

Abstract
Resistin-like molecule α (RELMα) has mitogenic, angiogenic, vasoconstrictive, and chemokine-like properties and is highly relevant in lung pathology. Here, we used RELMα knockout (Retnla−/−) mice to investigate the role of RELMα in pulmonary vascular remodeling after intermittent ovalbumin (OVA) challenge. We compared saline- and OVA-exposed wild-type (WT) mice and found that OVA induced significant increases in right ventricular systolic pressure, cardiac hypertrophy, pulmonary vascular remodeling of intra-alveolar arteries, goblet cell hyperplasia in airway epithelium, and intensive lung inflammation, especially perivascular inflammation. Genetic ablation of Retnla prevented the OVA-induced increase in pulmonary pressure and cardiac hypertrophy seen in WT mice. Histological analysis showed that Retnla−/− mice exhibited less vessel muscularization, less perivascular inflammation, reduced medial thickness of intra-alveolar vessels, and fewer goblet cells in upper airway epithelium (250–600 μm) than did WT animals after OVA challenge. Gene expression profiles showed that genes associated with vascular remodeling, including those related to muscle protein, contractile fibers, and actin cytoskeleton, were expressed at a lower level in OVA-challenged Retnla−/− mice than in similarly treated WT mice. In addition, bronchoalveolar lavage from OVA-challenged Retnla−/− mice had lower levels of cytokines, such as IL-1β, -1 receptor antagonist, and -16, chemokine (C-X-C motif) ligand 1, -2, -9, -10, and -13, monocyte chemoattractant protein-1, macrophage colony-stimulating factor, TIMP metallopeptidase inhibitor-1, and triggering receptor expressed on myeloid cells-1, than did that from WT mice when analyzed by cytokine array dot blots. Retnla knockout inhibited the OVA-induced T helper 17 response but not the T helper 2 response. Altogether, our results suggest that RELMα is involved in immune response–induced pulmonary vascular remodeling and the associated increase in inflammation typically observed after OVA challenge.
Keywords: resistin-like molecule α, pulmonary hypertension, vascular remodeling, inflammation, T helper 17
Clinical Relevance
We have demonstrated that resistin-like α (RELMα) knockout mice inhibited pulmonary vascular remodeling and the increase in pulmonary pressure and cardiac hypertrophy in a model of allergen-induced pulmonary vascular remodeling. Further studies show that this prevention is associated with suppressed inflammation in the lung, involving a T helper 17 immune response. Our study reveals a novel function of RELMα on immune regulation in pulmonary hypertension and shows that it could be a novel therapeutic target for pulmonary hypertension.
Resistin and resistin-like molecules (RELMs) have been identified as a family of cysteine-rich small proteins that share no homology to any known hormone or cytokine (1–4). The RELM family has four murine isoforms (RELMα, RELMβ, resistin, and RELMγ) and two human isoforms (resistin and RELMβ), all of which have a structurally conserved 10-cysteine residue motif.
The RELM family members have distinctive tissue distribution, implying that they have distinct functions (2, 4–6). Among the four murine members, RELMα, which is also known as found in inflammatory zone 1 (FIZZ1) and hypoxia-induced mitogenic factor based on independent discoveries, has mitogenic, angiogenic, vasoconstrictive, and chemokine-like properties (7–9). It is also closely associated with the pathologic conditions of pulmonary hypertension (PH), pulmonary fibrosis, and allergic lung inflammation in mice (6, 10, 11). RELMα is highly up-regulated in the lung in response to chronic hypoxia (11), ovalbumin (OVA)-induced allergic inflammation (6), or bleomycin-induced lung injury (6, 10, 11). We have shown previously that RELMα is associated with vascular remodeling in the chronic hypoxia–induced model of PH (11), and that adeno-associated virus–RELMα gene transfer to overexpress RELMα in the rat lung recruits bone marrow–derived cells, causes an elevation of mean pulmonary artery pressure, and induces substantial vascular remodeling in the lung (12). Both human isoforms, resistin and RELMβ, have been implicated in human PH. We have found that increased resistin gene expression in circulating white blood cells of patients with scleroderma (SSc)-associated PH and idiopathic pulmonary arterial hypertension correlates with severity of PH (unpublished data). We also have found that human RELMβ is up-regulated in the lung hypertrophic vasculature, plexiform lesions, and myofibroblasts of patients with idiopathic pulmonary arterial hypertension and SSc-associated PH (13).
Besides hypoxia, T helper (Th) 2 stimuli can also induce RELMα. Allergic inflammation substantially up-regulates RELMα in the lung in an IL-4/IL-13 and Signal Transducer and Activator of Transcription (STAT)-6–dependent manner (14, 15). In a recent model of Th2 immune response, prolonged and intermittent challenge with allergen via the airway induced pulmonary artery remodeling, which was associated with increased RELMα expression (16, 17), although PH did not develop. Given the role of RELMα in lung inflammation, the vascular remodeling associated with RELMα expression after chronic hypoxia and pulmonary adeno-associated virus–RELMα gene transfer, and recent reports of Th2-induced pulmonary vascular muscularization, ample evidence suggests that RELMα is involved in the development of PH (11, 12, 17). However, it is still unknown whether RELMα has a specific role in Th2-induced pulmonary vascular muscularization, either as a cause or consequence of the condition. Thus, in this study, we challenged RELMα knockout (KO) and wild-type (WT) mice with OVA and evaluated the lung inflammation profile, Th1/Th2/Th17 inflammation, and vascular remodeling to ascertain the role of RELMα in this model of pulmonary vascular remodeling secondary to chronic immune activation.
Materials and Methods
A detailed description of methods is provided in the online supplement.
Experimental Animals
Adult, male, 6- to 8-week-old WT BALB/c mice (Hilltop Lab Animals, Scottsdale, PA) were used for all experiments. Mice deficient in RELMα (Retnla−/−) on BALB/c background (>10 generations) were generated by VelociGene technology (18). Animal housing and experimental protocols were approved by the Animal Care and Use Committee of Johns Hopkins University.
OVA-Induced Pulmonary Vascular Remodeling
Animals were primed and challenged with OVA according to the method described previously (16, 17).
Hemodynamic Measurement
Right ventricular systolic pressure (RVSP) was measured by a method similar to that described previously (19), and data were collected with the AD Instruments Powerlab 8/35 (AD Instruments, Colorado Springs, CO) and Millar MPVS Ultra (Millar Instruments, Houston, TX).
Measurement of Fulton Index
After determining RVSP, we flushed the mouse heart and lungs with cold PBS. The heart was then dissected free of all major vessels, separated into right ventricle (RV) and left ventricle (LV) plus septum (S), and weighed. The Fulton index (RV/LV + S) was determined for each animal.
Collection of Bronchoalveolar Lavage and Preparation of Lung Tissue Homogenates
We collected the bronchoalveolar lavage (BAL) in three lung flushes by delivering 800 μl of sterile saline through a catheter inserted into the trachea. Cells in BAL flushes for each animal were collected and used for cell quantification and differentiation. After BAL fluid collection, the lung was removed and used for preparation of lung tissue block, RNA extraction, and homogenates.
RNA Isolation and Real-Time RT-PCR
RNA was extracted and cDNA obtained using commercial kits according to the manufacturer’s protocol (see the online supplement for details). RT-PCR was performed on an ABI Prism 7300 Sequence Detection System (Life Technologies, Foster City, CA).
Assessment of Pulmonary Vascular Remodeling
We assessed pulmonary vascular remodeling as previously described (12, 16).
Airway Goblet Cell Staining and Quantification
To detect mucus-producing goblet cells, we stained mouse lung sections with periodic acid–Schiff (PAS). Images were captured by an Olympus-BHS microscope under 20× objective (Olympus, Tokyo, Japan). To minimize the error that might come from tangential sectioning and bronchial branching, we considered only those bronchi with a circular appearance for counting the number of PAS-positive goblet cells in the epithelium. PAS-positive goblet cells were counted and expressed as a percentage of the total number of cells in the epithelium per airway. We assessed at least three lung sections per mouse.
Histology and Perivascular Inflammation
Lung sections were stained with hematoxylin and eosin before perivascular inflammation was evaluated on peribronchial and intra-alveolar vessels. A treatment-blind observer graded the degree of perivascular inflammation on a scale of 0–4 according to the following criteria (20): 0, no cells; 1, few isolated cells; 2, a ring of inflammatory cells one cell deep; 3, a ring of inflammatory cells two to four cells deep; and 4, a ring of inflammatory cells more than four cells deep. Three to four lung sections per mouse were graded, and perivascular inflammation was expressed as the average of the inflammation scores.
Western Blotting
We loaded 20 μg of lung lysate or 15 μl of BAL per animal onto 4–20% gradient SDS-polyacrylamide gels (Bio-Rad, Hercules, CA). After separation, the proteins were transferred to nitrocellulose membranes. Membranes were immunoblotted with anti-RELMα, anti-RELMβ, or anti-RELMγ antibody (1:1,000) and probed with horseradish peroxidase–conjugated goat anti-rabbit or sheep anti-goat (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) secondary antibody. The blotting was then visualized by chemiluminescence (ECL; Amersham Pharmacia Biotech, Arlington Heights, IL).
Antibodies
Goat anti-mouse RELMα/hypoxia-induced mitogenic factor/FIZZ1 antibody was purchased from R&D Systems (Minneapolis, MN). Rabbit anti-mouse RELMβ antibody was obtained from Thermo Scientific (Rockford, IL). Rabbit anti-mouse RELMγ/FIZZ4 antibody was prepared by using a synthesized peptide (EKKVKELLANRDDC) antigen specific for mouse RELMγ (AbMax Biotech, Beijing, China). The titers of the antibodies were determined by ELISA (final titer was 1:100,000), and the specificity of the antibodies was examined by lung tissue homogenate and recombinant RELMγ/FIZZ4 protein (21).
Microarray Analysis
RNA samples were labeled according to the chip manufacturer’s recommended protocols. Microarray was performed and quantitated on a BeadStation 500GX Genetic Analysis Systems scanner (Illumina Inc., San Diego, CA).
Mouse Cytokine Array
To detect cytokines in mouse BAL, we used a proteome profiler array kit (Mouse Cytokine Array Panel A; R&D Systems) according to the manufacturer’s instructions with a minor modification. This array detects 40 cytokines. After ECL reaction and exposure to HyBlot CL film (Denville Scientific, Metuchen, NJ), the dot blots generated were analyzed by ImageJ 1.47v (available at rsb.info.nih.gov/ij/). Cytokine changes of ±1.5-fold or greater in at least two animals per group were considered significant. Three animals from each group were tested.
Statistical Analysis
Experimental results are expressed as mean (±SEM). A Student’s t test was used to determine the statistical significance between groups. A P value of less than 0.05 was considered statistically significant.
Results
OVA Challenge Induces the Expression of Murine RELM Isoforms in the Lung
The four isoforms of murine RELM have high similarity in their transcriptional and amino acid sequences. RELMβ, RELMα, and RELMγ genes are aligned sequentially on chromosome 16B5 as Retnlb, Retnla, and Retnlg, whereas resistin (Retn) is located separately on chromosome 8A1. To test the possible complementary expression of other isoforms after Retnla KO, we performed PCR and Western blotting. PCR analysis of lung cDNA with specific primers for Retnla, Retnlb, Retn, and Retnlg showed that the expression of RELMα and RELMγ was up-regulated after OVA challenge, but not after saline challenge (Figure 1A). No resistin was detected in either saline-challenged or OVA-challenged lung (Figure 1A). We also found de novo expression of RELMβ in mouse lung after animals were challenged with OVA, whereas RELMβ was undetectable in saline controls (Figure 1A). To compare the changes of RELM gene expression in WT and KO mice after OVA challenge, we performed real-time PCR to quantify the expression levels of RELM genes in the lung. We found that the expression of Retnla, Retnlb, and Retnlg genes was significantly increased in OVA-challenged WT mice (Figure 1B). In KO mice, OVA challenge significantly induced the expression of Retnlb only; Retnlg expression was increased, but the difference was not significant (Figure 1B). Gene array analysis of lung messenger RNA was consistent with individual PCR results for expression of the four isoforms in WT mice (Table 1). In parallel to the gene expression, Western blotting revealed that OVA challenge caused an increase in the expression of secretory RELMα, RELMβ, and RELMγ in BAL of WT animals (Figure 1C). The expression of RELMβ and RELMγ in BAL from Retnla−/− mice also increased after OVA challenge, but the levels were lower than those of WT mice. Retnla KO did not induce a compensatory increase in the expression of other RELM isoforms in the lung in this model, as has been shown by others (22).
Figure 1.
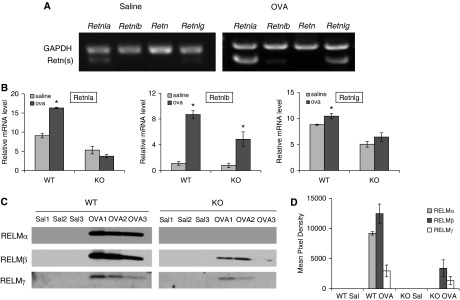
Expression of murine resistin (Retn)-like molecules (RELMs) in the lung after ovalbumin (OVA) challenge. (A) PCR analysis of wild-type (WT) mice showed that RELMα (Retnla) and RELMγ (Retnlg) had low baseline expression in control mice; RELMα, RELMβ (Retnlb), and RELMγ were up-regulated in lung after mice were challenged with OVA. Retn was not detected in lung from saline- or OVA-challenged mice. (B) Real-time PCR showed that the expression of RELMα and RELMβ genes was significantly increased in OVA-challenged WT and Retnla knockout (KO) mice (OVA versus saline, *P < 0.05). RELMγ expression was significantly increased by OVA challenge in WT mice but not in KO mice. (C) Western blotting detected the de novo expression of RELMα, RELMβ, and RELMγ in bronchoalveolar lavage (BAL) from the OVA group. (D) Densitometric quantification of RELM protein levels in BAL shown on Western blot (C) (n = 3). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; mRNA, messenger RNA; Sal, saline. Data are presented as mean (±SEM).
Table 1.
Fold Changes of RELM Family Genes by Gene Array Analysis in Wild-Type and Resistin-Like Molecule α Knockout Mice after OVA Treatment
| Gene | WT OVA/Saline |
Retnla−/− OVA/Saline |
||
|---|---|---|---|---|
| P Value | Fold Change | P Value | Fold Change | |
| RETNLA | 0.0001 | 51.51 | 1 | −1.37 |
| RETNLG | 0.0004 | 6.45 | 0.1124 | 2.72 |
| RETNLB | 0.0425 | 3.82 | 1 | 1.42 |
| RETN | 1 | 1.02 | 1 | −1.07 |
Definition of abbreviations: OVA, ovalbumin; RELM, Retn-like molecule; Retnla−/−, resistin-like molecule α knockout; WT, wild type.
Lung cDNA from each group (n = 5) was run on microarray, and the expression of individual resistin-like molecule genes was compared between OVA-treated and saline-treated groups. P value, OVA versus saline by Welch t test.
RELMα KO Prevents an Increase in Pulmonary Pressure and Cardiac Hypertrophy Induced by OVA Challenge
After 3 weeks of intermittent OVA challenge, RVSP, an indicator of pulmonary pressure, increased in WT mice compared with that in saline controls (OVA, 28.60 ± 1.69 mm Hg versus Sal, 23.87 ± 0.23 mm Hg; P = 0.05; Figure 2A). However, Retnla KO abolished the effect of OVA on RV pressure (OVA, 23.20 ± 1.15 mm Hg versus Sal, 24.51 ± 1.25 mm Hg). Accompanying the change in RVSP, the Fulton index, another indicator of PH and cardiac hypertrophy, was significantly increased in WT animals after OVA challenge (OVA, 0.348 ± 0.016 versus Sal, 0.284 ± 0.012; P < 0.05; Figure 2B). The Fulton index did not change in Retnla−/− mice (OVA, 0.292 ± 0.016 versus Sal, 0.315 ± 0.010). Taken together, Retnla KO prevented the increase in pulmonary pressure and cardiac hypertrophy.
Figure 2.

RELMα KO prevents OVA challenge–induced increases in pulmonary pressure and cardiac hypertrophy. (A) Right ventricular systolic pressure (RVSP), which was measured 1 day after the last OVA challenge, increased significantly in WT mice after 3 weeks of intermittent OVA challenge (OVA versus saline [Sal], P = 0.05). Retnla KO abolished the effect of OVA on pulmonary artery pressure. (B) Fulton index was determined as the ratio of right ventricle (RV) weight to left ventricle plus septum (LV + S) weight. The Fulton index increased significantly in WT animals (OVA versus Sal, *P < 0.05) but not in Retnla−/− animals after OVA challenge. Data are presented as mean (±SEM).
RELMα KO Inhibits Pulmonary Vascular Remodeling Induced by OVA Challenge
Muscularization of small vessels in the lung is a characteristic feature of vascular remodeling that results in increased pulmonary pressure and PH. Intra-alveolar small vessels (<80 μM), which contribute significantly to pulmonary vascular resistance, are normally nonmuscularized (NM), consisting primarily of a single layer of endothelial cells. Examination of the lung sections from OVA-challenged WT mice revealed muscularization of the small pulmonary vessels, including increases in partially muscularized (PM) and fully muscularized (FM) vessels (PM, 8.23 ± 2.26% versus 30.62 ± 4.27%; P < 0.05; FM, 0.84 ± 0.39% versus 54.18 ± 3.37%; P < 0.05), and a decrease in the percentage of NM vessels (90.93 ± 2.44% versus 15.20 ± 1.51%; P < 0.05), compared with those in saline-treated control animals (Figures 3A and 3B). This result is consistent with those of our previous studies in which up-regulation of RELMα was associated with chronic hypoxia–induced pulmonary vascular remodeling. To delineate the role of RELMα in this vascular remodeling, we examined the small vessels in Retnla−/− mice after OVA challenge. The results were similar to those of WT mice. After OVA challenge, Retnla−/− mice exhibited fewer NM small vessels and more PM and FM vessels (NM, 85.52 ± 2.81% versus 27.82 ± 5.28%; P < 0.05; PM, 12.70 ± 2.39% versus 35.00 ± 3.67%; P < 0.05; FM, 1.78 ± 0.61% versus 37.19 ± 5.30%; P < 0.05; Figures 3A and 3B) than did saline-treated KO mice. However, KO mice had less vascular remodeling than did the WT mice, as indicated by more NM and fewer FM small vessels after OVA challenge (P < 0.05).
Figure 3.
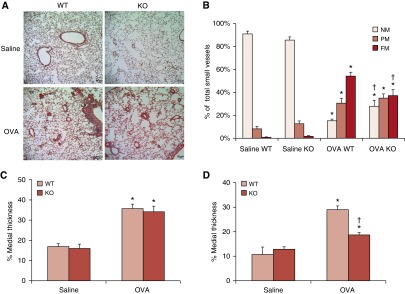
RELMα KO inhibits pulmonary vascular remodeling induced by OVA challenge. Lung sections were dual stained for von Willebrand factor (endothelium) and α-smooth muscle actin (vascular smooth muscle) to define nonmuscularized (NM), partially muscularized (PM), and fully muscularized (FM) intra-alveolar small vessels. (A) PM and FM small vessels were sparse in saline-treated WT and Retnla−/− mice. OVA challenge induced a significant increase in vessel muscularization, visible as an increase in red staining for α-smooth muscle actin. (B) Percent muscularization of small pulmonary vessels in mouse lung. OVA challenge induced a significant increase in the percentage of PM and FM small vessels and a decrease in the percentage of NM small vessels in the lungs of WT and KO mice (OVA versus saline, *P < 0.05). However, RELMα deficiency significantly attenuated OVA-induced muscularization in small artery vessels compared with that in WT mice (KO OVA versus WT OVA, †P < 0.05). (C) The percent medial thickness (%MT) of peribronchial vessels was significantly increased in WT and KO mice after OVA challenge (OVA versus saline, *P < 0.05). (D) The %MT of intra-alveolar vessels was significantly increased in both WT and KO mice after OVA challenge (OVA versus saline, *P < 0.05), but the degree of increase was significantly less in KO mice than in WT mice (†P < 0.05; n = 5 for each group). Data are presented as mean (±SEM).
OVA challenge also increased the thickness of the vascular media, another indicator for vascular remodeling (Figures 3C and 3D). The percent medial thickness (%MT) of peribronchial vessels significantly increased in both WT and Retnla−/− mice compared with that in the corresponding saline controls (Figure 3C). OVA challenge also increased %MT of intra-alveolar vessels in both WT and Retnla−/− mice. The %MT for intra-alveolar vessels increased in WT mice from 10.32 (±2.39)% to 29.26 (±0.75)% (P < 0.05) and in Retnla−/− mice from 13.94 (±0.75)% to 17.92 (±0.75)% (P < 0.05). Compared with WT mice, Retnla−/− mice had a smaller increase in medial thickness of intra-alveolar vessels (P < 0.05; Figure 3D), indicating that Retnla KO protects against remodeling of small, intra-alveolar vessels in the lung after OVA challenge.
We used a gene array to characterize the gene expression profiles after OVA challenge in WT and KO mice. Heat map (see Figure E1 in the online supplement) analysis revealed the pattern of gene expression in KO mice to be quite similar to that in WT mice, with 641 genes in common that were up-regulated and 413 genes in common that were down-regulated after OVA challenge. Tables E1 and E2 show results of the functional analysis of genes similarly affected by OVA in WT and KO mice, as determined by Database for Annotation, Visualization, and Integrated Discovery (DAVID; National Institute of Allergy and Infectious Diseases, National Institutes of Health) (23, 24). In addition, 109 genes were expressed at a lower level in Retnla−/− mice after OVA challenge compared with levels in OVA-challenged WT mice. These genes are involved in muscle protein, contractile fibers, and actin cytoskeleton (Table 2), suggesting that RELMα KO inhibits vascular remodeling via down-regulation of muscle- and structure-related genes.
Table 2.
Functional Annotation of Down-Regulated Genes after OVA Challenge in Resistin-Like Molecule α Knockout versus Wild-Type Mice
| Cellular Function | Genes (n) | Gene Names | Benjamini |
|---|---|---|---|
| Muscle protein | 7 | MYL7, MYL4, TNNC1, MYL3, MYL1, MYH6, MB | 3.78E−05 |
| Contractile fiber part | 7 | ATP2A1, HSPB1, ACTN2, MYH6, MYOZ2, TTN, CSRP3 | 2.20E−04 |
| Actin cytoskeleton | 9 | PKNOX2, MYL7, MYL4, MYL3, MYL1, ACTN2, MYH6, MYOZ2, TTN | 1.32E−04 |
Definition of abbreviation: OVA, ovalbumin.
RELMα KO Attenuates OVA-Induced Inflammation in the Lung
Repeated OVA challenge induced intensive inflammation in the lung that was particularly localized around vessels in both WT and KO mice. Inflammatory cells were more numerous near peribronchial and intra-alveolar vessels in the lungs from OVA-challenged mice than in those from saline-treated control mice (Figure 4A). OVA challenge induced significant peribronchial vascular inflammation compared with saline treatment (WT: OVA, 3.73 ± 0.03 versus saline, 0.87 ± 0.11; P < 0.05; KO: OVA, 3.53 ± 0.08 versus saline 0.63 ± 0.05; P < 0.05), but there was no significant difference between OVA-challenged WT and OVA-challenged KO mice (Figure 4A). Although OVA challenge also induced significant perivascular inflammation around intra-alveolar vessels (Figure 4A), we observed fewer inflammatory cells near these parenchymal vessels in KO mice than in WT mice (WT: OVA, 3.23 ± 0.10 versus saline, 0.88 ± 0.11; P < 0.05; KO: OVA, 2.40 ± 0.16 versus saline, 0.76 ± 0.07; P < 0.05; WT versus KO, P < 0.05).
Figure 4.

RELMα KO attenuates OVA-induced inflammation in the lung. (A) Hematoxylin and eosin staining revealed extensive perivascular inflammation in OVA-challenged lung from both WT and Retnla KO mice. Quantification and statistical analysis revealed significant increases in perivascular inflammation around peribronchial and intra-alveolar vessels after OVA challenge in WT and KO mice compared with that in saline-treated controls (OVA versus saline, *P < 0.05). There were fewer inflammatory cells around the intra-alveolar vessels in KO mice than in WT mice (KO OVA versus WT OVA, †P < 0.05). (B) Cell number and differentiation in BAL after OVA challenge. OVA challenge significantly increased immune cell recruitment in the lung. Total cell numbers were quantified by cell counter after BAL was collected and centrifuged. Percentage of lymphocytes, neutrophils, and eosinophils in BAL from WT and Retnla KO mice after OVA challenge were quantified after Diff-quick staining. Compared with WT OVA-challenged mice, KO OVA-challenged mice had fewer neutrophils and more eosinophils (*P < 0.05). (C) OVA challenge induced goblet cell hyperplasia in the airway, indicated by periodic acid–Schiff (PAS) staining; PAS-positive cells were rarely seen in saline-treated mice. Pearson correlation test showed a positive correlation between the percentage of PAS-positive goblet cells in epithelium of OVA-challenged WT and KO mice and bronchial diameter. Statistical analysis showed that the number of goblet cells in airway epithelium was lower in KO mice than in WT mice only in larger bronchi (diameter, 250–600 μm; *P < 0.05). The number of goblet cells did not differ significantly in bronchioles of 120–250 μm in diameter. Data are presented as mean (±SEM).
Total cell counts in BAL significantly increased in mice subjected to OVA challenge (Figure 4B, P < 0.05) and did not differ significantly between WT OVA and KO OVA groups. Differential cell counts in BAL of the saline control mice revealed a predominance of macrophages in both groups, without any significant difference between WT and Retnla−/− mice (data not shown). After OVA challenge, we observed a significant increase in lymphocytes, neutrophils, and eosinophils in mice of both genotypes (Figure 4B). Compared with WT OVA-challenged mice, KO OVA-challenged mice had fewer neutrophils and more eosinophils (Figure 4B, P < 0.05). These findings indicate that mice deficient in RELMα remain able to recruit significant numbers of inflammatory cells into the lung after OVA challenge, albeit in a distinctly different pattern than that in WT mice.
Pulmonary inflammation induced by allergic challenge is also associated with increased numbers of mucin-positive goblet cells, which we identified by PAS staining. Although few PAS-positive cells were present in saline-treated WT or KO mice, a considerable number of PAS-positive goblet cells were observed in mice of both genotypes after OVA challenge (Figure 4C). A plot of goblet cell percentage versus bronchial diameter showed that goblet cell numbers increased with increasing airway size (Figure 4C), with a positive association by Pearson correlation test. According to the 95% confidence interval from the correlation plot, we divided bronchi into two groups according to size for statistical analysis (Figure E2). The regression line fell close to an airway diameter of 250 μm. We found that KO mice had fewer goblet cells than did WT mice in epithelium from bronchi with diameters of 250–600 μm (WT, 75.07 ± 7.46% versus KO, 48.33 ± 8.80%) after OVA challenge; however, there was no significant difference in % PAS-positive cells in bronchioles of diameters of 120–250 μm (WT, 14.34 ± 4.00% versus KO, 10.97 ± 3.76%; Figure 4C). Retnla KO partially prevented the goblet cell differentiation in the upper bronchiole after allergen challenge.
RELMα KO Inhibits the Expression and Secretion of Cytokines Related to Inflammatory Response
Proteome profiler arrays were used to screen for cytokines that might contribute to the lung inflammatory response after OVA challenge. Figure 5A shows representative dot blots of three cytokine array experiments. We found that the protein levels of IL-1β, IL-1 receptor antagonist (ra), IL-16, IL-17, chemokine (C-X-C motif) ligand (CXCL) 1, CXCL2, CXCL9, CXCL10, CXCL13, monocyte chemoattractant protein-1 (MCP-1)/CCL2, macrophage colony-stimulating factor (M-CSF), TIMP metallopeptidase inhibitor-1 (TIMP-1), and triggering receptor expressed on myeloid cells-1 (TREM-1) increased in BAL fluid of WT mice after OVA challenge. Similarly, OVA challenge to Retnla−/− mice increased BAL cytokine levels of IL-1ra, IL-16, CXCL1, CXCL10, M-CSF, TIMP-1, and TREM-1, but to a lesser extent (at least 1.5-fold lower than in WT OVA-challenged mice; Figure 5B). Furthermore, cytokines IL-1β, IL-17, MCP-1/CCL2, CXCL9, and CXCL13 did not change after OVA challenge in Retnla−/− mice (Figure 5C). IL-13 was the only cytokine that greatly increased in BAL from OVA-challenged Retnla−/− mice (versus saline control), but not in that from OVA-challenged WT mice (Figure 5D). For a guide to reference individual cytokines in the dot blots, see Table E3.
Figure 5.

Retnla KO inhibits the expression and secretion of cytokines related to inflammatory response. (A) Cytokines in BAL were analyzed by the proteome profiler array. Dot blots represent 200 μL of BAL from each of three mice (per group) tested separately. (B) Levels of IL-1 receptor antagonist (IL-1ra), IL-16, chemokine (C-X-C motif) ligand (CXCL) 2, CXCL1, CXCL10, macrophage colony-stimulating factor (M-CSF), TIMP metallopeptidase inhibitor-1 (TIMP-1), and triggering receptor expressed on myeloid cells-1 (TREM-1) increased in BAL after OVA challenge in both WT and KO mice. However, levels were lower in KO mice than in WT mice. (C) IL-1β, IL-17, CXCL9, CCL2, and CXCL13 increased in BAL from WT mice after OVA challenge but were not significantly increased in BAL from Retnla KO mice. (D) IL-13 (label 5 in A) was the only protein that was elevated to a greater extent in BAL from OVA-challenged Retnla−/− mice than in that from OVA-challenged WT mice.
After analyzing the expression of cytokines on gene array, we found that a series of ILs and chemokines related to inflammatory response were up-regulated in OVA-challenged WT mice, but were expressed at relatively low levels in Retnla−/− mice (Table E4). Of these genes, the changes in IL-1ra, CXCL1, CXCL9, and TIMP1 were consistent with the changes detected by cytokine protein array. The findings from cytokine protein array and gene array indicate that Retnla KO suppresses the expression and secretion of cytokines related to inflammatory response.
Discussion
RELMα is a Th2-inducible molecule, the expression of which is dependent on IL-4, IL-13, and STAT-6 pathways (14, 15, 22, 25). In this study, we investigated the role of RELMα in a model of Th2 immune response–induced pulmonary artery remodeling, and found that RELMα markedly contributes to the pulmonary vascular remodeling process. Our data show that lack of RELMα prevented the elevation of RVSP, cardiac hypertrophy, and vascular remodeling, suppressed perivascular inflammation, and resulted in diminished secretion and/or expression of inflammatory cytokines.
The finding that RELMα is associated with pulmonary vascular remodeling is consistent with results from our previous studies. In models of chronic hypoxia–induced PH and RELMα overexpression, our group discovered that RELMα participates in the development of PH and vascular remodeling (11, 12). In addition to preventing vascular remodeling by inhibiting neomuscularization and thickening of small pulmonary arterioles (Figure 3), RELMα deficiency diminished the elevation of pulmonary artery pressure and right ventricular hypertrophy (Figure 2). These results differ slightly from those of our earlier study in which we compared pulmonary vascular remodeling induced by chronic hypoxia and by intermittent OVA challenge. In that study, the OVA model induced only medial thickening of peribronchial vessels, but not vascular remodeling of small, intra-alveolar vessels or cardiac hypertrophy (16).
Mouse strain is a factor that should be considered in an allergic study. In a study comparing allergic lung disease in three mouse strains, Zhu and Gilmour (26) found that FVB/NJ and BALB/cJ mice are higher Th2 responders than are C57BL/6J mice. In a study by Daley and colleagues (17), C57BL/6 mice developed severe pulmonary vascular remodeling in a Th2 immune response, but exhibited no increase in RVSP or RV hypertrophy, consistent with our previous study. The difference in genetic background of these mouse strains may account for their failure to show elevations in RVSP or develop intra-alveolar vascular remodeling; it might also explain discrepancies with other studies of pulmonary allergen challenge in Retnla−/− mice (16, 17).
Chronic allergic inflammation causes vascular remodeling and PH. In a model of home dust mite–induced pulmonary allergy, mice developed pulmonary vascular remodeling after exposure for 7 weeks, and elevated RVSP at 20 weeks (27). We believe that RELMα is a chemokine with proinflammatory properties, the inflammatory effects of which on small, intra-alveolar vessels cause pulmonary vascular remodeling that results in the elevation of pulmonary artery pressure and right heart remodeling. In our study, OVA challenge of WT mice induced intensive inflammation in the lung, and large numbers of inflammatory cells were visible around vessels. Although inflammatory cell recruitment around peribronchial vessels was pronounced in both Retnla−/− and WT mice, Retnla−/− mice showed less inflammation in the smaller, intra-alveolar vessels (Figure 4A). This difference might account for the reduction in vascular remodeling of small, intra-alveolar vessels, but not peribronchial vessels, in these KO mice (Figure 3).
The proinflammatory role of RELMα was also shown in a model of Citrobacter-induced colitis, in which Citrobacter exposure induced RELMα expression in intestinal epithelial cells, infiltrating macrophages, and eosinophils of the infected colon (28). In that study, Citrobacter-infected RELMα−/− mice exhibited less infection-induced intestinal inflammation than did WT mice, as evidenced by decreased leukocyte recruitment to the colon and reduced immune cell activation. In our study, inflammatory cell recruitment significantly increased in the lung after OVA challenge in both WT and Retnla−/− mice; however, the absence of RELMα resulted in recruitment of more macrophages and eosinophils, and significantly fewer neutrophils (Figure 4B). We observed this preference for leukocyte recruitment in a previous study that used a mouse dorsal air pouch model of inflammation. In that study, monocytes and neutrophils were recruited to the air pouch after 4 hours of induction with recombinant RELMα (unpublished data). The chemotactic effect of RELMα extends beyond these examples, as we have demonstrated that RELMα can stimulate the migration of bone marrow–derived cells in vitro (7). It was also a strong attractant for bone marrow–derived cells in a bone marrow transplant model in which overexpression of RELMα induced recruitment of previously transplanted cells to the pulmonary vasculature (13).
RELMα appeared to regulate inflammation either directly or indirectly by regulating inflammatory cytokines via distinct pathways. Gene array analysis of cytokine expression (Table E4) and dot blots of cytokine arrays for cytokines secreted into BAL (Figure 5) revealed that lack of RELMα markedly suppressed a series of inflammatory cytokines and chemokines (e.g., ILs, CCL, and CXCL) in the lung. Of the cytokines up-regulated by OVA challenge, many have chemotactic activity to recruit cells to the site of inflammation. For example, CXCL1 has chemotactic activity for neutrophils, and CXCL2 activates neutrophils and is chemotactic for polymorphonuclear leukocytes. CXCL9 is a T cell chemoattractant, whereas CXCL13 is selectively chemotactic for B cells. CXCL10 and MCP-1/CCL2 recruit monocytes, memory T cells, and dendritic cells to the sites of inflammation. In addition to attracting activated T cells, IL-16 can recruit and activate many other cells that express the CD4 molecule, including monocytes, eosinophils, and dendritic cells (29). M-CSF, which influences hematopoietic stem cell differentiation and functions at multiple levels within the innate and adaptive immune systems, regulates the migration, proliferation, function, and survival of macrophages and monocytes (30). Working coordinately, the cytokines affected in this study direct leukocyte recruitment and migration during the immune response. The result is an increase in the immune cell population of the lung after OVA challenge. Compared with OVA-challenged WT mice, OVA-challenged Retnla−/− mice secreted low levels of such chemotactic cytokines. The lower levels might account for the predominance of neutrophils in WT lung and eosinophils in Retnla−/− lung after OVA challenge.
Neither gene nor cytokine array revealed changes in Th1 cytokines, such as IFN-γ, TNF-α, IL-2, IL-6, or IL-12. These results imply that Th1 response may not contribute to the inflammatory response of OVA-induced vascular remodeling. Recent studies on helminth-induced Th2-type immunity showed that RELMα acts as a negative immunoregulator by suppressing Th2 immunity, as pulmonary granuloma formation and inflammation induced by Schistosoma mansoni eggs were exacerbated in Retnla−/− mice (22). Similar results were reported by Nair and colleagues (25). Our results from gene array and cytokine array showed that Th2 cytokines IL-4, IL-5, and IL-10 did not change in Retnla−/− mice after allergen induction. However, this result does not indicate a lower level of Th2 response. Instead, we found that IL-13, another Th2 cytokine, was up-regulated in OVA-challenged Retnla−/− mice (Figure 5). The increase in IL-13 expression with RELMα depletion is consistent with studies from other investigators (31, 32). In a footpad inflammatory model induced by Nocardia cyriacigeorgica (32), IL-13 expression was elevated in the chronic inflammatory phase and inversely correlated with RELMα/FIZZ1 expression. Munitz and colleagues (31) also found up-regulation of IL-13 in BAL during OVA-induced acute lung inflammation. A recent study with IL-13 transgenic mice showed that IL-13 overexpression can induce PH and vascular remodeling via arginase 2 and nitric oxide synthase 3 (33). Our current data do not support a role for IL-13 in inducing pulmonary vascular remodeling, and IL-13 is not likely vital to eosinophil recruitment. However, we provide additional evidence that the Th2 response can remain robust after OVA challenge, even when RELMα is deficient, as also reported by Munitz and colleagues (31). They found no difference in inflammatory cell recruitment in Retnla−/− mice after IL-13 challenge compared with that in WT mice. Although eosinophils are necessary for pulmonary vascular remodeling in adiponectin-deficient mice after OVA challenge (34), eosinophils do not contribute to pulmonary vascular remodeling in RELMα-deficient mice. Thus, our data support RELMα regulation of the Th2 response, but the Th2 immune response might not be responsible for pulmonary vascular remodeling in this model.
Th2 cells and Th2-related cytokines generally are believed to play a central role in the pathophysiology of asthma. Recent research on Th17 cells suggests that asthma is not exclusively driven by Th2 cytokines (35, 36). IL-17 expression was elevated in the inflamed lung of mice sensitized with OVA, and was correlated with enhanced neutrophil recruitment in the airway (37). IL-17F is also chemotactic for neutrophils, and increases the expression of genes for CXC and CCL chemokines and inflammatory cytokines in the lung (38). The Citrobacter-infected colitis model showed that the IL-23/Th17 axis is a critical mediator of RELMα-induced inflammation (28). In that model, infected Retnla−/− mice exhibited reduced intestinal inflammation, less recruitment of neutrophils to the colon, and fewer Th17 cells than did WT mice. IL-17A−/− mice were protected from RELMα-induced intestinal inflammation. In our study, IL-17 expression and secretion in BAL was significantly increased after OVA challenge of WT mice, but not Retnla−/− mice, indicating that RELMα regulates expression of IL-17. The regulation of Th17 can come from cytokines that mediate immunity. IL-1 receptor is required for Th17 response in allergic airway disease (39), and IL-1β can induce IL17 production in T cells, amplifying Th17 responses (40). The increase of IL-1β in the lung after OVA challenge may up-regulate IL-17 cytokine and Th17 responses. From our results, RELMα appears to regulate Th17 response. However, we do not yet have direct evidence that the Th17 response is associated with pulmonary vascular remodeling.
In conclusion, RELMα is a proinflammatory mediator that contributes to allergen-induced pulmonary vascular remodeling by inducing an inflammatory response in the small, intra-alveolar vessels.
Footnotes
This work was supported by National Institutes of Health (NIH) Specialized Centers of Clinically Oriented Research grant P50 084946 (R.A.J.) and NIH Centers for Advanced Diagnostics and Experimental Therapeutics in Lung Diseases grant P50HL 107182 (R.A.J.).
Author Contributions: Conception and design—C.F., R.A.J., Q.S., D.J.A., M.E.R., I.K., and K.Y.-K.; analysis and interpretation—C.F., C.C., A.Z., and O.D.M.; drafting the manuscript for important intellectual content—C.F., L.W.M., R.A.J., and I.K.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0322OC on January 8, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Banerjee RR, Lazar MA. Dimerization of resistin and resistin-like molecules is determined by a single cysteine. J Biol Chem. 2001;276:25970–25973. doi: 10.1074/jbc.M103109200. [DOI] [PubMed] [Google Scholar]
- 2.Chumakov AM, Kubota T, Walter S, Koeffler HP. Identification of murine and human XCP1 genes as C/EBP-epsilon–dependent members of FIZZ/resistin gene family. Oncogene. 2004;23:3414–3425. doi: 10.1038/sj.onc.1207126. [DOI] [PubMed] [Google Scholar]
- 3.Kim KH, Lee K, Moon YS, Sul HS. A cysteine-rich adipose tissue–specific secretory factor inhibits adipocyte differentiation. J Biol Chem. 2001;276:11252–11256. doi: 10.1074/jbc.C100028200. [DOI] [PubMed] [Google Scholar]
- 4.Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, Enders GH, Silberg DG, Wen X, Wu GD, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA. 2001;98:502–506. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerstmayer B, Küsters D, Gebel S, Müller T, Van Miert E, Hofmann K, Bosio A. Identification of RELMgamma, a novel resistin-like molecule with a distinct expression pattern. Genomics. 2003;81:588–595. doi: 10.1016/s0888-7543(03)00070-3. [DOI] [PubMed] [Google Scholar]
- 6.Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, Nelson C, Lowman HB, Wright BD, Skelton NJ, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–4055. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Q, Zhou Y, Johns RA. Bruton’s tyrosine kinase (BTK) is a binding partner for hypoxia induced mitogenic factor (HIMF/FIZZ1) and mediates myeloid cell chemotaxis. FASEB J. 2007;21:1376–1382. doi: 10.1096/fj.06-6527com. [DOI] [PubMed] [Google Scholar]
- 8.Yamaji-Kegan K, Su Q, Angelini DJ, Champion HC, Johns RA. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1159–L1168. doi: 10.1152/ajplung.00168.2006. [DOI] [PubMed] [Google Scholar]
- 9.Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol. 2005;33:425–431. doi: 10.1165/rcmb.F307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, McKenzie A, Lukacs NW, Phan SH. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–3431. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 11.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res. 2003;92:1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 12.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Champion HC, Crow MT, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296:L582–L593. doi: 10.1152/ajplung.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angelini DJ, Su Q, Kolosova IA, Fan C, Skinner JT, Yamaji-Kegan K, Collector M, Sharkis SJ, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM alpha) recruits bone marrow–derived cells to the murine pulmonary vasculature. PLoS One. 2010;5:e11251. doi: 10.1371/journal.pone.0011251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol Lett. 2003;85:173–180. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 15.Stütz AM, Pickart LA, Trifilieff A, Baumruker T, Prieschl-Strassmayr E, Woisetschläger M. The Th2 cell cytokines IL-4 and IL-13 regulate found in inflammatory zone 1/resistin-like molecule alpha gene expression by a STAT6 and CCAAT/enhancer-binding protein–dependent mechanism. J Immunol. 2003;170:1789–1796. doi: 10.4049/jimmunol.170.4.1789. [DOI] [PubMed] [Google Scholar]
- 16.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Poloczek A, El-Haddad H, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir Res. 2013;14:1. doi: 10.1186/1465-9921-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, et al. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munitz A, Waddell A, Seidu L, Cole ET, Ahrens R, Hogan SP, Rothenberg ME. Resistin-like molecule alpha enhances myeloid cell activation and promotes colitis. J Allergy Clin Immunol. 2008;122:1200–1207.e1. doi: 10.1016/j.jaci.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA. Measurement of cardiac function using pressure–volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann Bowman MA, Heydemann A, Gawdzik J, Shilling RA, Camoretti-Mercado B. Transgenic expression of human S100A12 induces structural airway abnormalities and limited lung inflammation in a mouse model of allergic inflammation. Clin Exp Allergy. 2011;41:878–889. doi: 10.1111/j.1365-2222.2011.03714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan C, Johns BA, Su Q, Kolosova IA, Johns RA. Choosing the right antibody for resistin-like molecule (RELM/FIZZ) family members. Histochem Cell Biol. 2013;139:605–613. doi: 10.1007/s00418-012-1042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pesce JT, Ramalingam TR, Wilson MS, Mentink-Kane MM, Thompson RW, Cheever AW, Urban JF, Jr, Wynn TA. Retnla (RELMalpha/FIZZ1) suppresses helminth-induced Th2-type immunity. PLoS Pathog. 2009;5:e1000393. doi: 10.1371/journal.ppat.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 24.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, Swain GP, Yancopoulos GD, Valenzuela DM, Murphy A, et al. Alternatively activated macrophage-derived RELM-alpha is a negative regulator of type 2 inflammation in the lung. J Exp Med. 2009;206:937–952. doi: 10.1084/jem.20082048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu W, Gilmour MI. Comparison of allergic lung disease in three mouse strains after systemic or mucosal sensitization with ovalbumin antigen. Immunogenetics. 2009;61:199–207. doi: 10.1007/s00251-008-0353-8. [DOI] [PubMed] [Google Scholar]
- 27.Mushaben EM, Hershey GK, Pauciulo MW, Nichols WC, Le Cras TD. Chronic allergic inflammation causes vascular remodeling and pulmonary hypertension in BMPR2 hypomorph and wild-type mice. PLoS One. 2012;7:e32468. doi: 10.1371/journal.pone.0032468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Osborne LC, Joyce KL, Alenghat T, Sonnenberg GF, Giacomin PR, Du Y, Bergstrom KS, Vallance BA, Nair MG. Resistin-like molecule α promotes pathogenic Th17 cell responses and bacterial-induced intestinal inflammation. J Immunol. 2013;190:2292–2300. doi: 10.4049/jimmunol.1200706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conti P, Kempuraj D, Kandere K, Di Gioacchino M, Reale M, Barbacane RC, Castellani ML, Mortari U, Boucher W, Letourneau R, et al. Interleukin-16 network in inflammation and allergy. Allergy Asthma Proc. 2002;23:103–108. [PubMed] [Google Scholar]
- 30.Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- 31.Munitz A, Cole ET, Karo-Atar D, Finkelman FD, Rothenberg ME. Resistin-like molecule-α regulates IL-13–induced chemokine production but not allergen-induced airway responses. Am J Respir Cell Mol Biol. 2012;46:703–713. doi: 10.1165/rcmb.2011-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meester I, Rosas-Taraco AG, Salinas-Carmona MC. Retnla down-regulation and IL-13–rich environment correlate with inflammation severity in experimental actinomycetoma by Nocardia brasiliensis. Pathog Dis. 2013;67:214–220. doi: 10.1111/2049-632X.12036. [DOI] [PubMed] [Google Scholar]
- 33.Cho WK, Lee CM, Kang MJ, Huang Y, Giordano FJ, Lee PJ, Trow TK, Homer RJ, Sessa WC, Elias JA, et al. IL-13 receptor α2–arginase 2 pathway mediates IL-13–induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2013;304:L112–L124. doi: 10.1152/ajplung.00101.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weng M, Baron DM, Bloch KD, Luster AD, Lee JJ, Medoff BD. Eosinophils are necessary for pulmonary arterial remodeling in a mouse model of eosinophilic inflammation-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2011;301:L927–L936. doi: 10.1152/ajplung.00049.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 cells: new players in asthma pathogenesis. Allergy. 2011;66:989–998. doi: 10.1111/j.1398-9995.2011.02576.x. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima H, Hirose K. Role of IL-23 and Th17 cells in airway inflammation in asthma. Immune Netw. 2010;10:1–4. doi: 10.4110/in.2010.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 38.Oda N, Canelos PB, Essayan DM, Plunkett BA, Myers AC, Huang SK. Interleukin-17F induces pulmonary neutrophilia and amplifies antigen-induced allergic response. Am J Respir Crit Care Med. 2005;171:12–18. doi: 10.1164/rccm.200406-778OC. [DOI] [PubMed] [Google Scholar]
- 39.Martin RA, Ather JL, Lundblad LK, Suratt BT, Boyson JE, Budd RC, Alcorn JF, Flavell RA, Eisenbarth SC, Poynter ME. Interleukin-1 receptor and caspase-1 are required for the Th17 response in nitrogen dioxide–promoted allergic airway disease. Am J Respir Cell Mol Biol. 2013;48:655–664. doi: 10.1165/rcmb.2012-0423OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]