

Abstract
Strategies for the treatment of bacterial pneumonia beyond traditional antimicrobial therapy have been limited. The recently discovered novel genus of lipid mediators, coined “specialized proresolving mediators” (SPMs), which orchestrate clearance of recruited leukocytes and restore epithelial barrier integrity, have offered new insight into the resolution of inflammation. We performed lipid mediator (LM) metabololipidomic profiling and identification of LMs on peripheral blood leukocytes and plasma from a baboon model of Streptococcus pneumoniae pneumonia. Leukocytes and plasma were isolated from whole blood of S. pneumoniae–infected (n = 5–6 per time point) and control, uninfected baboons (n = 4 per time point) at 0, 24, 48, and 168 hours. In a subset of baboons with pneumonia (n = 3), we administered inhaled carbon monoxide (CO) at 48 hours (200–300 ppm for 60–90 min). Unstimulated leukocytes from control animals produced a proresolving LM signature with elevated resolvins and lipoxins. In contrast, serum-treated, zymosan-stimulated leukocytes and leukocytes from baboons with S. pneumoniae pneumonia produced a proinflammatory LM signature profile with elevated leukotriene B4 and prostaglandins. Plasma from baboons with S. pneumoniae pneumonia also displayed significantly reduced LM–SPM levels, including eicosapentaenoic acid–derived E-series resolvins (RvE) and lipoxins. CO inhalation increased levels of plasma RvE and lipoxins relative to preexposure levels. These results establish the leukocyte and plasma LM profiles biosynthesized during S. pneumoniae pneumonia in baboons and provide evidence for pneumonia-induced dysregulation of these proresolution programs. Moreover, these SPM profiles are partially restored with inhaled low-dose CO and SPM, which may shorten the time to pneumonia resolution.
Keywords: inflammation resolution, therapeutic carbon monoxide, pneumonia, bacterial
Clinical Relevance
Lipid mediator signature profiles were measured in a reported baboon model of pneumococcal pneumonia. Baboons with pneumonia displayed significantly reduced levels of specialized proresolving mediators (SPMs) relative to controls. Levels could be partially restored by administering inhaled low-dose CO. This study advances prior observations in mice to nonhuman primates and is the first to identify SPM and CO regulation of SPMs in baboons. These findings can offer insight into impaired host resolution responses during bacterial pneumonia.
Pneumococcal pneumonia remains a leading cause of death worldwide despite the use of powerful antimicrobial agents (1–5). These agents work to limit the infection but may not be sufficient for resolution of severe pneumonia with acute lung injury (6–9). Lipid mediators (LMs) are recently recognized to govern the initiation and resolution of inflammation and infections in mice (10, 11).
These mediators are produced via stereospecific enzymatic conversion of essential fatty acids in humans (11, 12). If the inflammation is self-limited, leukocytes undergo LM class switching and produce specialized proresolving mediators (SPMs) (13), a recently discovered genus of LMs that actively promote resolution of inflammation (12, 14). SPMs include five main families of mediators: the arachidonic acid–derived lipoxins, the eicosapentaenoic acid–derived resolvins (E-series; RvE) (15), the docosahexaenoic acid–derived resolvins (D-series; RvD), the protectins (14), and the maresins (16).
SPMs and their bioactive metabolomes display potent stereoselective actions that reduce further leukocyte recruitment, stimulate clearance of apoptotic cells and debris, and promote epithelial barrier integrity (6–8, 17–19). Each SPM displays additional characteristic actions. For example, RvD1, RvD2, and RvD5 activate host defenses in mice during bacterial infection (18, 19), promoting microbial containment and clearance and thereby reducing antibiotic requirements (18). SPMs also regulate the production of antimicrobial peptides by human mucosal epithelial cells (20). In this regard, SPMs are defensive rather than immunosuppressive by enhancing innate responses. However, if LM class switching is dysregulated, there is excessive leukocyte recruitment, tissue destruction, and eventually chronic inflammation (6–8, 11).
The production of SPMs can be stimulated by induction of the heme oxygenase-1/carbon monoxide (HO-1/CO) system (21). HO-1 is an inducible enzyme that degrades heme to bilivirdin, CO, and free ion, thereby acting as the endogenous source for CO production during inflammation. HO-1/CO is rapidly up-regulated in the lung (22) during murine bacterial pneumonia, where it displays cytoprotective actions; mice with impaired HO-1 activation display worsened acute lung injury (23). CO administered exogenously by inhalation also has proresolving effects. CO gas up-regulates the expression of 15-lipoxygenase type 1, a key enzyme in SPM biosynthesis, and prolongs SPM bioactivity by inhibiting inactivation (21). Moreover, inhaled CO has been shown to accelerate the resolution of acute inflammation in murine peritonitis (21, 24) and in a nonhuman primate model of lung inflammation (25). However, the effects of inhaled CO on SPM production during bacterial pneumonia are of interest in baboons.
The complete stereochemistry of the main bioactive mediators and their related biosynthetic isomers are established and were confirmed using total organic synthesis of the potent bioactive products (17) (see Table E1 in the online supplement). Using an automated approach, authenticated standards for SPMs, their related pathway markers and known proinflammatory LMs, and enhanced sensitivity, we can now perform targeted LM metabololipidomic profiling of each of these autacoid pathways (26). In the present report, we identified SPMs in a baboon model of Streptococcus pneumoniae pneumonia (9), the most common cause of bacterial pneumonia in humans (27, 28). This model was used to investigate the temporal SPM production, regulation, and relationship to other inflammatory LMs during acute inflammation to assess processes that may lead to failed lipid mediator class switching and chronic inflammation. We established the baseline LM profiles of baboon peripheral blood leukocytes and plasma during the course of infection and found that these profiles are significantly altered. In a subset of infected baboons, these SPM programs could be partially increased with low-dose CO inhalation.
Materials and Methods
Materials
Liquid chromatography (LC)-grade solvents were purchased from Fisher Scientific (Pittsburgh, PA), Eclipse Plus C18 column (100 × 4.6 mm × 1.8 μm) was obtained from Agilent (Santa Clara, CA), C18 SPE columns were from Waters (Milford, MA), and synthetic standards for LC tandem mass spectrometry (LC-MS-MS) quantitation and deuterated (d) internal standards (d5–RvD2, d5–lipoxin [LX]A4, d4–prostaglandin [PG]E2, d4–LTB4, and d8–5S-HETE) were purchased from Cayman Chemicals (Ann Arbor, MI)
Animal Model
Adult male colony-bred baboons (Papio cynocephalus) (n = 15), 7.5 ± 1.7 years of age (mean ± SD) and weighing 26 ± 2.4 kg, were purchased from Texas Biomedical Research Institute (San Antonio, TX). All animals were housed in the Duke University Vivarium (Durham, NC) and handled in accordance with American Association for Accreditation of Laboratory Animal Care guidelines. The experimental protocol was approved by the Duke University Institutional Animal Care and Use Committee.
On Day 1, an individual animal was sedated, intubated, and ventilated mechanically. After a baseline bronchoalveolar lavage (BAL) of the left lower lobe with 20 ml 0.9% NaCl (saline) using a fiberoptic bronchoscope (Pentax, Montvale, NJ), the animals were randomly assigned to receive saline vehicle (n = 4) or S. pneumoniae at 109 CFU (n = 8). The bacterial suspension (10 ml) was divided and instilled equally between the left lower lobe and lingula via bronchoscope. After 6 hours, the animals were recovered, extubated, and placed in isolation. At 24 hours, the animals were sedated briefly to collect blood samples. At 48 hours, the animals were sedated, intubated, and ventilated similar to Day 1. Samples were collected, including a repeat BAL of the left lower lobe, and the animals were extubated and returned to isolation. After the 48-hour samples were obtained, 1 g ceftriaxone (Hospira Inc., Lake Forest, IL) was administered to all animals once daily for 3 days (9).
CO Inhalation
A second group of animals (n = 3) underwent a modified experimental protocol as follows. On Day 1, S. pneumoniae (8 × 108 to 2 × 109 CFU) was instilled equally via bronchoscope into both lower lobes with or without instillation into the right middle lobe/lingula. After 1 hour, the animals were recovered, extubated, and placed in isolation. After collection of the 48-hour samples, inhaled CO gas was administered through the ventilator circuit at 200 to 300 ppm for 60 to 90 minutes. Arterial carboxyhemoglobin levels were followed serially during the CO exposure and remained below 10%. Immediately after the CO exposure (∼50 h after inoculation), blood samples were collected, and the animals were given FiO2 1.0 until carboxyhemoglobin values returned to near baseline levels. The rest of the protocol was unchanged.
Sample Collection
Vital sign data (heart rate, respiratory rate, blood pressure, pulse oximetry, and temperature) were collected periodically throughout the experiment. Peripheral blood was collected in EDTA (for plasma and complete blood counts) at 0, 6 (not in the CO experiment), 24, 48, 50 (for the CO experiment only), and 168 hours and in a heparin-primed syringe (for leukocyte isolation) using a 19- to 20-gauge needle at 0, 24, 48, and 168 hours. Urine was collected at 0, 6 (not in the CO experiment), 48, 50 (in the CO experiment only), and 168 hours. Chest radiographs and BAL fluid (BALF) were collected at 0, 48, and 168 hours. All samples were stored at −80°C. “Leukocytosis” is defined as a white blood cell (WBC) count ≥2-fold elevation above baseline or absolute WBC count >15,000 cells/μl. “Leukopenia” is defined as absolute WBC count <4,000 cells/μl.
Targeted Lipid Mediator Metabololipidomics
All samples for LC-MS-MS–based metabololipidomics were extracted with solid-phase extraction columns as reported (26). Before sample extraction, deuterated internal standards representing each region in the chromatographic analysis (500 pg each) were added to facilitate quantification. Extracted samples were analyzed by a LC-MS-MS system (Qtrap 6500; AB Sciex, Framingham, MA) equipped with a SIL-20AC autoinjector and LC-20AD binary pump (Shimadzu Corp., Kyoto, Japan). Further details are provided in the online supplement.
Statistical Analysis
Grouped data were expressed as mean ± SD or as median with interquartile range (IQR) where specified. Part of the physiological and microbiological data used to define the pneumonia model was reported in an earlier paper (9). Data were analyzed using Student’s t test with Welch’s correction (Excel; Microsoft, Redmond, WA). Partial least squares-discrimination analysis (PLS-DA) (26, 29) was performed using SIMCA 13.0.3 software (Umetrics, Umea, Sweden).
Additional methodological details are provided in the online supplement.
Results
SPMs Are Produced by Baboon Peripheral Blood Leukocytes
Using targeted LM metabololipidomics, we investigated the LM profiles of peripheral blood leukocytes isolated from uninfected baboons (n = 7). We identified mediators from both lipoxygenase- and cyclooxygenase-dependent pathways, including RvD1, RvE2, PGD2, and PGF2α. We also identified biosynthetic isomers for select SPM markers, including 10S,17S-diHDHA, a protectin biosynthetic isomer, and 4S,14S-diHDHA, a maresin biosynthetic isomer (Figure 1A), each produced by double lipoxygenation reactions. These were identified in accordance with published criteria (26) that included matching retention times (Figure 1A), fragmentation patterns, and at least six characteristic and diagnostic ions for each. This is illustrated for RvD1 (375 = M-H; 357 = M-H-H2O; 339 = M-H-2H2O; 331 = M-H-CO2; 313 = M-H-H2O-CO2; 295 = M-H-2H2O-CO2; 215 = 233-H2O) and RvE2 (Figure 1B, insets).
Figure 1.

Specialized proresolving lipid mediator (LM) biosynthesis by baboon peripheral blood leukocytes. Baboon peripheral blood leukocytes were isolated from heparinized whole blood. A total of 1 × 107 cells/ml were incubated at 37°C for 30 minutes with or without 0.1 mg serum-treated zymosan (STZ). LMs were assessed using LM metabololipidomics after solid-phase extraction (see Materials and Methods). (A) Representative multiple reaction monitoring (MRM) chromatograms of selected ion pairs for arachidonic acid–, eicosapentaenoic acid–, and docosahexaenoic acid–derived LMs (a = 6-trans-leukotriene [LT]B4; b = 6-trans,12-epi-LTB4). (B) Representative tandem mass spectrometry spectra with diagnostic ions used for the identification of resolvin (Rv)D1 and RvE2. (C) Cumulative levels for D-series resolvins, protectins, and maresins and E-series resolvins, lipoxins, LTB4 metabolome, and prostaglandins. Results are mean ± SEM (n = 3–4 cell isolations). *P < 0.05 versus peripheral blood leukocytes (control incubations). (D) Pie charts depicting the relative LM levels in leukocytes without further agonists (left panel) and STZ-stimulated (right panel) peripheral blood leukocytes. Data are representative of seven baboons. Da = dalton; LX = lipoxin; M-H = molecular ion; m/z = mass-to-charge ratio; PG = prostaglandin; SPM = specialized proresolving mediator.
We next assessed the LM profiles of baboon leukocytes stimulated with serum-treated zymosan (STZ) and found increased proinflammatory LM levels, including LTB4 and its omega hydroxylation metabolite 20-OH-LTB4, and PGE2, PGD2, and PGF2α (Table E2), which were reflected in significant increases in the overall levels of the leukotriene (LT)B4 metabolome and prostaglandins (Figure 1C). These results demonstrate that unstimulated baboon leukocytes produce SPMs but undergo proinflammatory class switching after STZ stimulation (Figure 1D).
Animals Inoculated with S. pneumoniae Developed Lobar Pneumonia with Bacteremia
Animals infected with S. pneumoniae (n = 11) met our preestablished criteria for pneumonia (9). In contrast to the control animals (n = 4), by 24 to 48 hours the S. pneumoniae–infected animals had developed fever (temperature, 38.8 ± 0.7 versus 37.3 ± 0.4°C), tachycardia (heart rate, 122 ± 20 versus 80 ± 4 beats/min), and tachypnea (respiratory rate, 47 ± 12 versus 23 ± 7 breaths/min) (all P ≤ 0.02). Each S. pneumoniae–infected animal also displayed one or more of the following WBC count abnormalities: leukocytosis (range, 12.4–38.4 × 103 cells/μl) (n = 9), leukopenia (range, 0.9–2.9 × 103 cells/μl) (n = 2), and/or neutrophilia ≥90% on WBC differential (n = 9) (Figure 2A). BALF cell counts were significantly increased in the S. pneumoniae–infected animals relative to controls at 48 hours (488 ± 512 × 104 versus 14 ± 13 × 104 cells/ml; P = 0.01) and 168 hours (133 ± 151 × 104 versus 17 ± 8 × 104 cells/ml; P = 0.065) (Figure 2B). BALF cell differentials in the control animals remained at baseline throughout the experiment (81–86% mononuclear; 13–19% neutrophils), whereas in the S. pneumoniae–infected animals, the percentage of neutrophils rose at 48 hours to 69 ± 13% (P = 0.054) and returned to baseline by 168 hours. Chest radiographs at 48 hours demonstrated left lung parenchymal opacities of variable density (n = 9) (Figures 2C and 2D). The experimental S. pneumoniae strain was isolated from blood at 24 and/or 48 hours (n = 9) (median [IQR], 64 [234] CFU/ml) and from BALF (n = 10) at 48 hours (median [IQR], 1.3 × 105 [3.8 × 105] CFU/ml) in the S. pneumoniae–infected group. These sites had been sterilized after antibiotic administration by 168 hours, whereas control group samples remained sterile throughout the experiment.
Figure 2.

Animals inoculated with Streptococcus pneumoniae displayed signs and symptoms of pneumonia. (A) Box-and-whisker plot of white blood cell (WBC) count (×103 cells/μl) at 0, 24, 48, and 168 hours after inoculation with S. pneumoniae (gray; n = 11). Control values (black; n = 4) shown for comparison. (B) Bronchoalveolar lavage fluid (BALF) cell counts at 0, 48, and 168 hours after inoculation with S. pneumoniae (gray; n = 8–11 per time point). Control animals (black; n = 3–4 per time point) shown for comparison. BALF percent neutrophils are given for each time point and are higher in S. pneumoniae–infected animals compared with uninfected controls at 48 hours (P = 0.054). *P = 0.01; #P = 0.065. (C and D) Representative chest radiographs at baseline (C) and 48 hours after inoculation with 109 CFU demonstrating left lower lobe alveolar consolidation (D). Results are representative of n = 4 for control and n = 11 for infected baboons.
Plasma LM-SPMs Are Reduced after S. pneumoniae Pneumonia
We next investigated the plasma LM-SPM profiles in control and S. pneumoniae–infected animals. At baseline, we identified 22 bioactive mediators and pathway products from the docosahexaenoic acid (DHA), arachidonic acid (AA), and eicosapentaenoic acid (EPA) bioactive metabolomes in plasma using previously published criteria (26). These included RvD2, RvD3, RvE1, PGD2, PGE2, and LTB4 (Figures 3A and 3B) and the 17-R epimers of RvD1 and RvD3.
Figure 3.


Dysregulated LM-specialized proresolving mediator levels in baboon plasma during pneumonia. Plasma samples were obtained from S. pneumoniae–infected or control animals at 0, 24, 48, and 168 hours after inoculation. Ceftriaxone (1 g daily for 3 d) was administered at 48 hours after sample collection. LMs were assessed using LM metabololipidomics after solid-phase extraction (see Materials and Methods). (A) Representative MRM chromatograms of selected ion pairs for arachidonic acid–, eicosapentaenoic acid–, and docosahexaenoic acid–derived LMs. a = 6-trans-LTB4; b = 6-trans,12-epi-LTB4. (B) Representative tandem mass spectrometry spectra with diagnostic ions used for the identification of LXB4 and RvD1. (A and B) Representative of 40 samples. (C) Cumulative levels for D-series resolvins, E-series resolvins, lipoxins, LTB4 metabolome, and prostaglandins. Results are mean ± SEM (n = 4 for control baboons; n = 6 for infected baboons for each time point). *P < 0.05 versus control values at respective time point; #P < 0.05 versus the 0-hour S. pneumoniae group.
LM quantification achieved using multiple reaction monitoring of signature ion pairs Q1 (parent ion) and Q3 (characteristic daughter ion) is shown in Table E2. Compared with baseline values, S. pneumoniae inoculation (n = 6) reduced overall plasma LM levels at 48 and 168 hours after inoculation (Figure 3C). DHA-derived (D-series) resolvins decreased from 28.8 ± 3.8 pg/ml at baseline to 4.0 ± 0.3 pg/ml at 48 hours; E-series resolvins RvE2 and RvE3 were identified in plasma from infected baboons, and their levels decreased from 50.3 ± 11.2 pg/ml at baseline to 19.8 ± 4.7 pg/ml at 48 hours; and lipoxins decreased from 188.6 ± 71.1 pg/ml at baseline to 22.8 ± 3.1 pg/ml at 48 hours (Figure 3C). LM levels did not significantly change in plasma from uninfected control baboons throughout this time course (Figure 3C). Compared with values obtained from control baboons, S. pneumoniae–inoculated animals demonstrated significantly reduced levels of plasma D-series (3.9 ± 0.7 versus 9.7 ± 1.9 pg/ml) and E-series (17.8 ± 2.1 versus 41 ± 8.7 pg/ml) resolvins, lipoxins (28.2 ± 3.7 versus 78.5 ± 17.0 pg/ml), LTB4 metabolome (1.5 ± 0.4 versus 5.9 ± 0.5 pg/ml), and prostaglandins (12.4 ± 2.1 versus 69.7 ± 17.9 pg/ml) at 48 and 168 hours. These results demonstrated that there was a temporal shift in the plasma LM profiles after infection (Figure E1).
We next investigated the plasma LM profiles of control (n = 4) and S. pneumoniae–infected animals (n = 6) using PLS-DA. LM profiles from control and infected animals produced separate, distinct clusters at 24, 48, and 168 hours (Figure 4A). Compared with controls, profiles of S. pneumoniae–infected animals displayed tight clusters at 24 and 48 hours, indicating that these plasma LM-SPM profiles were highly homogeneous (Figure 4A). The gray zone of the score plots depicts outliers with 95% confidence (26).
Figure 4.

S. pneumoniae pneumonia yields distinct plasma LM profiles. Plasma samples were obtained from S. pneumoniae–infected or control animals at 24, 48, and 168 hours after inoculation. Ceftriaxone (1 g daily for 3 d) was administered at 48 hours after the sample collection. LMs were assessed using LM metabololipidomics after solid-phase extraction (see Materials and Methods). (A) Partial least-squares discriminant analysis 2D score plots (left panels) and (B) the corresponding loading plots (right panels) for plasma samples. The gray zone in the score plots depicts outliers with 95% confidence. Results are representative of n = 4 for control baboons and n = 6 for infected baboons for each time point.
Loading plots depict associations between the identified LM-SPMs and clusters displayed in the score plots (26, 29). The loading plot at 24 hours demonstrates an association between plasma RvD1, RvD6, RvE3, LXB4, LTB4, and PGF2α and the S. pneumoniae–infected animals (Figure 4B). Statistical analysis of this select group of mediators demonstrated significantly higher plasma RvD6 (13.9 ± 2.7 versus 1.5 ± 0.5 pg/ml), RvE3 (35.3 ± 7.5 versus 7.9 ± 3.2 pg/ml), and LXB4 (112.8 ± 28.4 versus 19.1 ± 6.9 pg/ml) levels in S. pneumoniae–infected animals compared with control animals (Table E3). In contrast, loading plots at 48 and 168 hours demonstrate an association between the identified plasma LM-SPMs and the control cluster, indicating a decrease in plasma LM-SPM levels in S. pneumoniae–infected animals (Figure 4B; Table E3). Together, these results indicate that there is temporal regulation of plasma LM-SPM levels after inoculation with S. pneumoniae leading to dysregulated LM-SPM profiles.
Peripheral Blood Leukocytes Display a Proinflammatory LM-SPM Profile during S. pneumoniae Pneumonia
Having found that plasma LM profiles were altered by pneumonia, we assessed the effects of pneumonia on intact leukocyte LM profiles. Before infection, baboon peripheral blood leukocytes (n = 5) displayed a proresolving LM phenotype with elevated levels of SPMs, including D- and E-series resolvins and lipoxins (Figure 5A; Table E4; Figure E2). By 24 hours after S. pneumoniae inoculation, there was a reduction in the leukocyte proresolving mediator levels, including lipoxins, and an increase in classic eicosanoid levels, including prostaglandins (Figure 5B; Table E4; Figure E2). At 48 and 168 hours, leukocytes from baboons with pneumonia showed an increase in products from the LTB4 metabolome (Figures 5C and 5D; Table E4; Figure E2). These results indicate that peripheral blood leukocytes display a proinflammatory LM profile after S. pneumoniae infection.
Figure 5.
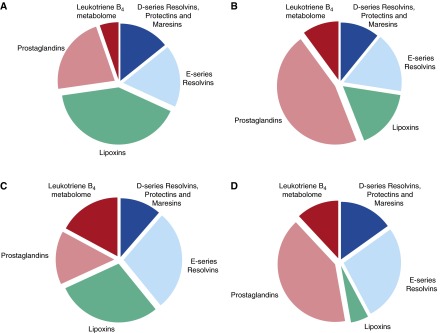
Infection alters LM profiles of circulating leukocytes. Blood was collected before and after S. pneumoniae inoculation at the indicated intervals. Leukocytes were isolated by density centrifugation (see Materials and Methods). LM profiles were investigated by targeted LM metabololipidomics. Pie charts depicting the relative LM levels in leukocytes isolated at (A) 0, (B) 24, (C) 48, and (D) 168 hours after inoculation. Results are representative of four baboons at 0 hours and six baboons at 24, 48, and 168 hours.
Infection Leads to a Temporal Regulation in the BAL LM-SPM Profile
We investigated the LM-SPM profiles of BALF from infected baboons to determine the temporal relationships in the biosynthesis of SPM and classic eicosanoids at the site of infection. In BAL before bacterial inoculation, we identified mediators from all three bioactive metabolomes, including the D-series resolvins, E-series resolvins, lipoxins, and prostaglandins (Figure 6A). Forty-eight hours after S. pneumoniae inoculation, we found an increase in all BAL LM and SPM levels. Here we found significant increases in LXB4 levels (Table E5) and in prostaglandins and LTB4 metabolome levels when compared with preinfection levels (Figure 6A). At 168 hours, SPM levels had decreased except for LXA4, which was found to be significantly higher than levels measured at 48 hours. We also found significantly higher levels of PGD2 and PGF2α in these BAL samples (Figure 6A; Table E5). Assessment of the overall LM-SPM profile in uninfected BAL was proresolving, with higher overall levels of proresolving mediators when compared with classic proinflammatory eicosanoids (Figure 6B). However, this changes upon infection, with prostaglandins playing a dominant role at 168 hours when compared with SPMs.
Figure 6.

Altered LM profile in BALF during Infection. BALF was collected before and after S. pneumoniae inoculation at the indicated intervals (see Materials and Methods). LM profiles were investigated by targeted LM metabololipidomics. (A) Cumulative levels for D-series resolvins, protectins, and maresins; E-series resolvins; lipoxins; LTB4 metabolome; and prostaglandins. Results are mean ± SEM (n = 4 for control baboons; n = 6 for infected baboons for each time point). *Statistical significance versus Time 0. (B) Pie charts depicting the relative LM levels in BALF preinoculation (top left), 48 hours after inoculation (top right), and 168 hours after inoculation (bottom) at the indicated intervals. Results are representative of three or four baboons for each time point.
CO Regulates Plasma Lipid Mediator Profiles during Pneumonia
Inhaled CO has been shown to promote LM-SPM biosynthesis during murine peritonitis (21, 24). Therefore, we investigated whether inhaled CO regulates proresolving programs in baboons during S. pneumoniae pneumonia. Inhaled CO (200–300 ppm for 60–90 min) was administered to a subset of animals with pneumonia (n = 3) at 48 hours after inoculation. There were no differences in the pre- and post-CO WBC counts (17.9 ± 5 × 103 versus 18.2 ± 4.7 × 103 cells/μl), percent neutrophils on differential (90 ± 3 versus 88 ± 5%), or platelet count (333 ± 19 × 103 versus 323 ± 37 × 103 cells/μl).
Before CO exposure, we identified plasma mediators from the DHA, EPA, and AA bioactive metabolomes, including RvD1, RvD2, RvE1, RvE2, LXA4, LXB4, LTB4, and thromboxane (Tx)B2, using published criteria (26, 30). CO exposure significantly increased plasma lipoxins (78.5 ± 11.8 versus 53.6 ± 13.2 pg/ml) and E-series resolvins (27.6 ± 7.8 versus 18.4 ± 5.9 pg/ml) and reduced TxB2 (1.5 ± 0.2 versus 2.4 ± 0.1 pg/ml) compared with pre-CO exposure plasma levels (Figure 7; Table E6). Thus, these results suggest that low-dose inhaled CO regulates plasma LMs by increasing circulating SPM levels and decreasing proinflammatory mediators (Figure 7D). This is illustrated in Figure 7D, where there is an increase in the overall proresolving LM (highlighted in blue) contribution to the LM profile when compared with animals exposed to ambient air (right panel) that displayed a higher proinflammatory mediator contribution (highlighted in red; left panel).
Figure 7.
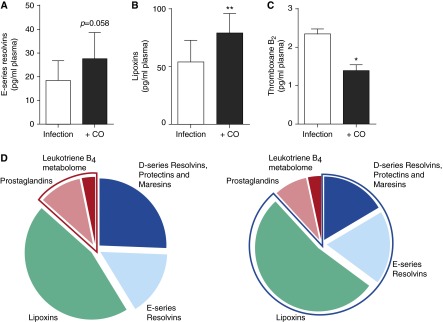
Inhaled CO elevates plasma SPMs during bacterial pneumonia. Baboons were inoculated with S. pneumoniae (8 × 108 to 2 × 109 CFU) and then exposed to inhaled CO (200–300 ppm for 60–90 min) 48 hours later. Plasma samples were collected 48 hours after infection (pre-CO) and immediately after CO exposure (∼50 h after infection). LMs were assessed using LM metabololipidomics after solid-phase extraction (see Materials and Methods). Plasma E-series resolvins (A), lipoxins (B), and thromboxane B2 (C). (D) Pie charts depicting the relative LM-SPM levels identified in plasma samples after infection (left panel) and after CO exposure (right panel). Highlighted areas display the proinflammatory (left panel) and proresolving (right panel) mediator contribution to the plasma LM-SPM profile. Results for A, B, and C are mean ± SEM (n = 3 animals per group). Results for D are representative of three animals per group. *P < 0.05 and **P < 0.01 versus the Pre-CO group.
Discussion
In the present report, we document the first identification of SPMs in baboons and LM-SPM signatures of plasma and peripheral blood leukocytes of baboons with S. pneumoniae pneumonia. We recently established a targeted LM metabololipidomics and functional approach for profiling active SPM pathways (26). Using synthetic and authentic standards, we can identify bioactive mediators and pathway markers with enhanced sensitivity to provide LM-SPM signatures that reflect the status of a particular tissue and/or isolated cell type(s) (26, 30). Here, compared with control leukocytes, STZ-stimulated leukocytes and leukocytes isolated from animals with S. pneumoniae pneumonia displayed a proinflammatory LM profile. Plasma SPM levels were also significantly reduced in baboons with S. pneumoniae pneumonia compared with values at baseline and in control animals. This was partially corrected by administering low-dose inhaled CO, which elevated plasma SPM levels.
Leukocyte-derived proresolving mediators have been identified at sites of inflammation in multiple species, including humans, mice, and trout (14, 30, 31). In response to local and systemic infections, leukocytes of nonhuman primates produce classic eicosanoids, including PGE2 and TxB2 (32, 33). Building on this work, we establish for the first time that baboon leukocytes also produce specialized proresolving mediators, including RvD1, RvD2, and RvE2. Additionally, leukocyte stimulation with STZ, a proinflammatory, yeast-derived antigen (30, 34), increased proinflammatory LM levels, including LTB4 and PGE2. Similar findings were obtained with leukocytes from animals with S. pneumoniae pneumonia. These results are consistent with those from studies with human peripheral blood neutrophils (30) and establish the LM profiles of baboon peripheral blood leukocytes in the absence and presence of further agonists.
We recently found that, during experimental pneumonia in baboons initiated by S. pneumoniae inoculation, there is a significant regulation of plasma cytokine levels (9). Using LM metabololipidomics in the present report, we identified mediators from the arachidonic bioactive metabolome, including the proinflammatory eicosanoids LTB4 and PGF2α. In plasma from these animals, we also identified mediators from the DHA and EPA bioactive metabolomes, including RvD1, RvD6, and RvE3. PLS-DA of the lipid mediator profiles identified in plasma from infected animals demonstrated that at 24 hours after inoculation these lipid mediator profiles clustered separately from those of control animals, suggesting that plasma LM signature profiles may provide an indicator of pneumonia. In addition, animals with pneumonia displayed reduced circulating SPMs, including RvD1, RvD2, and RvD5, at 48 and 168 hours when compared with control values. The reduction in plasma SPMs may reflect ongoing, dysregulated systemic inflammation (35–38). This is in line with findings made with mice and human primary cells where these mediators display potent protective actions in infections. In these settings, SPMs counterregulate proinflammatory mediator production, including cytokines (e.g., TNF-α and IL-6) and eicosanoids (e.g., LTB4 and TxB2), and promote the host response to expedite resolution of infections. SPMs also expedite the clearance of bacteria by promoting leukocyte phagocytosis and intraphagolysosomal reactive oxygen species production. By definition, SPMs stimulate macrophage phagocytosis of apoptotic cells and tissue debris, regulate neutrophil recruitment to the site, and promote macrophage phenotype switch to a resolution phenotype, lowering antibiotic requirements and restoring tissue homeostasis (18, 19, 37).
The biosynthesis of LMs involves a series of highly controlled enzymatic steps that dictate the stereochemistry, and therefore the potency, of each mediator (11, 26). Even subtle differences in double-bond geometry or hydroxyl group chirality can be significant (17). In the present study, we identified 17R-containing epimers of RvD1 and RvD3 and found these were also down-regulated during the course of pneumonia. In humans, in the presence of low-dose aspirin, acetylated COX-2 initiates the biosynthesis of aspirin-triggered, 17R-containing epimers of the resolvins and protectins. These 17R-containing epimers exhibit sustained proresolving actions because they are less susceptible to local inactivation (17). These 17R-containing epimers can also be produced via cytochrome p450 enzymes present in a number of cell types, including endothelial cells, leukocytes, and bacteria (15, 34); this is a likely source of the 17R-containing epimers in baboons because these animals did not receive aspirin. These results demonstrate that SPM biosynthesis is highly conserved (i.e., bacteria, mice, baboons, and humans). Moreover, the down-regulation of these potent mediators postinoculation suggests they may be additional indicators of pneumonia progression.
Studies across species have demonstrated the antiinflammatory actions of low-dose inhaled CO for ventilator- or LPS-induced lung inflammation and injury (39–42). In non-human primates with LPS-induced lung inflammation, low-dose CO reduces BALF TNF-α and neutrophil levels (25). In studies in mice and human cells, low-dose CO up-regulated SPM levels by increasing lipoxygenase expression, the initiating enzymes for SPM biosynthesis, and inhibiting enzyme(s) involved in the further conversion of SPMs. Low-dose CO displays leukocyte-directed actions, regulating leukocyte–platelet interactions via down-regulation of adhesion molecule expression, which leads to a reduction of proinflammatory cysteinyl-leukotrienes (43) that stimulate bronchoconstriction and airway edema (11). CO also stimulates macrophage phagocytosis and promotes resolution of acute inflammation (21). In the present report, we found that inhalation of low-dose CO (200–300 ppm for 60–90 min) significantly increased the circulating levels of lipoxins in S. pneumoniae–infected animals. This was coupled with a reduction in circulating TxB2 levels. Given the high binding affinity of CO for heme molecules, the reduction in TxB2 may be explained by the CO-mediated inhibition of the heme-containing enzyme Tx synthase, an effect previously shown to reduce platelet aggregation (44, 45) during acute inflammation (21).
These results are consistent with observations in mice demonstrating that low-dose inhaled CO increases LM-SPM levels via a HO-1–SPM circuit (21). Furthermore, recent studies demonstrate that the fine regulation of human peripheral blood LM-SPM levels may have significant functional consequences on bacterial clearance. In healthy volunteers, up-regulation of select SPMs in peripheral blood by omega-3 supplementation increased ex vivo bacterial phagocytosis by leukocytes (26). The functional impact of these changes in peripheral blood LM-SPM levels on leukocyte function and bacterial clearance remains to be established in baboons during S. pneumoniae infection. In this context, results with human whole blood (26) suggest that the increases in peripheral blood SPM levels found herein after CO administration may also lead to improved leukocyte responses in baboons after bacterial infection.
Low-dose CO may also engage hypoxia-dependent proresolving pathways via stabilization of hypoxia inducible factor-1α that in turn may regulate the expression of netrin-1 (46), an axonal guidance molecule that also displays potent host-protective actions and stimulates SPM production (47). We have recently found that low-dose CO displays additive actions with RvD1 in protection against neutrophil-mediated lung injury from ischemia reperfusion (43) and that inhaled RvD1 protects against cigarette smoke–mediated lung injury (48). Thus, these results indicate that inhaled low-dose CO and SPMs each hold potential as a novel therapeutic approach in the treatment of acute (e.g., ventilator-induced lung injury) as well as chronic lung inflammation, such as chronic obstructive pulmonary disease resulting from cigarette smoke. These will need to be addressed in more detail in future studies to establish clinical efficacy and dosing strategies.
In summation, we systematically identified baboon LMs and investigated the LM profiles of baboon leukocytes using targeted LM metabololipidomics. Our results demonstrate that these cells have proresolving LMs and classic eicosanoids. Furthermore, in vivo and in vitro challenge of these cells changed the LM-SPM profiles from proresolving to proinflammatory. We also report the plasma LM-SPM profiles before and after S. pneumoniae inoculation and found that SPM levels are reduced during pneumonia. In addition, we found that SPM levels could be partially restored by administering inhaled low-dose CO, which suggests it may exert protective actions in bacterial pneumonia. Taken together, these results indicate that resolution programs appear to become dysregulated during S. pneumoniae pneumonia in baboons, which is partially rescued by low-dose CO inhalation via activation of local SPM biosynthesis.
Acknowledgments
Acknowledgments
The authors thank Mary H. Small for expert assistance in manuscript preparation, Craig Marshall for assistance with the baboon studies, Olga Better for assistance with microbiology work, Ali Ulrich for assistance with leukocyte preparations, and Sonny Boso, Aaron Walker, and John Davies for assistance with CO exposures.
Footnotes
This work was supported by National Institutes of Health grants P01-HL108801 and P01-GM095467 and by the Gates Foundation Global Health Institute.
Author Contributions: J.D., R.A.C., M.S., and N.C. performed metabololipidomics and lipid mediator analysis. B.D.K., K.W.-W., and C.A.P. performed animal work and, along with L.E.F. and D.R.H., performed CO exposures. B.D.K. performed leukocyte isolations/stimulations. A.M.C. contributed to experimental design and manuscript preparation. All authors contributed to experimental design, data interpretation, and manuscript preparation. C.N.S. and C.A.P. conceived overall experimental design and research plan.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0299OC on January 8, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rudan I, Boschi-Pinto C, Biloglav Z, Mulholland K, Campbell H. Epidemiology and etiology of childhood pneumonia. Bull World Health Organ. 2008;86:408–416. doi: 10.2471/BLT.07.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heron M. Deaths: leading causes for 2010. National vital statistics reports: from the Centers for Disease Control and Prevention, National Center for Health Statistics. Natl Vital Stat Rep. 2013;62:1–96. [Google Scholar]
- 3.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 4.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE. Global burden of childhood pneumonia and diarrhoea. Lancet. 2013;381:1405–1416. doi: 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fullerton JN, O'Brien AJ, Gilroy DW. Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. 2014;35:12–21. doi: 10.1016/j.it.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 9.Kraft BD, Piantadosi CA, Benjamin AM, Lucas JE, Zaas AK, Betancourt-Quiroz M, Woods CW, Chang AL, Roggli VL, Marshall CD, et al. Development of a novel preclinical model of pneumococcal pneumonia in nonhuman primates. Am J Respir Cell Mol Biol. 2014;50:995–1004. doi: 10.1165/rcmb.2013-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flower RJ, Vane JR. Inhibition of prostaglandin biosynthesis. Biochem Pharmacol. 1974;23:1439–1450. doi: 10.1016/0006-2952(74)90381-5. [DOI] [PubMed] [Google Scholar]
- 11.Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. J Biol Chem. 2012;287:10070–10080. doi: 10.1074/jbc.X112.351437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, Petasis NA. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26:1755–1765. doi: 10.1096/fj.11-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canny G, Levy O, Furuta GT, Narravula-Alipati S, Sisson RB, Serhan CN, Colgan SP. Lipid mediator-induced expression of bactericidal/ permeability-increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci USA. 2002;99:3902–3907. doi: 10.1073/pnas.052533799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang N, Shinohara M, Dalli J, Mirakaj V, Kibi M, Choi AM, Serhan CN. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J Immunol. 2013;190:6378–6388. doi: 10.4049/jimmunol.1202969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53:1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2012;185:851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell LA, Channell MM, Royer CM, Ryter SW, Choi AM, McDonald JD. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L891–L897. doi: 10.1152/ajplung.00366.2009. [DOI] [PubMed] [Google Scholar]
- 26.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307:C39–C54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cevey-Macherel M, Galetto-Lacour A, Gervaix A, Siegrist CA, Bille J, Bescher-Ninet B, Kaiser L, Krahenbuhl JD, Gehri M. Etiology of community-acquired pneumonia in hospitalized children based on WHO clinical guidelines. Eur J Pediatr. 2009;168:1429–1436. doi: 10.1007/s00431-009-0943-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Said MA, Johnson HL, Nonyane BA, Deloria-Knoll M, O'Brien KL, Team AAPBS, Andreo F, Beovic B, Blanco S, Boersma WG, et al. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. PLoS One. 2013;8:e60273. doi: 10.1371/journal.pone.0060273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat Rev Mol Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 30.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:e60–e72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowley AF, Lloyd-Evans P, Barrow SE, Serhan CN. Lipoxin biosynthesis by trout macrophages involves the formation of epoxide intermediates. Biochemistry. 1994;33:856–863. doi: 10.1021/bi00170a002. [DOI] [PubMed] [Google Scholar]
- 32.Camporesi EM, Oda S, Fracica PJ, Griebel JA, Shook JE, Piantadosi CA, Watkins WD. Eicosanoids and the hemodynamic course of live Escherichia coli-induced sepsis in baboons. Circ Shock. 1989;29:229–244. [PubMed] [Google Scholar]
- 33.Ebersole JL, Steffen MJ, Holt SC, Kesavalu L, Chu L, Cappelli D. Systemic inflammatory responses in progressing periodontitis during pregnancy in a baboon model. Clin Exp Immunol. 2010;162:550–559. [Google Scholar]
- 34.Divanovic S, Dalli J, Jorge-Nebert LF, Flick LM, Galvez-Peralta M, Boespflug ND, Stankiewicz TE, Fitzgerald JM, Somarathna M, Karp CL, et al. Contributions of the three CYP1 monooxygenases to pro-inflammatory and inflammation-resolution lipid mediator pathways. J Immunol. 2013;191:3347–3357. doi: 10.4049/jimmunol.1300699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–475. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. 2014;76:467–492. doi: 10.1146/annurev-physiol-021113-170408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolinay T, Szilasi M, Liu M, Choi AM. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med. 2004;170:613–620. doi: 10.1164/rccm.200401-023OC. [DOI] [PubMed] [Google Scholar]
- 40.Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, Alber S, Kaynar AM, Schmidt R, Ryter SW, et al. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med. 2008;177:1223–1232. doi: 10.1164/rccm.200708-1265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu SH, Ma K, Xu B, Xu XR. Carbon monoxide inhalation protects lung from lipopolysaccharide-induced injury in rat. Sheng Li Xue Bao. 2006;58:483–489. [PubMed] [Google Scholar]
- 42.Mazzola S, Forni M, Albertini M, Bacci ML, Zannoni A, Gentilini F, Lavitrano M, Bach FH, Otterbein LE, Clement MG. Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J. 2005;19:2045–2047. doi: 10.1096/fj.05-3782fje. [DOI] [PubMed] [Google Scholar]
- 43.Shinohara M, Kibi M, Riley IR, Chiang N, Dalli J, Kraft BD, Piantadosi CA, Choi AM, Serhan CN. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am J Physiol Lung Cell Mol Physiol. 2014;307:L746–L757. doi: 10.1152/ajplung.00166.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brune B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol. 1987;32:497–504. [PubMed] [Google Scholar]
- 45.Mansouri A, Perry CA. Alteration of platelet aggregation by cigarette smoke and carbon monoxide. Thromb Haemost. 1982;48:286–288. [PubMed] [Google Scholar]
- 46.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 47.Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med. 2014;211:1037–1048. doi: 10.1084/jem.20132103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, Olsen KC, Pollock SJ, Serhan CN, Phipps RP, et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One. 2013;8:e58258. doi: 10.1371/journal.pone.0058258. [DOI] [PMC free article] [PubMed] [Google Scholar]