) macrophages (βENaC, P < 0.001; SCN, P > 0.05; interaction, P > 0.05), neutrophils (βENaC, P < 0.001; SCN, P < 0.05; interaction, P < 0.05), and lymphocytes (βENaC, P < 0.001; SCN, P < 0.05; interaction, P > 0.05) were assessed in relative proportion by differential stain and normalized to the total number of leukocytes quantified. (B) Keratinocyte chemoattractant (KC; also known as C-X-C motif chemokine ligand 1) (βENaC, P < 0.001; SCN, P < 0.05; interaction, P > 0.05), (C) IL-1β (two-way ANOVA impossible), (D) TNF-α (βENaC, P < 0.001; SCN, P > 0.05; interaction, P > 0.05), (E) IL-6 (βENaC, P > 0.05; SCN, P > 0.05; interaction, P > 0.05), and (F) IL-4 (βENaC, P > 0.05; SCN, P > 0.05; interaction, P > 0.05) were assessed in BAL fluid (BALF). Other cytokines in the panel were not detectable. Reduced glutathione (GSH) and oxidized glutathione (GSSG) were measured in (G) lung tissue (GSH: βENaC P > 0.05, SCN P > 0.05; interaction P > 0.05; GSSG: βENaC P < 0.001, SCN P < 0.01; interaction P < 0.01) and (H) epithelial lining fluid (ELF) (GSH: βENaC P > 0.05, SCN P > 0.05; interaction P > 0.05; GSSG: βENaC P > 0.05, SCN P > 0.05; interaction P > 0.05). ND, not detectable. One-way ANOVA: *P < 0.05, ***P < 0.001 compared with WT within a treatment; ##P < 0.01, ###P < 0.001 comparing SCN and vehicle within a genotype. Data are expressed as mean (±SEM).
Nebulized SCN Improves Lung Infection Outcomes in βENaC and WT Mice
Spontaneous P. aeruginosa infection was examined in 6-day-old pups from our βENaC colony. Lung homogenates from either WT or βENaC mice lacked any growth for P. aeruginosa. To investigate the effect of SCN on infection outcomes in βENaC mice, we intratracheally infected mice with a clinical isolate of P. aeruginosa, intervening 24 hours later with vehicle or SCN. Body weights declined by a mean value of 4.6% in the first 24 hours with infection (Figure 4A), but, by 48 hours, WT mice that received SCN were stabilizing (cumulative mean = −5.9%), whereas βENaC mice receiving vehicle were doing significantly worse than other groups (cumulative mean = −13.9%). Vehicle-treated WT and βENaC + SCN mice were in between. By 72 hours, SCN-treated mice were significantly less morbid than vehicle-treated mice (Figure 4A). At 96 hours, all groups inflected positive slopes for weight gain, but vehicle-treated βENaC mice were significantly more morbid than all other groups (Figure 4A). Mean weight loss by 96 hours was 0.3% for WT + SCN, 7.4% for vehicle-treated WT, 7.3% for βENaC + SCN and 14.0% vehicle-treated βENaC mice.
Figure 4.

Nebulized SCN rescues βENaC and WT mice from morbidity, lung infection, and inflammation. Mice were infected with 3.0 × 108 CFUs of clinical Pseudomonas aeruginosa strain and followed for 96 hours, including 72 hours of twice-daily intervention with vehicle or SCN. Two-way ANOVA results are given in parentheses here. (A) Mouse body weight was monitored daily at 7:00 a.m. (24 h: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05; 48 h: βENaC P < 0.001, SCN P < 0.05, interaction P > 0.05; 72 h: βENaC P < 0.001, SCN P < 0.001, interaction P > 0.05; 96 h: βENaC P < 0.001, SCN P < 0.001, interaction P > 0.05). (B) Lung bacterial burden was assessed by colonies that grew from lung tissue homogenate (βENaC, P < 0.001; SCN, P < 0.01; interaction, P > 0.05). (C) BAL macrophages (βENaC, P > 0.05; SCN, P < 0.001; interaction, P > 0.05), neutrophils (βENaC, P < 0.05; SCN, P < 0.01; interaction, P > 0.05), and lymphocytes (βENaC, P < 0.01; SCN, P > 0.05; interaction, P > 0.05) were assessed by differential stain as in Figure 3. One-way ANOVA: *P < 0.05, ***P < 0.001 (A) compared with WT + Pa + SCN or (B and C) compared with WT + Pa; #P < 0.05, ##P < 0.01, ###P < 0.001 (A) compared with βENaC + Pa or (B and C) comparing SCN and vehicle within a genotype. Pa, P. aeruginosa. Data are expressed as mean (±SEM).
Lung homogenate was plated on LB agar to culture P. aeruginosa. Paralleling trends observed in morbidity, WT mice that received SCN had 70% less bacterial burden than vehicle-treated WT (Figure 4B). βENaC mice cultured 80-fold more bacteria than WT, but SCN treatment significantly decreased bacterial burden by 92% (Figure 4B). Infection with P. aeruginosa caused an influx of neutrophils into the airway in both WT and βENaC mice, which was significantly decreased 66% in WT and 72% in βENaC mice nebulized with SCN (Figure 4C). Macrophages were significantly increased in WT and βENaC mice given SCN, whereas lymphocytes were significantly increased over threefold in βENaC mice compared with WT with no SCN effect. Infection also increased expression of cytokines related to inflammation in WT and βENaC mice, including five that were not detected in uninfected mice (IFN-γ, IL-12 p70, IL-10, IL-2, and IL-5; Figure 5). Infected βENaC mice expressed 189% more KC than WT, whereas SCN significantly decreased KC expression 82 and 75% in both WT and βENaC mice, respectively (Figure 5A). Similar trends were observed in IL-1β, TNF-α, IL-6, IFN-γ, IL-12 p70, and IL-10 (Figures 5B–5G). IL-4 was significantly decreased in infected βENaC mice and adjusted to WT values with SCN treatment (Figure 5H). IL-2 and IL-5 did not show significant changes in either direction (Figures 5I and 5J). GSH and GSSG were assessed in lung tissue and ELF, but, in contrast to the uninfected mice, no significant changes due to genotype or treatment were observed (Figure E3). Although ELF SCN was decreased in infected βENaC mice compared with WT infected mice, unlike in uninfected mice, there was not a significant difference (Figure E2).
Multiple Evidences of Antioxidant, but Not Antisecretion, Effects of SCN
HOCl is difficult to detect in vivo, because the products of its reaction with thiols (sulfenic, sulfinic, and sulfonic acids) lack specificity for this oxidant. For example, H2O2 and HOSCN also oxidize thiols, although the rate constants differ by orders of magnitude (30, 31). However, GSA is a metabolite of GSH oxidation specific to HOCl that has been identified and characterized by liquid chromatography–mass spectrometry (28, 32, 33). GSA is a cyclic compound formed when 3 mol of HOCl oxidize 1 mol of GSH, with the oxidized cysteine sulfur forming a sulfonamide bond with the amino group of glutamate. Unlike GSSG, GSA is not a substrate of glutathione reductase, which may enhance its accumulation in biologic fluids. GSA was significantly increased 5-fold in the ELF of uninfected βENaC mice and 6- to 8-fold in mice infected with P. aeruginosa (Figure 6A). SCN decreased ELF GSA 40–70% in βENaC mice with or without infection, and decreased basal ELF GSA 60% in WT mice (Figure 6A). MPO is the major biological driver of HOCl formation in vivo, and was significantly increased in the BALF of all infected mice, but was also detectable in uninfected βENaC mice (Figure 6B). Inflammatory peroxidase activity is also associated with nitrogen dioxide radical formation, which can react with tyrosine to form 3-nitrotyrosine (3-NT) (12). 3-NT (expressed as a ratio of tyrosine) was increased in infected WT and βENaC mice, and decreased with SCN in infected WT mice (Figure 6C). Lung tissue protein-bound glutathione (PSSG) was also significantly increased in all infected mice by 3- to 4-fold over control, but neither mouse strain nor SCN treatment affected the results (Figure 6D). Finally, MUC5B, a major component of airway mucins, which may be induced by the pseudomonal toxin pyocyanin (34), was significantly increased by infection and unaffected by SCN treatment (Figure 6E). These changes were also mirrored in BALF protein that was increased by infection and unaffected by SCN treatment (data no shown).
Figure 6.

Markers of inflammation and oxidative stress in inflamed and infected mice. Analytes associated with inflammation, oxidative stress, and airway mucin secretion were measured in BALF or lung tissue lysate. Two-way ANOVA results are given separately for uninfected and infected data sets. (A) Glutathione sulfonamide (GSA) was measured in BALF (corrected to ELF) by liquid chromatography–mass spectrometry (uninfected: βENaC P < 0.01, SCN P < 0.01, interaction P > 0.05; infected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05). (B) MPO was quantified in BALF using an ELISA method (uninfected: two-way ANOVA impossible; infected: βENaC P < 0.05, SCN P > 0.05, interaction P > 0.05). Uninfected WT values of MPO were below the lower limit of detection of the assay (19 ng ml−1). (C) The 3-nitrotyrosine (3-NT) from lung tissue lysate was quantified by electrochemical HPLC, and is expressed as a ratio of native tyrosine (mmol/mol) (uninfected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05; infected: βENaC P > 0.05, SCN P < 0.01, interaction P < 0.05). (D) Protein-bound glutathione was measured from lung tissue lysate (uninfected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05; infected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05). (E) MUC5B was measured in BALF (uninfected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05; infected: βENaC P > 0.05, SCN P > 0.05, interaction P > 0.05). ND, not detectable; PSSG, protein-glutathione disulfides. One-way ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle-treated WT. ##P < 0.01, ###P < 0.001 comparing vehicle and SCN, holding genotype and infection status the same. Data are expressed as mean (±SEM).
Mammalian TrxR Is Induced by Airway Inflammation
Mammalian TrxR is important in metabolizing HOSCN generated during inflammation (15). We hypothesized that an in vivo function of mammalian TrxR is to protect tissue from neutrophilic inflammation by adaptively responding and dampening oxidative species (e.g., HOSCN). Lung tissue from uninfected and infected mice was compared for TrxR activity and protein expression. Infection increased TrxR activity 60% in WT and 44% in βENaC lung tissue, although the increase was only statistically significant in WT mice (Figure 7A). Furthermore, basal TrxR activity in uninfected lung tissue was significantly increased 43% in βENaC compared with WT mice. Lung tissue from mice treated with SCN did not significantly differ in TrxR activity from vehicle-treated mice (Figure E4). Changes in activity were paralleled by trends in protein expression, as measured by Western blot, where infection significantly increased cytosolic TrxR1 expression 185% in WT and 67% in βENaC mice, whereas uninfected βENaC mice expressed 120% more TrxR1 compared with WT mice (Figure 7B; see representative blot in Figure 7C).
Figure 7.
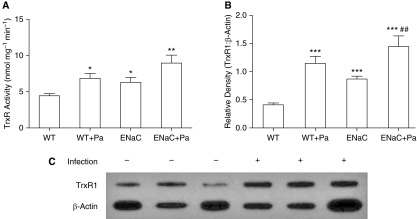
TrxR is induced by inflammation and infection in mouse lung tissue. Mouse lung tissue was homogenized and lysate isolated to assess TrxR activity and protein expression. (A) TrxR activity from lung tissue lysate was normalized to tissue protein (βENaC, P < 0.01; infection, P < 0.01; interaction, P > 0.05). (B) TrxR protein expression was measured by Western blot (βENaC, P < 0.001; infection, P < 0.001; interaction, P < 0.01). (C) Representative blot of TrxR1 and β-actin blots (developed simultaneously) from lung tissue of WT mice with and without infection. *P < 0.05, **P < 0.01, ***P < 0.001 compared with uninfected WT; ##P < 0.01 comparing βENaC uninfected and infected. Data are expressed as mean (±SEM).
Discussion
We previously observed that nebulized SCN improves P. aeruginosa lung infection outcomes in WT mice (10) and that TrxR confers resistance to HOSCN in mammalian cells enhancing selective biocide against pathogens (15). In this study, we observed new findings that SCN improves morbidity, bacterial load, neutrophilia, and proinflammatory cytokines in constitutively inflamed βENaC mice infected intratracheally with P. aeruginosa. SCN also had anti-inflammatory effects in uninfected βENaC mice, including decreased neutrophilia, decreased BALF KC, restoration of GSH:GSSG, and decreased GSA. Novel adaptive increases in lung TrxR were observed in response to both infection and inflammation. Together, these data suggest that inhaled SCN may improve host defense against pathogens, such as P. aeruginosa, promote resolution of inflammation, and restore redox tone. SCN is a favored substrate of chordate peroxidases that perturbs generation of HOX in favor of HOSCN (11, 12). Reductions of HOCl, hypobromous acid, and H2O2 to HOSCN are also antioxidant reactions (change in standard biological reduction potential [ΔE°′] = 570–760 mV) (35). At a concentration of 400 μM SCN, most of the H2O2 consumed by MPO forms HOSCN in vitro (8, 15). Postnebulization ELF SCN peak concentration was approximately 2 mM, and remained at or above 400 μM for the majority of the 12-hour treatment interval, suggesting that HOSCN is the major (pseudo)-HOX species in the inflammatory milieu in these animals instead of HOCl. Replacement of HOX with HOSCN decreases mammalian cell death while promoting bacterial clearance (15), likely decreasing pathogen-initiated cell behaviors in turn (e.g., neutrophil infiltration and cytokine signaling). However, SCN also inhibited airway neutrophilia independent of infectious stimuli, and was associated with better redox tone. Both mechanisms are not mutually exclusive, and comport with enhanced HOCl scavenging, supported by decreased ELF GSA.
The bactericidal function of SCN as a peroxidase substrate was first identified in milk almost 50 years ago (36). However, the antimicrobial properties of exogenously administered SCN in vivo have not been studied as closely. In this study, SCN was administered to mice infected with an acute titer of P. aeruginosa (108 CFU). Previous research has established the bactericidal efficacy of HOSCN as inversely proportional to bacterial load (37), so equal or greater efficacy of this therapy is expected at lower bacterial titers. Morbidity, bacterial burden, and airway inflammation were exacerbated in βENaC mice compared with WT mice infected with equal bacterial load, with significant improvement of these endpoints in both groups given SCN. This suggests that supraphysiologic SCN enhances clearance and resolution of lung infection and inflammation, even in cases of exacerbated inflammatory response.
βENaC mice used in this study are a model of spontaneous CF-like proinflammatory lung disease (26). To the best of our knowledge, we are the first to report that βENaC mice exhibit increased GSSG and GSA and decreased SCN levels in airway compartments. However, this decrease is not as dramatic as that of CFTR KO mice, which exhibit an 80% decrease compared with WT mice (9). Uninfected βENaC mice that received SCN had fewer airway neutrophils and lower KC values. KC is analogous to human neutrophil chemokine, IL-8, which is implicated in the pathogenesis of patients with CF (38). Chronic inflammation and oxidative stress in βENaC mice implicates abnormal, injurious HOCl exposure, which accounts for the majority of H2O2 consumed by MPO when ELF SCN is concentrated at values we observed in unchallenged βENaC mice (39). HOCl has been implicated in oxidative stress in the airways of subjects with CF due to significant increases in the proportion and concentration of GSA (40). Enhanced HOCl scavenging by exogenous SCN should improve redox tone (indicated by decreased GSSG and GSA in SCN-treated mice), which regulates proinflammatory signaling and inflammation in human CF airway epithelial cells and CF mouse models (41, 42).
This study is the first to use direct HOX-targeting therapy in the βENaC mouse model, but other interventions have been tested. Sustained amiloride therapy is an effective preventive measure against early-life mortality, mucus obstruction, and airway inflammation in newborn βENaC mice, but does not improve symptoms in mice aged 5 days or older (43). Airway neutrophils, KC, TNF-α, IL-6, macrophage inflammatory protein-2, and MUC5B, do not respond to prednisolone treatment in βENaC mice, although eosinophils and mucus secretory cells decrease (44). Preventive treatment with intranasal hypertonic saline (HS) improves βENaC mouse survival, but also causes significant neutrophilic inflammation, whereas 7% HS at 4 weeks of age causes increased neutrophilia in WT littermates, but decreases KC in βENaC mice (45). Isotonic SCN used in our study appears more effective than HS in reducing neutrophilia, while also being less irritating to unchallenged mice. Furthermore, SCN was effective against parameters resistant to glucocorticoids (e.g., neutrophilia and cytokines) (44), and is effective as a late-intervention therapy, whereas amiloride and HS were either totally or mostly limited to preventive effects (43, 45). MUC5B protein in BALF from this study (adult mice) was not significantly responsive to SCN, suggesting that SCN may not alter airway mucus obstruction, particularly after onset of disease.
Oxidative stress imposed by excessive H2O2 in CF results in chronic production of IL-8 and IL-6 (42). The oxidizing GSH:GSSG redox ratio observed in βENaC mice may yield increased H2O2 and proinflammatory cytokines. Although uninfected βENaC mice did not show an increase in IL-6, they did express significantly more KC, IL-1β, and TNF-α. We have previously shown in J774A.1 cells that, unlike GSH (46), SCN does not directly inhibit LPS-induced p65 trafficking (10). However, in vivo administration of SCN decreased oxidation of GSH and repressed KC in the uninfected βENaC mice. Furthermore, KC, IL-1β, TNF-α, IL-12 p70, and IL-10 decreased in infected mice treated with SCN (most significant results by two-way ANOVA). This suggests that SCN indirectly inhibits expression of many cytokines via enhanced bacterial clearance and faster resolution of illness, whereas there may be a more direct relationship with SCN, KC, and neutrophils mediated by changes in redox tone. However, βENaC mice also expressed much more KC than other cytokines, which may have made it easier to detect changes in its concentration.
Although GSSG accumulated in uninfected βENaC mouse lung tissue, GSSG accumulation was not observed in infected βENaC mice. Likewise, there was no effect of SCN on GSSG in these mice. However, infection significantly increased lung tissue PSSG in both mouse strains. Preservation of soluble GSH:GSSG during infection and/or compartmentalizing glutathione in protein may be an adaptation to maintain or alter critical physiological functions dependent on this redox couple (47). S-glutathionylation of Fas receptor has recently been reported to aid in airway bacterial clearance (48). Furthermore, MPO- and the GSH-derived HOCl metabolite GSA (28) were increased in uninfected βENaC mice and infected mice of both strains. However, GSA was decreased with SCN treatment in both infected and uninfected βENaC mice. These results suggest that GSA is a sensitive marker of HOCl generated during inflammation, and provide in vivo evidence of the competition of SCN and chloride for MPO compound I (11). GSA is a relevant biomarker of inflammation and oxidative stress in CF children (40). Other markers of either oxidative (3-NT) or hypersecretory (MUC5B) phenotypes were not constitutively increased in the βENaC mouse strain. However, 3-NT was decreased by SCN therapy in infected WT mice.
Both strains in this study displayed a significant increase in airway monocytes and macrophages in SCN-treated infected animals, but not in SCN-treated uninfected animals (10). Monocytes and macrophages are vital to the resolution of lung inflammation, engulfing dead cells and bacteria after the initial immune response (49, 50). The accelerated decline in neutrophil infiltrates in infected, SCN-treated mice may also accelerate monocyte recruitment to begin inflammatory resolution. The exclusivity of this phenomenon to infected animals suggests that it is driven by accelerated bacterial clearance and neutrophil removal with SCN, or enhancement of a specific signaling factor. HOSCN increases the expression of cell adhesion molecules and leukocyte attachment to endothelial cells through NF-κB signaling (51). Although SCN treatment decreased neutrophilia, increased site-specific cell adhesion molecule expression in HOSCN-exposed endothelia may enhance circulating leukocyte trafficking to sites of infection and, thus, may explain improved bacterial clearance with SCN treatment.
J774A.1 macrophage-like cells have been shown to apoptose more after HOSCN exposure than after HOCl (52). However, we found that 400 μM SCN, which rapidly converts HOCl to HOSCN (13), was protective against HOCl-mediated necrosis. Differences in experimental methods (e.g., exposure length, HOSCN generation method, and postexposure conditions) may account for the disagreement in these studies. We have also noted decreased TrxR activity in J774A.1 cells compared with other cell lines (data not shown), which may predispose them to the HOSCN sensitivity reported in some publications.
Metabolism of HOSCN by mammalian cells is crucial to its function as a selective biocide, while avoiding host cytotoxicity. Cells of the oral cavity must tolerate constant exposure to 40–60 μM HOSCN (53). Although HOSCN is much less oxidizing than HOCl (HOCl, E°′ = 1.28 V; HOSCN, E°′ = 0.56 V [35]), it could, nevertheless, perturb cellular processes if unchecked. However, chemical selectivity makes HOSCN an ideal substrate of mammalian TrxR, which we previously identified as the first known direct HOSCN-metabolizing enzyme in mammals (15). HOSCN formation is linked to inflammation, and it follows that mammalian TrxR might also be up-regulated in response to inflammation. We observed that TrxR activity and expression increased in WT and βENaC lung tissue during inflammation, independent of infection. This supports the importance of TrxR in the cell’s ability to tolerate and resolve inflammation, mediated, at least in part, by HOSCN metabolism.
The ultimate goal of this work is to evaluate SCN as a potential therapy for clinical CF. Other redox active thiol-bearing small molecules have already been tested as inhaled therapeutics in CF, including GSH, N-acetyl L-cysteine, and 2-mercaptothane sulfonate (54). These studies showed little or no efficacy in prevention of pulmonary exacerbations and quality-of-life improvement, and few improvements in FEV1 (55, 56). However, SCN has unusual chemical properties compared with thiols, including high redox potential (HOSCN + H+ + 2 e− → SCN + H2O, E°′ = 560 mV compared with GSSG + 2 H+ + 2 e− → 2 GSH, E°′ = −240 mV [35, 57]). This effectively means that SCN only reduces certain very strong oxidants (i.e., HOX and peroxidase compound I), resulting in a mechanism of action much more focused than reducing thiols that produces an oxidant retaining antimicrobial properties (HOSCN) instead of quenching all potential microbicides. Nasal lining fluid SCN positively correlates with FEV1 in patients with CF (25), and the results of this study suggest that exogenous SCN may also decrease pulmonary exacerbations and improve quality of life.
These findings demonstrate novel antiinflammatory effects and expand on antimicrobial potential of SCN in lung infection. SCN decreases BALF KC, IL-1β, TNF-α, and airway neutrophil infiltrate concurrent with infectious stimulus, in addition to enhancing bacterial clearance in βENaC and WT mice. Uninfected βENaC mice exhibited airway neutrophilia and KC expression that was decreased by SCN. Increased TrxR expression was observed in mouse lung tissue with inflammation and infection, implicating a role for the HOSCN-metabolizing enzyme in modulating inflammation. Taken together, these results support the potential of SCN to serve as a novel therapeutic in pulmonary diseases characterized by bacterial infection and exuberant host inflammatory response.
Footnotes
This work was supported by National Institutes of Health grant R01 HL08469 (B.J.D.), Colorado Clinical and Translational Sciences Institute grant KL2 TR000156 (D.P.N.), and the Cystic Fibrosis Foundation (B.J.D. and D.P.N.).
Author Contributions: conception and study design—J.D.C., D.P.N., and B.J.D.; data acquisition, analysis, and interpretation—J.D.C., E.M., J.H., C.S.M., N.D., T.M., A.A.F., C.M.E., L.L., M.P., A.J.K., D.P.N., and B.J.D.; drafting of the manuscript for important intellectual content—J.D.C., C.M.E., M.P., A.J.K., D.P.N., and B.J.D.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0208OC on December 9, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol. 2013;93:185–198. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klebanoff SJ. Myeloperoxidase: Friend and Foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 3.Souza CE, Maitra D, Saed GM, Diamond MP, Moura AA, Pennathur S, Abu-Soud HM. Hypochlorous acid-induced heme degradation from lactoperoxidase as a novel mechanism of free iron release and tissue injury in inflammatory diseases. PLoS One. 2011;6:e27641. doi: 10.1371/journal.pone.0027641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stacey MM, Vissers MC, Winterbourn CC. Oxidation of 2-cys peroxiredoxins in human endothelial cells by hydrogen peroxide, hypochlorous acid, and chloramines. Antioxid Redox Signal. 2012;17:411–421. doi: 10.1089/ars.2011.4348. [DOI] [PubMed] [Google Scholar]
- 5.Chandler JD, Day BJ. Thiocyanate: a potentially useful therapeutic agent with host defense and antioxidant properties. Biochem Pharmacol. 2012;84:1381–1387. doi: 10.1016/j.bcp.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aune TM, Thomas EL. Accumulation of hypothiocyanite ion during peroxidase-catalyzed oxidation of thiocyanate ion. Eur J Biochem. 1977;80:209–214. doi: 10.1111/j.1432-1033.1977.tb11873.x. [DOI] [PubMed] [Google Scholar]
- 7.Thomas EL, Fishman M. Oxidation of chloride and thiocyanate by isolated leukocytes. J Biol Chem. 1986;261:9694–9702. [PubMed] [Google Scholar]
- 8.Xu Y, Szép S, Lu Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc Natl Acad Sci USA. 2009;106:20515–20519. doi: 10.1073/pnas.0911412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gould NS, Gauthier S, Kariya CT, Min E, Huang J, Day BJ. Hypertonic saline increases lung epithelial lining fluid glutathione and thiocyanate: two protective CFTR-dependent thiols against oxidative injury. Respir Res. 2010;11:119. doi: 10.1186/1465-9921-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandler JD, Min E, Huang J, Nichols DP, Day BJ. Nebulized thiocyanate improves lung infection outcomes in mice. Br J Pharmacol. 2013;169:1166–1177. doi: 10.1111/bph.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu W, Chen Y, Hazen SL. Eosinophil peroxidase nitrates protein tyrosyl residues. J Biol Chem. 1999;274:25933–25944. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- 13.Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- 14.Xulu BA, Ashby MT. Small molecular, macromolecular, and cellular chloramines react with thiocyanate to give the human defense factor hypothiocyanite. Biochemistry. 2010;49:2068–2074. doi: 10.1021/bi902089w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandler JD, Nichols DP, Nick JA, Hondal RJ, Day BJ. Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J Biol Chem. 2013;288:18421–18428. doi: 10.1074/jbc.M113.468090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilliard JB, Konstan MW, Davis PB. Inflammatory mediators in CF patients. Methods Mol Med. 2002;70:409–431. doi: 10.1385/1-59259-187-6:409. [DOI] [PubMed] [Google Scholar]
- 17.Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- 18.Fragoso MA, Fernandez VE, Forteza R, Randell SH, Salathe M, Conner GE. Transcellular thiocyanate transport by human airway epithelia. J Physiol. 2004;561:183–194. doi: 10.1113/jphysiol.2004.071548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia MAS, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator–dependent bicarbonate secretion. J Clin Invest. 2009;119:2613–2622. doi: 10.1172/JCI38662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gould NS, Min E, Martin RJ, Day BJ. CFTR is the primary known apical glutathione transporter involved in cigarette smoke–induced adaptive responses in the lung. Free Radic Biol Med. 2012;52:1201–1206. doi: 10.1016/j.freeradbiomed.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13:231–240. doi: 10.1016/j.molmed.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Moskwa P, Lorentzen D, Excoffon KJDA, Zabner J, McCray PB, Jr, Nauseef WM, Dupuy C, Banfi B. A Novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–183. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007;581:271–278. doi: 10.1016/j.febslet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minarowski Ł, Sands D, Minarowska A, Karwowska A, Sulewska A, Gacko M, Chyczewska E. Thiocyanate concentration in saliva of cystic fibrosis patients. Folia Histochem Cytobiol. 2008;46:245–246. doi: 10.2478/v10042-008-0037-0. [DOI] [PubMed] [Google Scholar]
- 25.Lorentzen D, Durairaj L, Pezzulo AA, Nakano Y, Launspach J, Stoltz DA, Zamba G, McCray PB, Jr, Zabner J, Welsh MJ, et al. Concentration of the antibacterial precursor thiocyanate in cystic fibrosis airway secretions. Free Radic Biol Med. 2011;50:1144–1150. doi: 10.1016/j.freeradbiomed.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis–like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 27.Wine JJ. The development of lung disease in cystic fibrosis pigs. Sci Transl Med. 2010;2:29ps20. doi: 10.1126/scitranslmed.3001130. [DOI] [PubMed] [Google Scholar]
- 28.Harwood DT, Kettle AJ, Winterbourn CC. Production of glutathione sulfonamide and dehydroglutathione from GSH by myeloperoxidase-derived oxidants and detection using a novel LC–MS/MS method. Biochem J. 2006;399:161. doi: 10.1042/BJ20060978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rennard SI, Basset G, Lecossier D, O'Donnell KM, Pinkston P, Martin PG, Crystal RG. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol. 1986;60:532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 30.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 31.Skaff O, Pattison DI, Davies MJ. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: absolute rate constants and assessment of biological relevance. Biochem J. 2009;422:111–117. doi: 10.1042/BJ20090276. [DOI] [PubMed] [Google Scholar]
- 32.Winterbourn CC, Brennan SO. Characterization of the oxidation products of the reaction between reduced glutathione and hypochlorous acid. Biochem J. 1997;326:87–92. doi: 10.1042/bj3260087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harwood DT, Kettle AJ, Brennan S, Winterbourn CC. Simultaneous determination of reduced glutathione, glutathione disulphide and glutathione sulphonamide in cells and physiological fluids by isotope dilution liquid chromatography–tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3393–3399. doi: 10.1016/j.jchromb.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Hao Y, Kuang Z, Xu Y, Walling BE, Lau GW. Pyocyanin-induced mucin production is associated with redox modification of FOXA2. Respir Res. 2013;14:82. doi: 10.1186/1465-9921-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnhold J, Monzani E, Furtmüller PG, Zederbauer M, Casella L, Obinger C. Kinetics and thermodynamics of halide and nitrite oxidation by mammalian heme peroxidases. Eur J Inorg Chem. 2006;2006:3801–3811. [Google Scholar]
- 36.Klebanoff SJ, Clem WH, Luebke RG. The peroxidase-thiocyanate-hydrogen peroxide antimicrobial system. Biochim Biophys Acta. 1966;117:63–72. doi: 10.1016/0304-4165(66)90152-8. [DOI] [PubMed] [Google Scholar]
- 37.Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, Salathe M, Conner GE. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–212. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- 38.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 39.Morgan PE, Pattison DI, Talib J, Summers FA, Harmer JA, Celermajer DS, Hawkins CL, Davies MJ. High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic Biol Med. 2011;51:1815–1822. doi: 10.1016/j.freeradbiomed.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Kettle AJ, Turner R, Gangell CL, Harwood DT, Khalilova IS, Chapman AL, Winterbourn CC, Sly PD AREST CF. Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J. 2014;44:122–129. doi: 10.1183/09031936.00170213. [DOI] [PubMed] [Google Scholar]
- 41.Nichols DP, Ziady AG, Shank SL, Eastman JF, Davis PB. The triterpenoid CDDO limits inflammation in preclinical models of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol. 2009;297:L828–L836. doi: 10.1152/ajplung.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Kinter M, Shank S, Cotton C, Kelley TJ, Ziady AG. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS One. 2008;3:e3367. doi: 10.1371/journal.pone.0003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Z, Treis D, Schubert SC, Harm M, Schatterny J, Hirtz S, Duerr J, Boucher RC, Mall MA. Preventive but not late amiloride therapy reduces morbidity and mortality of lung disease in betaENaC-overexpressing mice. Am J Respir Crit Care Med. 2008;178:1245–1256. doi: 10.1164/rccm.200803-442OC. [DOI] [PubMed] [Google Scholar]
- 44.Livraghi A, Grubb BR, Hudson EJ, Wilkinson KJ, Sheehan JK, Mall MA, O'Neal WK, Boucher RC, Randell SH. Airway and lung pathology due to mucosal surface dehydration in β-epithelial Na+ channel–overexpressing mice: role of TNF-α and IL-4R signaling, influence of neonatal development, and limited efficacy of glucocorticoid treatment. J Immunol. 2009;182:4357–4367. doi: 10.4049/jimmunol.0802557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graeber SY, Zhou-Suckow Z, Schatterny J, Hirtz S, Boucher RC, Mall MA. Hypertonic saline is effective in the prevention and treatment of mucus obstruction, but not airway inflammation, in mice with chronic obstructive lung disease. Am J Respir Cell Mol Biol. 2013;49:410–417. doi: 10.1165/rcmb.2013-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gould NS, Min E, Day BJ. Macropinocytosis of extracellular glutathione ameliorates tumor necrosis factor α release in activated macrophages. PLoS One. 2011;6:e25704. doi: 10.1371/journal.pone.0025704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kemp M, Go Y-M, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anathy V, Aesif SW, Hoffman SM, Bement JL, Guala AS, Lahue KG, Leclair LW, Suratt BT, Cool CD, Wargo MJ, et al. Glutaredoxin-1 attenuates S-glutathionylation of the death receptor fas and decreases resolution of Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2014;189:463–474. doi: 10.1164/rccm.201310-1905OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 50.Filep JG, Kebir DE. Neutrophil apoptosis: a target for enhancing the resolution of inflammation. J Cell Biochem. 2009;108:1039–1046. doi: 10.1002/jcb.22351. [DOI] [PubMed] [Google Scholar]
- 51.Wang J-G, Mahmud SA, Nguyen J, Slungaard A. Thiocyanate-dependent induction of endothelial cell adhesion molecule expression by phagocyte peroxidases. J Immunol. 2006;177:8714–8722. doi: 10.4049/jimmunol.177.12.8714. [DOI] [PubMed] [Google Scholar]
- 52.Lloyd MM, van Reyk DM, Davies MJ, Hawkins CL. Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem J. 2008;414:271. doi: 10.1042/BJ20080468. [DOI] [PubMed] [Google Scholar]
- 53.Pruitt KM, Tenovuo J, Mansson-Rahemtulla B, Harrington P, Baldone DC. Is thiocyanate peroxidation at equilibrium in vivo? Biochim Biophys Acta. 1986;870:385–391. doi: 10.1016/0167-4838(86)90245-1. [DOI] [PubMed] [Google Scholar]
- 54.Tam J, Nash EF, Ratjen F, Tullis E, Stephenson A. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev. 2013;7:CD007168. doi: 10.1002/14651858.CD007168.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griese M, Ramakers J, Krasselt A, Starosta V, van Koningsbruggen S, Fischer R, Ratjen F, Müllinger B, Huber RM, Maier K, et al. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- 56.Griese M, Kappler M, Rietschel E, Hartl D, Hector A. Inhalation treatment with glutathione in patients with cystic fibrosis: a randomized clinical trial. Am J Respir Crit Care Med. 2013;188:83–89. doi: 10.1164/rccm.201303-0427OC. [DOI] [PubMed] [Google Scholar]
- 57.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]