

Abstract
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is responsible for respiratory burst in immune cells. Chloride channel 3 (CLC3) has been linked to the respiratory burst in eosinophils and neutrophils. The effect of cytokines and the involvement of CLC3 in the regulation of NADPH-dependent oxidative stress and on cytokine-mediated migration of eosinophils are not known. Human peripheral blood eosinophils were isolated from healthy individuals and from individuals with asthma by negative selection. Real-time PCR was used to detect the expression of NADPH oxidases in eosinophils. Intracellular reactive oxygen species (ROS) measurement was done with flow cytometry. Superoxide generation was measured with transforming growth factor (TGF)-β, eotaxin, and CLC3 blockers. CLC3 dependence of eosinophils in TGF-β– and eotaxin-induced migration was also examined. The messenger RNA (mRNA) transcripts of NADPH oxidase (NOX) 2, dual oxidase (DUOX) 1, and DUOX2 were detected in blood eosinophils, with very low expression of NOX1, NOX3, and NOX5 and no NOX4 mRNA. The level of NOX2 mRNA transcripts increased with disease severity in the eosinophils of subjects with asthma compared with healthy nonatopic volunteers. Change in granularity and size in eosinophils, but no change in intracellular ROS, was observed with phorbol myristate acetate (PMA). PMA, TGF-β, and eotaxin used the CLC3-dependent pathway to increase superoxide radicals. TGF-β and eotaxin induced CLC3-dependent chemotaxis of eosinophils. These findings support the requirement of CLC3 in the activation and migration of human blood eosinophils and may provide a potential novel therapeutic target to regulate eosinophil hyperactivity in allergic airway inflammation in asthma.
Keywords: airway inflammation, allergic asthma, CLC3 channels, eosinophils, NADPH oxidase
Clinical Relevance
We, for the first time, report a critical role of chloride channel 3 (CLC3) channels in the migration and activation of human blood eosinophils. We also report, in addition to the known presence of nicotinamide adenine dinucleotide phosphate oxidase (NOX) 2, the existence of subtypes of NOX, including NOX1, NOX3, NOX5, dual oxidase (DUOX) 1, and DUOX2, albeit at low levels, in human blood eosinophils. However, very high levels of NOX2 messenger RNA transcripts were found in the eosinophils of subjects with moderate to severe asthma compared with subjects with mild to moderate asthma and healthy subjects, suggesting a positive correlation between the expression of NOX2 messenger RNA transcripts and disease severity. These findings provide a novel target in regulating eosinophil infiltration and activation in allergic airway inflammation in asthma.
Allergic asthma is a complex and heterogeneous disease. Eosinophils play a major role in the exacerbation of allergic asthma by migrating to the site of inflammation and releasing manifold granules with preformed and newly formed mediators that propagate excessive damage and repair cycle in the airways (1, 2). The preformed mediators consist of major basic protein, eosinophil-derived neurotoxin, eosinophil peroxidase, and eosinophil cationic protein. The newly formed mediators include cytokines and chemokines (IL-2, IL-4, IL-6, IL-10, IL-12, IL-13, IL-16, IL-18, IFN-γ, and TNF-α), prostaglandins, leukotrienes (mostly LTC4), reactive oxygen species (ROS), and growth factors, including transforming growth factor (TGF)-β (2, 3).
Although major basic protein is highly toxic, the production and release of ROS from activated eosinophils elicit severe damage to the airway epithelium. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a membrane-bound oxidase that triggers respiratory burst. A typical NADPH oxidase in phagocytes consists of membrane-bound gp91phox (NOX2) and p22phox in addition to cytosolic p47phox, p67phox, and p40phox and a small GTPase Rac. Nonphagocytic NADPH oxidase (NOX) consists of different homologs of NOX2, namely NOX1, NOX3, NOX4, and NOX5, and dual oxidase (DUOX) (4–6). NADPH oxidase generates superoxide radicals (O2∙−), which are converted to H2O2 by superoxide dismutase. Unlike superoxide radicals, H2O2 can freely cross plasma membrane but has less oxidative power. Through a chemical reaction called Fenton’s reaction, H2O2 can readily react with iron to convert it from ferrous (Fe2+) to ferric (Fe3+) and generate hydroxyl radicals (·OH). Alternatively, H2O2 can react with H+ and Cl− to make hypochlorous acid (HOCl) in the presence of myeloperoxidase (7) or, in the case of eosinophils, eosinophil peroxidase (8, 9), which is a powerful oxidizing agent capable of oxidizing proteins and cell membranes, leading to damage and inflammation in the airways. Chloride channel 3 (CLC3) has been shown to regulate the production of O2∙− in human eosinophils (9). However, the precise mechanism involved in the activation of eosinophils is unclear.
Eosinophil migration and activation are carefully controlled by cytokines and chemokines that are released from damaged epithelium or other immune cells, including Th2 cells. TGF-β (10, 11), eotaxin (12, 13), and IL-5 (14, 15) are major cytokines and chemokines controlling eosinophils from their differentiation through maturity and activation in a time-, space-, and concentration-dependent manner. Ion channels could also be involved in such processes. Indeed, chloride channels are critical players in a variety of physiological and cellular processes, including pH regulation, volume homeostasis, organic solute transport, cell migration, and cell proliferation and differentiation (16–18). CLC3, a chloride channel and H+/Cl− antiporter, has been shown to modulate respiratory burst in eosinophils (9) and regulates the migration and activation of neutrophils (19, 20). This double-barreled antiporter (21–23) has been shown to be expressed ubiquitously in a variety of cells. Its expression has been detected on plasma membrane and intracellularly on lysosomes and nucleus (21, 24). In this study we examined the role of CLC3 on eosinophil activation involving NADPH oxidase system and on eosinophil migration.
Materials and Methods
Human Subjects
The Institutional Review Board of Creighton University approved the protocol. Healthy volunteers and volunteers with asthma (19–65 yr of age) provided informed consent. Inclusion criteria were avoidance of short-acting β-agonists for 8 hours, long-acting β-agonists and caffeine for 2 days, antihistamines for 3 days, corticosteroids, cromolyn/nedocromil for 2 weeks, and astemizole for 3 months. Healthy subjects had no history of allergies or recent infections. Subjects with mild to moderate asthma had greater than or equal to 70% FEV1 with no medications. Subjects with moderate to severe asthma had FEV1 of <70% and were taking a daily dose of their prescribed Advair (GlaxoSmithKline, Philadelphia, PA) (250/50 or 500/50 μg fluticasone propionate/salmeterol). Due to the health concerns of the patients, subjects with mild to severe asthma were still taking their asthma control medicine at the time of recruitment (Table 1).
Table 1.
Information on the Recruited Volunteers for Nicotinamide Adenine Dinucleotide Phosphate Oxidase Expression Analysis
| Volunteers/Parameters | Healthy Subjects | Subjects with Mild to Moderate Asthma | Subjects with Moderate to Severe Asthma |
|---|---|---|---|
| Age, yr | 21–62 | 19–54 | 30–63 |
| Sex, M/F | 3/3 | 0/6 | 3/4 |
| Atopy | No | Yes | Yes |
| Symptoms | None | Up to twice per week | Daily |
| Lung function | N/A | FEV1 ≥70% | FEV1 <70% |
| FEV1/FVC normal | FEV1/FVC reduced ≥5% | ||
| Medications (at the time of blood draw) | None | None | Advair 250/50 or 500/50 |
Definition of abbreviation: N/A, not applicable.
Isolation and Culture of Human Blood Eosinophils
Eosinophils were isolated by density gradient centrifugation and dextran sedimentation, followed by hypotonic lysis of venous blood with negative selection using an eosinophil isolation kit (Miltenyi Biotec, Auburn, CA) in autoMACS (Miltenyi Biotec). Purity (>99%) and viability (>98%) of eosinophils were examined by staining with Hema-Diff (StatLab Medical Products Inc., Lewisville, TX) and trypan blue (Sigma-Aldrich, St. Louis, MO), respectively.
The purified eosinophils were cultured in RPMI complete medium (RPMI + 10% FBS) at 37°C with 5% CO2.
RNA Isolation and Reverse Transcription
Total RNA was isolated using Ambion mirVana miRNA Isolation Kit (Life Technologies, Grand Island, NY). Total RNA (500 ng) was reverse transcribed with the ImProm-II Reverse Transcription System (Promega, Madison, WI).
Real-Time PCR
Quantitative PCR was performed using a CFX96 thermocycler (Bio-Rad, Hercules, CA) with specific primers for NADPH oxidases (Table 2) and iQ SYBR Green Supermix (Bio-Rad). Fold-change in the gene expression was calculated as 2−ΔΔcq from healthy compared with asthmatic eosinophils or relative to the respective healthy untreated eosinophils in treatment groups, where ΔΔCq is (Cqasthmatic − Cqhealthy)CqNOX2 − (Cqasthmatic − Cqhealthy)Cq18S, or (Cqtreatment − Cqcontrol)CqNOX2 − (Cqtreatment − Cqcontrol)CqGAPDH/18S.
Table 2.
Primer Pairs Used in Quantitative PCR Analysis for Nicotinamide Adenine Dinucleotide Phosphate Oxidases
| Primer | Accession (NCBI) | Sequence (5′–3′) | Product Size (bp) | Temperature (°C) |
|---|---|---|---|---|
| NOX1-F | NM_007052.4 | TGCGCTGGACAAATGTTCCATTCC | 192 | 60.3 |
| NOX1-R | NM_007052.4 | TCAGGAAGGCATCCACAAACAGGA | 192 | 60.5 |
| NOX2-F | NM_000397.3 | AGACTTTGTATGGACGGCCCAACT | 103 | 60.4 |
| NOX2-R | NM_000397.3 | AAGGCTTCAGGTCCACAGAGGAAA | 103 | 60.2 |
| NOX3-F | NM_015718.2 | ACCGTGGAGGAGGCAATTAGACAA | 109 | 60.2 |
| NOX3-R | NM_015718.2 | TTCCAGGTTGAAGAAATGCGCCAC | 109 | 60.3 |
| NOX4-F | NM_001143836.1 | AGCAGAGCCTCAGCATCTGTTCTT | 115 | 60.4 |
| NOX4-R | NM_001143836.1 | TGGTTCTCCTGCTTGGAACCTTCT | 115 | 60.2 |
| NOX5-F | NM_001184779.1 | TGCTGCTCCTCCTCATGTTCATCT | 135 | 60.0 |
| NOX5-R | NM_001184779.1 | TCCAGAAGTTGGGCCCATGAAAGA | 135 | 60.5 |
| DUOX1-F | NM_017434.3 | AACAATTTGTGCGGCTACGGGATG | 109 | 60.3 |
| DUOX1-R | NM_017434.3 | TCCTGCAGGGTGGTATTTCGGATT | 109 | 60.3 |
| DUOX2-F | NM_014080.4 | AGTACAAGCGCTTCGTGGAGAACT | 85 | 60.3 |
| DUOX2-R | NM_014080.4 | TCTGCAAACACGCCAACACAGATG | 85 | 60.3 |
| GAPDH-F | NM_002046.3 | TCGACAGTCAGCCGCATCTTCTTT | 94 | 60.6 |
| GAPDH-R | NM_002046.3 | ACCAAATCCGTTGACTCCGACCTT | 94 | 60.5 |
| 18S-F | NR_003286.2 | TCAACTTTCGATGGTAGTCGCCGT | 108 | 60.3 |
| 18S-R | NR_003286.2 | TCCTTGGATGTGGTAGCCGTTTCT | 108 | 60.2 |
Definition of abbreviations: bp, base pair; NCBI, National Center for Biotechnology Information.
Measurement of Intracellular ROS
Cells were treated with 1 μM phorbol myristate acetate (PMA) (Sigma-Aldrich) for 30 minutes followed by 30 minutes with CellROX Deep Red Reagent (Life Technologies, Grand Island, NY). Intracellular ROS was measured with FACS analysis in the Flow Cytometry Core Facility of Creighton University.
Measurement of Extracellular ROS
After adding the cytokines and blockers, O2∙− was measured every 5 minutes for 4 hours as the intensity of luminescence with Enspire plate reader (PerkinElmer, Waltham, MA) using a superoxide anion assay kit (Sigma-Aldrich).
Chemotaxis
A Transwell system (Costar, Corning, NY) with a 5.0-μm polycarbonate membrane was used with respective chemoattractants and blockers. Equal numbers of eosinophils were put in the upper chamber and kept in the incubator for 90 minutes at 37°C with 5% CO2. Ten random high-power fields were chosen to count the migrating eosinophils on membranes stained with Hema-Diff (StatLab Medical Products Inc.) using a bright-field microscope (Olympus, Center Valley, PA) with 20× objective.
Data Analysis
Statistical differences of data were evaluated by Student’s t test or by one-way or two-way ANOVA with Tukey’s post hoc test. Data were considered significant at P < 0.05. Data represent means ± SEM.
Results
NADPH Oxidase Expression in Human Peripheral Blood Eosinophils
Very low levels of NOX1, NOX3, and NOX5 were detected, with no expression of NOX4 (Figure 1A). Relatively higher levels of DUOX1, DUOX2, and NOX2 messenger RNA (mRNA) transcripts were found in eosinophils, but TGF-β did not have any effect on the transcript levels of any of the NADPH isoforms (Figure 1A). Eotaxin-1 (10 ng/ml) decreased NOX2 mRNA transcripts in a time-dependent manner (Figure 1B).
Figure 1.

Expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) in blood eosinophils. (A) Real-time PCR exhibiting expression of NOX2, dual oxidase (DUOX) 1, and DUOX2 in blood eosinophils with low levels of NOX1, NOX3, and NOX5. No expression of NOX4 was detected. Transforming growth factor (TGF)-β1 did not have significant effect on the expression levels of the NADPH oxidase isoforms (n = 4). (B) Decreased NOX2 expression with eotaxin-1 in a time-dependent manner (n = 4). (C) The effect of IL-5, IL-13, IL-25, and IL-3 on the expression of NOX2 (n = 5). (D) A high level of NOX2 messenger RNA (mRNA) transcripts was seen in the eosinophils of subjects with moderate to severe asthma (n = 7) compared with subjects with mild to moderate asthma (n = 6) or healthy subjects (n = 6). There was no significant difference between eosinophils from healthy subjects and from subjects with mild to moderate asthma. *P < 0.05, ***P < 0.001, and ****P < 0.0001. GAPDH, glyceraldehyde phosphate dehydrogenase.
Treatment of eosinophils with IL-33 (10 ng/ml) significantly increased NOX2 mRNA transcripts (∼4-fold) compared with respective controls. However, there was no significant change in the NOX2 mRNA levels when eosinophils were treated with 10 ng/ml IL-5, IL-13, or IL-25 (Figure 1C). NOX2 mRNA transcripts in the eosinophils were 7-fold higher but were insignificantly different in subjects with mild to moderate asthma and 75-fold higher and significantly different in subjects with mild to severe asthma compared with healthy subjects (Figure 1D). These findings suggest a critical role of NOX2 in the activation of eosinophils in subjects with moderate to severe asthma.
Intracellular ROS in Human Peripheral Blood Eosinophils
PMA increased the granularity and cell size of eosinophils (Figures 2D, 2E, 2J, and 2K) compared with untreated eosinophils (Figures 2A, 2B, 2G, and 2H). Also, there was a remarkable increase in the cell debris (Figures 2D and 2J) with PMA outside the analysis gate compared with the unstimulated cells (Figures 2A 2G), suggesting degranulation of eosinophils. Fluorescent micrographs of CellROX- and 4′,6-diamidino-2-phenylindole–stained eosinophils further strengthened the observation of degranulating eosinophils in response to PMA (Figure 2N). Compared with the unstimulated eosinophils (Figures 2B and 2C), PMA induced and increase in ROS generation (Figures 2E and 2F) in a subset of healthy subjects but exhibited no change or decrease in intracellular ROS in the eosinophils of other volunteers (Figures 2K and 2L). Although no significant change in the intracellular ROS was observed with PMA stimulation among subjects (Figure 2O), there was a heterogeneity to the PMA-stimulated change in intracellular ROS in eosinophils, as shown by two distinct populations of eosinophils upon PMA stimulation (Figures 2K and 2L). This suggests a possibility of segregation of the eosinophil population to hypo- and normo-dense eosinophils.
Figure 2.

Intracellular reactive oxygen species (ROS) in blood eosinophils. Flow cytometry analysis of eosinophils stained with CellROX showed change in cell size and granularity with PMA. (A–C) Data obtained from the eosinophils from one subject. (G–H) Data obtained from another subject. In both cases, eosinophils were stained with CellROX without stimulation. (D–F and J–L) The corresponding data in PMA-treated and CellROX-stained eosinophils of these two individuals (n = 12). (E, F, K, and L) Data from two subjects with different degrees of segregation of two populations of eosinophils with respect to their ROS content after PMA stimulation. (M and N) Fluorescent micrographs of unstimulated and PMA-stimulated eosinophils, respectively, stained with CellROX and 4′,6-diamidino-2-phenylindole. (O) There was no change in the intracellular ROS in PMA-induced eosinophils (n = 12). FSC-A, forward scatter; PB Eos, peripheral blood eosinophils; PMA, phorbol myristate acetate; SSC, side scatter.
Extracellular ROS
NADPH oxidase requires an ion channel to compensate the difference in the membrane potential caused by superoxide release. CLC3 has been found to assist NADPH oxidase in the superoxide generation and charge compensation (9). Measurement of superoxide generation in human blood eosinophils revealed the dependency of NADPH oxidase activity on CLC3 in the plasma membrane of eosinophils. PMA exhibited a quick burst of O2∙−, which was partially blocked by 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), a selective CLC3 blocker (Figure 3A). The cytokines TGF-β1, TGF-β2, eotaxin-1, and eotaxin-3 induced two different bursts of O2∙− in human peripheral blood eosinophils (Figure 3B). The first burst was generated at 20 minutes and was relatively lower in amplitude than the second burst, which was generated at 55 minutes after the addition of the respective cytokines to the eosinophils (Figure 3B). The TGF-β1–induced peak of extracellular O2∙− generation was lower than that induced by eotaxin-1 at 20 minutes. However, there was no statistically significant difference between O2∙− peaks induced by TGF-β1 and eotaxin-3 or O2∙− peaks induced by TGF-β2 and eotaxin-1/eotaxin-3 at 20 or 55 minutes (Figure 3B). The amplitude of O2∙− peak induced by all cytokines in the first burst was smaller than in the second burst.
Figure 3.

Extracellular ROS (superoxide [O2∙−]) in blood eosinophils. (A) Luminescence assay indicating a sharp increase in the O2∙− generation with PMA that was chloride channel 3 (CLC3) dependent. Control groups contained superoxide dismutase (SOD), no enhancer, or no PMA (n = 3). P < 0.05. (B) Eotaxin-1, eotaxin-3, TGF-β1, and TGF-β2 increased O2∙− release in a two-step process, which was lower in amplitude than PMA-induced O2∙− generation (n = 3). *P < 0.05 and ****P < 0.0001. NPPB, 5-nitro-2-(3-phenylpropylamino)benzoic acid.
NPPB inhibited the extracellular ROS (O2∙−) generation in response to the cytokines, suggesting the dependency of ROS generation on CLC3 (Figures 4A–4D). The NADPH oxidase inhibitor diphenyleneiodonium (DPI) also inhibited the extracellular ROS (O2∙−) generation due to TGF-β1, TGF-β2, and eotaxin-1 (Figures 4A–4C). NADPH oxidase inhibitor did not reduce the eotaxin-3–induced O2∙− to the baseline levels as it did in other groups. DPI significantly reduced the eotaxin-3–induced O2∙− generation in the first oxidative burst at 20 minutes. However, there was a slight delay in the peak time of eotaxin-3–induced second respiratory burst without any significant change the amplitude compared with the first respiratory burst (Figure 4D).
Figure 4.
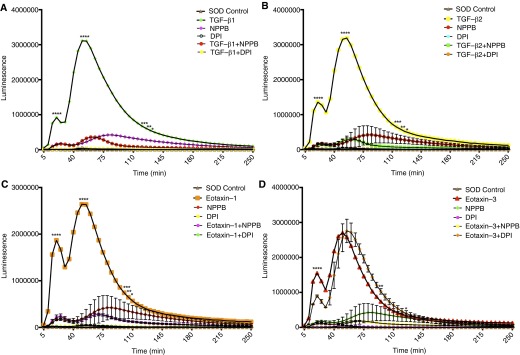
CLC3 dependence of cytokine-induced O2∙− release in blood eosinophils. (A–D) Effect of TGF-β1, TGF-β2, eotaxin-1, and eotaxin-3 on O2∙− release, respectively. The CLC3 blocker NPPB inhibited the increase in O2∙− release. However, diphenyleneiodonium (DPI) blocked the O2∙− release in all except eotaxin-3–induced eosinophils, where it partially blocked the 20-minute burst and delayed the second burst (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To determine if the O2∙− production was mediated through CCR3, a selective blocker of CCR3 (SB328437) was used. SB328437 was able to induce O2∙− release in eosinophils by itself. It did not change the time or amplitude of the first burst, but the second burst was even bigger than the eotaxin-1– and eotaxin-3–induced O2∙−. Additionally, the second burst was delayed, and there was significant difference in the O2∙− production induced by eotaxin compared with that in the presence of CCR3 blocker (Figure 5).
Figure 5.

Effect of chemokine (C-C motif) receptor 3 (CCR3) blocker on eotaxin-induced O2∙− release in blood eosinophils. CCR3 blocker (SB328437, 100 μM) had no effect on the first burst, but it increased the O2∙− release alone and in the presence of eotaxin-1 and eotaxin-3. However, there was a delay in the second oxidative burst, suggesting activation of an alternate pathway. At the peak of the eotaxin-induced second oxidative burst, there was significant difference in released O2∙− with the CCR3 blocker groups (n = 3). *P < 0.05, **P < 0.01, and ****P < 0.0001.
Eosinophil Chemotaxis
TGF-β and eotaxin increased the migration of eosinophils. Nonetheless, the effect of eotaxin-1 and eotaxin-3 on eosinophil migration was greater than that of TGF-β1 and TGF-β2 (Figures 6A and 6B). The CCR3 blocker SB328437 had no effect on the eosinophil migration by itself but inhibited the eotaxin-1– and eotaxin-3–induced migration of eosinophils, suggesting a direct involvement of the CCR3 in eosinophil migration. The CLC3 selective blocker NPPB by itself had no significant effect on eosinophil migration. However, NPPB significantly inhibited cytokine-induced migration of eosinophils, suggesting the involvement of CLC3 (Figures 6A and 6B). Phloretin, a known inhibitor of the PKC enzyme that inhibits CLC3, also inhibited cytokine-induced migration of eosinophils (Figure 6C). This further supports the role of CLC3 in chemokine-induced migration of eosinophils.
Figure 6.
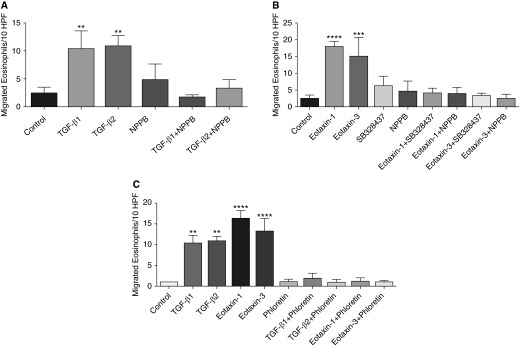
Chemotaxis of blood eosinophils. (A) TGF-β1 and TGF-β2 increased the migration of eosinophils that was blocked by the CLC3 inhibitor NPPB (n = 3). **P < 0.01. (B) Eotaxin-1 and eotaxin-3 increased the migration of blood eosinophils that was blocked by CCR3 and CLC3 inhibitors SB328437 and NPPB, respectively (n = 3). ***P < 0.001 and ****P < 0.0001. (C) The effect of phloretin, an inhibitor of CLC3/PKC, on TGF-β1–, TGF-β2–, eotaxin-1–, and eotaxin-3–induced chemotaxis of eosinophils (n = 4). **P < 0.01 and ****P < 0.0001. HPF, high-power field.
Discussion
Eosinophil respiratory burst generates 10-fold more extracellular O2∙− than neutrophils (25, 26). The current reports in the literature indicate the presence of only NOX2 among many NADPH oxidases in eosinophil (25). However, in addition to strong expression of NOX2 mRNA transcripts in our investigation, we found very low levels of NOX1, NOX3, and NOX5 and found no NOX4 in eosinophils. However, the low mRNA transcripts of NADPH oxidases may suggest no or a very minor role in the eosinophil respiratory burst. Unlike other NADPH oxidase homologs, DUOX1 and DUOX2 uniquely produce H2O2 without depending on superoxide dismutase to catalyze the reaction from O2∙− to H2O2 (27). Therefore, NOX2 is the major player in eosinophil respiratory burst. Eotaxin and TGF-β are known to activate eosinophils, leading to their degranulation (2, 3). TGF-β did not elicit any effect on the mRNA levels of NOX2, suggesting that it has no role in regulating gene expression. However, TGF-β may be an activator of the NADPH oxidase protein possibly through a PKC-dependent pathway. Eotaxin, on the other hand, decreased NOX2 mRNA transcripts in a time-dependent manner, suggesting the role of eotaxin in eosinophils in early stages of allergic airway inflammation, and may also control eosinophil activation. Under in vitro conditions, eosinophils survive for a limited time. Therefore, extrapolation of the findings from in vitro experiments to in vivo where eosinophils remain in contact with eotaxin and perhaps other cytokines for a longer period of time might be difficult. Additionally, the presence of multiple cytokines and their complex interactions with the signaling molecules might affect NOX2 expression in patients with asthma. The increase in the mRNA levels of NOX2 with IL-33 supports the theory that NOX2 expression is increased in patients with asthma and assists in the activation of eosinophils. IL-33 is a potent cytokine involved in the development of eosinophils from CD34+CD117+ progenitors and supports production of IL-13, TGF-β, and CCL17 in an ST2-dependent pathway in eosinophils (28). Because IL-33 potentiates the expression of CCR3 on differentiating eosinophils (28) through IL-33/ST2 signaling, it is possible that IL-33 is also involved in the regulation of NOX2 expression in eosinophils. However, the precise molecular mechanism of IL-33–induced NOX2 expression warrants further attention. Massive levels of NOX2 mRNA in moderate to severe asthmatic eosinophils in comparison to the eosinophils from healthy subjects and in subjects with mild to moderate asthma indicate an increased activity level of eosinophils based on the severity of asthma. The presence of more NOX2 in the eosinophils could indicate their ability to produce more O2∙− and hence more oxidative damage mediated by eosinophils in the airways of subjects with allergic asthma.
Sustained activation of NADPH oxidase depends on costimulation of ion channels that can compensate the charge imbalance generated by O2∙− production from NADPH oxidase. CLC3, a Cl−/H+ antiporter, works to balance the depolarization generated in the process (27, 29). Phorbol esters, like PMA, produce respiratory burst in cells, including eosinophils (9, 30, 31). PMA binds to the C1 domain of conventional protein kinase C (PKC) to change its conformation (32, 33). This signaling event leads to the activation of eosinophils and assists in degranulation and O2∙− generation. There is some evidence that PKC modulates the phosphorylation at the N-terminus of CLC3 at Ser 51 (34). Thus, the activation of PKC may be a critical step in eosinophil activation through NADPH oxidase and CLC3 activation.
Lysosomes contain CLC3 channels that are involved in their acidification via a Cl−/H+ antiporter mechanism (35, 36). The presence of NADPH oxidase on the phagosomes (37) and the interplay between CLC3 and NADPH oxidase in respiratory burst of neutrophils (38) and eosinophils (9) led us to examine the intracellular ROS generation in eosinophils. Although there was no significant effect of PMA in the generation of intracellular ROS, PMA-treated eosinophils exhibited heterogeneity within and between subjects (Figures 2F and 2L). This could relate to the presence of hypo- and normo-dense eosinophils in the blood (39). The heterogeneity in the eosinophil population may indicate their activated state and could be associated with increased susceptibility to the pathogenesis in allergic asthma. Additionally, an increase in the size and granularity of a subpopulation of PMA-stimulated eosinophils supports their PKC-dependent activity and further highlights the presence of multiple populations of eosinophils in a subject.
Extracellular O2∙− generation from NADPH oxidase showed a direct dependence on PKC activation. Treatment with PMA resulted in massive O2∙− release from eosinophils in 20 minutes, which was decreased to half in the presence of CLC3 inhibitor. However, O2∙− generation in response to TGF-β and eotaxin showed two peaks, suggesting a two-step release of O2∙−. This two-step release of O2∙− may relate to a sequential activation of NADPH oxidase and CLC3 in the presence of TGF-β and eotaxin. In all cases of O2∙− release, CLC3 dependence was reiterated by the CLC3 blocker NPPB (40). Generation of O2∙− in eosinophils is directly dependent on NADPH oxidase. DPI is an uncompetitive inhibitor of flavoenzymes (41) and therefore can only inhibit the enzyme when the substrate is bound to the active site of the enzyme. DPI significantly reduced the early peak of O2∙− generation, but there was no significant change in the amplitude in the second burst. However, eotaxin-3–induced O2∙− did not decrease to the baseline with the NADPH oxidase inhibitor in the first peak and delayed the second peak. This may suggest that eotaxin-3–induced NADPH oxidase activity supersedes the DPI blockade by restricting the DPI binding to the enzyme through conformational change or steric hindrance. The cause of the shift in the respiratory burst with DPI is not known but may be related to the partial masking of the DPI binding site. CLC3 blocker, on the other hand, is not affected by any of the cytokines and consistently blocks the O2∙− generation, confirming the dependence of O2∙− generation on CLC3. The CCR3 blocker SB328437, in the concentration that inhibited eosinophil migration, did not inhibit O2∙− production. However, it delayed the kinetics of the burst, indicating the involvement of additional pathways in the process. Because SB328437 induced O2∙− production by itself and in the presence of eotaxin-1 and eotaxin-3, it is possible that additional signaling pathways to induce O2∙− production are activated upon blocking of CCR3. Involvement of an alternative pathway in eosinophil activation could be further supported by the finding of delayed O2∙− release in the presence of SB328437. In addition, the possibility of eotaxin exerting its effect on O2∙− release in eosinophils by binding to a site other than CCR3 cannot be ruled out. In this regard, a direct interaction of eotaxin to CLC3 is a possibility and warrants further attention.
Eosinophils migrate to the site of inflammation and release cytokines, chemokines, growth factors, chemical mediators, and ROS to support the excessive damage and repair cycle in the airways in allergic asthma. Activation of CLC3 is required for the NADPH oxidase activity. At the same time, charge imbalance potentiated by the activation of NADPH oxidase requires activation of CLC3, which transports Cl− in and H+ out of the cells to maintain homeostasis. However, the underlying molecular signaling involved in the coactivation of closely associated NADPH oxidase and CLC3 is unknown and warrants further attention. In addition to activation, CLC3 has been implicated in the migration of neutrophils (19). To our knowledge, this is the first report to link CLC3 to the migration of eosinophils. Eotaxin is a potent chemoattractant for eosinophils. However, the potency and selectivity varies between eotaxin-1, eotaxin-2, and eotaxin-3. Eotaxin-1 and eotaxin-3 have been implicated in the pathogenesis of asthma, especially eosinophilic asthma (13). Although eotaxin-1 and eotaxin-3 showed similar overall increases in the migration of eosinophils, the effect of eotaxin-1 was consistent in all individuals, whereas the effect of eotaxin-3 varied between the individuals, suggesting heterogeneity in eotaxin-3–induced cellular response among the subjects. Because both these chemokines use the same receptor, CCR3 blocker (SB328437) markedly reduced the number of migrated cells in the eotaxin-1 and eotaxin-3 groups. TGF-β has also been related to the migration of eosinophils. Our findings on the chemotaxis suggest that the cytokines TGF-β and eotaxin use CLC3 to induce eosinophil migration. The role of PKC in eosinophil migration could be supported by the studies using phloretin, which selectively inhibits PKCs and CLC3 (20, 40).
Uptake of Cl− through CLC3 may result in the swelling of eosinophils, causing shape change that may lead to the cytoskeletal rearrangement and migration of eosinophils (Figure 7). Alternatively, CLC3 activation may cause migration of eosinophils through direct changes in the actin filaments. Studies have shown that hypotonic activation of CLC3b (a transcript variant) facilitates cell swelling–mediated remodeling of the actin cytoskeleton (42, 43). The cytosolic c-terminus of CLC3b directly binds to filamentous actin between amino acids 690 and 760 at proline 688 and leucine 734 (43). In addition to the direct modulation of actin, conformational changes initiated by phosphorylation/activation of CLC3 may lead to downstream signaling through cytoskeletal proteins like Rac and ROCK to cause shape change and migration of eosinophils.
Figure 7.

Mechanism of activation and migration of blood eosinophils. NOX2 generates O2∙−, causing a depolarization and charge imbalance in eosinophils, which can be compensated by the antiporter activity of the CLC3. Superoxide may dismutate to hydrogen peroxide (H2O2) at low pH or in the presence of SOD. Being permeable, H2O2 can travel through the membrane and become H2O in presence of catalase or transform to a more oxidative hypochlorous acid (HOCl) through eosinophil peroxidase (EPO) in eosinophils. The activation of CLC3 leads to Cl− influx, resulting in shape change and cytoskeletal rearrangement in eosinophils and supporting the migration of eosinophils in response to cytokines/chemokines. Physical interaction of CLC3 to filamentous actin (F-actin) may also result in the cytoskeletal rearrangement and migration of eosinophils upon channel activation/phosphorylation.
Novel findings related to the eotaxin and TGF-β–dependent activation and migration of eosinophils involving CLC3 in eosinophils provide a promising target for therapy in allergic asthma. More studies are required to identify the precise underlying molecular events involving the cytoskeletal proteins and their rearrangement for eosinophil transendothelial migration and in the activation of eosinophil in allergic asthma.
Acknowledgments
Acknowledgments
The authors thank Dr. Greg Perry, Flow Cytometry Core Facility of Creighton University, and volunteers who took part in this study.
Footnotes
This work was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases grant R01AI075315.
Author Contributions: Conception and design: R.G. and D.K.A. Analysis and interpretation: R.G., A.K.B., and D.K.A. Drafting the manuscript for important intellectual content: R.G. and D.K.A. Finalization and approval of the final version of the manuscript: R.G., A.K.B., and D.K.A.
Originally Published in Press as DOI: 10.1165/rcmb.2014-0300OC on December 16, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 2.Radonjic-Hösli S, Simon HU. Eosinophils. Chem Immunol Allergy. 2014;100:193–204. doi: 10.1159/000358735. [DOI] [PubMed] [Google Scholar]
- 3.Gaurav R, Agrawal DK. Clinical view on the importance of dendritic cells in asthma. Expert Rev Clin Immunol. 2013;9:899–919. doi: 10.1586/1744666X.2013.837260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Lambeth JD, Cheng G, Arnold RS, Edens WA. Novel homologs of gp91phox. Trends Biochem Sci. 2000;25:459–461. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- 6.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 7.Morgan D, Capasso M, Musset B, Cherny VV, Ríos E, Dyer MJ, DeCoursey TE. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc Natl Acad Sci USA. 2009;106:18022–18027. doi: 10.1073/pnas.0905565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh MT, Connell K, Sheahan AM, Gleich GJ, Costello RW. Eosinophil peroxidase signals via epidermal growth factor-2 to induce cell proliferation. Am J Respir Cell Mol Biol. 2011;45:946–952. doi: 10.1165/rcmb.2010-0454OC. [DOI] [PubMed] [Google Scholar]
- 9.Schwingshackl A, Moqbel R, Duszyk M. Involvement of ion channels in human eosinophil respiratory burst. J Allergy Clin Immunol. 2000;106:272–279. doi: 10.1067/mai.2000.107752. [DOI] [PubMed] [Google Scholar]
- 10.Luttmann W, Franz P, Matthys H, Virchow JC., Jr Effects of TGF-beta on eosinophil chemotaxis. Scand J Immunol. 1998;47:127–130. doi: 10.1046/j.1365-3083.1998.00298.x. [DOI] [PubMed] [Google Scholar]
- 11.Wenzel SE, Trudeau JB, Barnes S, Zhou X, Cundall M, Westcott JY, McCord K, Chu HW. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J Immunol. 2002;169:4613–4619. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol. 2003;111:227–242, quiz 243. doi: 10.1067/mai.2003.139. [DOI] [PubMed] [Google Scholar]
- 13.Chae SC, Lee YC, Park YR, Shin JS, Song JH, Oh GJ, Hong ST, Pae HO, Choi BM, Chung HT. Analysis of the polymorphisms in eotaxin gene family and their association with asthma, IgE, and eosinophil. Biochem Biophys Res Commun. 2004;320:131–137. doi: 10.1016/j.bbrc.2004.05.136. [DOI] [PubMed] [Google Scholar]
- 14.Kotsimbos AT, Hamid Q. IL-5 and IL-5 receptor in asthma. Mem Inst Oswaldo Cruz. 1997;92:75–91. doi: 10.1590/s0074-02761997000800012. [DOI] [PubMed] [Google Scholar]
- 15.Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med. 1995;182:1169–1174. doi: 10.1084/jem.182.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim MJ, Cheng G, Agrawal DK. Cl- channels are expressed in human normal monocytes: a functional role in migration, adhesion and volume change. Clin Exp Immunol. 2004;138:453–459. doi: 10.1111/j.1365-2249.2004.02635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMains E, Krishnan V, Prasad S, Gleason E. Expression and localization of CLC chloride transport proteins in the avian retina. PLoS One. 2011;6:e17647. doi: 10.1371/journal.pone.0017647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki M, Morita T, Iwamoto T. Diversity of Cl(-) channels. Cell Mol Life Sci. 2006;63:12–24. doi: 10.1007/s00018-005-5336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS. Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem. 2006;281:12277–12288. doi: 10.1074/jbc.M511030200. [DOI] [PubMed] [Google Scholar]
- 20.Volk AP, Heise CK, Hougen JL, Artman CM, Volk KA, Wessels D, Soll DR, Nauseef WM, Lamb FS, Moreland JG. ClC-3 and IClswell are required for normal neutrophil chemotaxis and shape change. J Biol Chem. 2008;283:34315–34326. doi: 10.1074/jbc.M803141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pusch M. Structural insights into chloride and proton-mediated gating of CLC chloride channels. Biochemistry. 2004;43:1135–1144. doi: 10.1021/bi0359776. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda JJ, Filali MS, Volk KA, Collins MM, Moreland JG, Lamb FS. Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol. 2008;294:C251–C262. doi: 10.1152/ajpcell.00338.2007. [DOI] [PubMed] [Google Scholar]
- 23.Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 24.Mao J, Li X, Chen W, Xu B, Zhang H, Li H, Wang L, Jin X, Zhu J, Lin G, et al. Cell cycle-dependent subcellular distribution of ClC-3 in HeLa cells. Histochem Cell Biol. 2012;137:763–776. doi: 10.1007/s00418-012-0937-0. [DOI] [PubMed] [Google Scholar]
- 25.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 26.Lacy P, Abdel-Latif D, Steward M, Musat-Marcu S, Man SF, Moqbel R. Divergence of mechanisms regulating respiratory burst in blood and sputum eosinophils and neutrophils from atopic subjects. J Immunol. 2003;170:2670–2679. doi: 10.4049/jimmunol.170.5.2670. [DOI] [PubMed] [Google Scholar]
- 27.Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185:3472–3480. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- 29.Shuvaev VV, Han J, Yu KJ, Huang S, Hawkins BJ, Madesh M, Nakada M, Muzykantov VR. Pecam-targeted delivery of sod inhibits endothelial inflammatory response. FASEB J. 2011;25:348–357. doi: 10.1096/fj.10-169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umei T, Ohhara N, Okamura S, Harada M, Nakao M, Shirai T, Niho Y. Activation of neutrophils NADPH oxidase by PMA: cytosol activity is translocated in phorbol-primed neutrophils. Int J Biochem. 1993;25:631–633. doi: 10.1016/0020-711x(93)90346-g. [DOI] [PubMed] [Google Scholar]
- 31.Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, Quinn MT, Gougerot-Pocidalo MA, Dang PM, El-Benna J. Regulation of the phagocyte nadph oxidase activity: phosphorylation of gp91phox/nox2 by protein kinase c enhances its diaphorase activity and binding to rac2, p67phox, and p47phox. FASEB J. 2009;23:1011–1022. doi: 10.1096/fj.08-114553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leonard TA, Różycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C βII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J Gen Physiol. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, Verkman AS. ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem. 2005;280:1241–1247. doi: 10.1074/jbc.M407030200. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Wang T, Zhao Z, Weinman SA. The ClC-3 chloride channel promotes acidification of lysosomes in CHO-K1 and Huh-7 cells. Am J Physiol Cell Physiol. 2002;282:C1483–C1491. doi: 10.1152/ajpcell.00504.2001. [DOI] [PubMed] [Google Scholar]
- 37.Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci USA. 2010;107:10496–10501. doi: 10.1073/pnas.0914867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreland JG, Davis AP, Matsuda JJ, Hook JS, Bailey G, Nauseef WM, Lamb FS. Endotoxin priming of neutrophils requires NADPH oxidase-generated oxidants and is regulated by the anion transporter ClC-3. J Biol Chem. 2007;282:33958–33967. doi: 10.1074/jbc.M705289200. [DOI] [PubMed] [Google Scholar]
- 39.Agrawal DK, Sarmiento EU, Nabe M, Miyagawa H, Townley RG. Increased hypodense eosinophils after activation with paf-acether and calcium ionophore in asthmatic subjects. J Asthma. 1996;33:213–219. doi: 10.3109/02770909609055362. [DOI] [PubMed] [Google Scholar]
- 40.Do CW, Lu W, Mitchell CH, Civan MM. Inhibition of swelling-activated Cl- currents by functional anti-ClC-3 antibody in native bovine non-pigmented ciliary epithelial cells. Invest Ophthalmol Vis Sci. 2005;46:948–955. doi: 10.1167/iovs.04-1004. [DOI] [PubMed] [Google Scholar]
- 41.Riganti C, Gazzano E, Polimeni M, Costamagna C, Bosia A, Ghigo D. Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J Biol Chem. 2004;279:47726–47731. doi: 10.1074/jbc.M406314200. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki S, Yui N, Noda Y. Actin directly interacts with different membrane channel proteins and influences channel activities: AQP2 as a model. Biochim Biophys Acta. 2014;1838:514–520. doi: 10.1016/j.bbamem.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 43.McCloskey DT, Doherty L, Dai YP, Miller L, Hume JR, Yamboliev IA. Hypotonic activation of short ClC3 isoform is modulated by direct interaction between its cytosolic C-terminal tail and subcortical actin filaments. J Biol Chem. 2007;282:16871–16877. doi: 10.1074/jbc.M700379200. [DOI] [PubMed] [Google Scholar]