

Abstract
Pompe disease results from a mutation in the acid α-glucosidase gene leading to lysosomal glycogen accumulation. Respiratory insufficiency is common, and the current U.S. Food and Drug Administration–approved treatment, enzyme replacement, has limited effectiveness. Ampakines are drugs that enhance α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor responses and can increase respiratory motor drive. Recent work indicates that respiratory motor drive can be blunted in Pompe disease, and thus pharmacologic stimulation of breathing may be beneficial. Using a murine Pompe model with the most severe clinical genotype (the Gaa−/− mouse), our primary objective was to test the hypothesis that ampakines can stimulate respiratory motor output and increase ventilation. Our second objective was to confirm that neuropathology was present in Pompe mouse medullary respiratory control neurons. The impact of ampakine CX717 on breathing was determined via phrenic and hypoglossal nerve recordings in anesthetized mice and whole-body plethysmography in unanesthetized mice. The medulla was examined using standard histological methods coupled with immunochemical markers of respiratory control neurons. Ampakine CX717 robustly increased phrenic and hypoglossal inspiratory bursting and reduced respiratory cycle variability in anesthetized Pompe mice, and it increased inspiratory tidal volume in unanesthetized Pompe mice. CX717 did not significantly alter these variables in wild-type mice. Medullary respiratory neurons showed extensive histopathology in Pompe mice. Ampakines stimulate respiratory neuromotor output and ventilation in Pompe mice, and therefore they have potential as an adjunctive therapy in Pompe disease.
Keywords: ampakines, Pompe disease, respiratory insufficiency, neuropathology, medulla
Clinical Relevance
Respiratory insufficiency occurs in both early- and late-onset Pompe disease and is often unresponsive to current therapeutic approaches, which focus on muscular pathology. Ampakines are drugs that can increase the responses of the glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, and they most effectively stimulate breathing when the neural input to respiratory muscles is blunted. We observed that an ampakine, CX717, robustly increased respiratory neuromotor output and ventilation in Pompe but had limited impact on wild-type mice. These data indicate that ampakines have potential as an adjunctive therapy for hypoventilation in Pompe disease.
Pompe disease occurs due to a mutation in the acid α-glucosidase (GAA) gene. The GAA protein is a hydrolase that degrades lysosomal glycogen, and its deficiency causes glycogen accumulation, disruption of cellular architecture, and neuromuscular impairments (1–3). Motor units controlling the diaphragm and pharyngeal airway are particularly affected (4), and severe respiratory insufficiency and ventilator dependence are common in both infantile and late-onset Pompe disease. Respiratory dysfunction in Pompe disease has been attributed to skeletal muscle pathology (5), but recent work from independent laboratories has raised awareness of a neural component (1, 3, 6–8). For example, respiratory motoneurons show profound histopathology in Pompe mice (1, 6, 7), and an autopsy report confirms pathology in the region of the phrenic motor nucleus in an infant with Pompe (1). Neuropathology is an important consideration, because the only U.S. Food and Drug Administration–approved pharmacologic treatment is intravenous infusion of recombinant GAA protein. Although this approach has modest functional benefit (9), it does not prevent the progression to respiratory failure in many patients (4), and the recombinant protein will not reach the central nervous system after intravenous delivery. Accordingly, in addition to therapies aimed at skeletal muscles, treatments targeting the central nervous system are needed to effectively manage respiratory insufficiency in Pompe disease. Here, we explore a pharmacologic approach aimed at enhancing neuromotor output to the respiratory muscles. Specifically, we tested the impact of ampakines on breathing in the Gaa−/− mouse (10), a Pompe model that recapitulates the most severe genotype with absence of functional GAA.
Ampakines enhance the function of glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (11, 12). These drugs successfully stimulated breathing after opiate-induced respiratory depression in clinical trials (13, 14). Importantly, preclinical studies show that ampakines most effectively stimulate breathing when the neural input to respiratory muscles is blunted (13, 15–17). Ampakines may therefore be a therapeutic option for respiratory insufficiency, particularly when neuromotor impairment is suspected. Accordingly, we tested the hypothesis that ampakines increase respiratory neuromotor output and ventilation in Pompe mice. In complementary studies, brainstem respiratory control neurons and motoneurons were histologically evaluated. Although motoneuron histopathology has been documented in Pompe tissues (1, 4, 7, 18, 19), currently, there is no information available regarding the impact of GAA gene deletion on the medullary neurons and networks that regulate breathing. This is a fundamental consideration, as delineation of the neuronal mechanisms contributing to respiratory failure in Pompe disease is a prerequisite for optimizing therapeutic strategies. Mechanistically, ampakines can act on both respiratory premotor circuits and motoneurons (13, 20), and, accordingly, we believed it was important to determine if pathology was present in premotor respiratory control neurons, and to compare and contrast the histological appearance of these cells with respiratory motoneurons.
Materials and Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Florida. 129SVE (wild-type) mice (n = 16; age, 13 ± 0.8 mo; 32 ± 0.9 g) were obtained from Taconic Inc. (Albany, NY). Gaa−/− mice (10) (n = 16; 14 ± 1.2 mo; 26 ± 0.9 g) were outbred to a 129SVE background (Taconic). The ampakine compound, CX717, was provided by Cortex Pharmaceuticals (Irvine, CA). Stock solutions were dissolved in 10% 2-hydroxypropyl-β-cyclodextran (HPCD; Sigma, St. Louis, MO).
Procedures for recording of respiratory nerve activity were adapted from recent reports (1, 7); see the online supplement for a comprehensive description. Mice were urethane anesthestized (1.6 g/kg, intraperitoneal; Sigma), vagotomized, and mechanically ventilated while hypoglossal and phrenic nerve activity was recorded with tungsten electrodes. Data were digitized and recorded using Spike2 v7.10 software (Cambridge Electronic Design, Cambridge, UK).
After a 10- to 20-minute period of stable baseline activity, mice received a sham (10% HPCD) intraperitoneal injection. Sham injection never evoked a persistent change in neural activity, and, 10 minutes later, an equivalent volume of CX717 solution (15 or 30 mg/kg) was administered. Inspiratory time (TI), expiratory time (TE), and the overall respiratory cycle duration (TTOT) were calculated from the integrated neurogram (21). Peak integrated hypoglossal and phrenic amplitude (∫hypoglossal and ∫phrenic) and burst frequency were averaged for 1 minute during baseline, after vehicle injection, and 30 minutes after CX717. Burst amplitude data are expressed in arbitrary units (a.u.; i.e., volts) and relative to the baseline condition (% baseline). Coefficient of variation was calculated as the ratio of the standard deviation and mean.
Ventilation and an estimate of the metabolic CO2 production (co2) were assessed using a whole-body plethysmograph (Buxco, Inc., Wilmington, NC) in unrestrained, unanesthetized mice (1) (n = 8 per group; see the online supplement for details). During a 30-minute acclimation period and subsequent 30- to 60-minute baseline period, mice were exposed to normoxic air (21% O2, 79% N2). Plethysmography measurements were taken after sham injection (10% HPCD, intraperitoneal) on three consecutive days for each mouse, and the average of these three trials was calculated. At 2 weeks after the final sham trial, mice received either 15 or 30 mg/kg CX717 (intraperitoneal), and the plethysmography trial was repeated.
Immunohistochemistry on medullary tissue sections and subsequent imaging were done using standard methods (22); see the online supplement. Statistical tests were performed using SigmaStat v12.0 software (Systat Software, Inc., San Jose, CA). For analysis of cross-sectional area, images taken from serial sections were analyzed with LSM 510 software (Zeiss, Toronto, ON, Canada). Cells with visible nuclei and proximal processes were identified and a cross-sectional area calculated. Averaged cell size measurements and raw XII and phrenic amplitudes from wild-type and Gaa−/− mice were compared using Student’s t test. Two-way repeated measures ANOVA was used to compare hypoglossal and phrenic activity and ventilation data. The Student-Newman-Keuls post hoc analysis was used for multiple pair-wise comparisons. Data are presented as mean (±SE) and were considered to be statistically different at a P value less than 0.05.
Results
Ampakine CX717 Increases Respiratory Neuromotor Output in Pompe Mice
Representative nerve recordings are provided in Figure 1. Baseline respiratory motor output was assessed before vehicle or CX717 delivery, and was more variable in anesthetized Gaa−/− mice, as evidenced by an increased coefficient of variation for TI, TE, and TTOT (P < 0.05 versus wild type; Figure 2). Baseline phrenic inspiratory burst amplitude averaged 0.16 (±0.03) a.u. and 0.58 (±0.18) a.u. for Gaa−/− and wild-type mice, respectively (P = 0.02). The XII burst amplitude (a.u.) was also reduced in Gaa−/− (0.11 ± 0.05) versus wild-type mice (0.39 ± 0.12) (P = 0.03). Note, however, that the relative change in burst amplitude (% baseline) after ampakine or sham treatment was the primary dependent variable in these studies (see the next paragraph).
Figure 1.

Representative examples of hypoglossal and phrenic nerve discharge. The top of A demonstrates the respiratory neuromotor response to delivery of vehicle (left arrow) and then ampakine CX717 (15 mg/kg, right arrow) in an acid α-glucosidase (Gaa)−/− mouse. Approximately 30 minutes of inspiratory bursting are depicted; expanded time scales, traced in which individual inspiratory bursts are discernable, are also provided for the areas indicated by a and b. Note that the vehicle injection had minimal impact on inspiratory bursting, but CX717 injection resulted in a relatively rapid and sustained increase in both phrenic and hypoglossal burst amplitude. (B) A typical response to both the vehicle and CX717 in wild-type mice. The scale bars on the frequency traces indicate breaths per minute.
Figure 2.
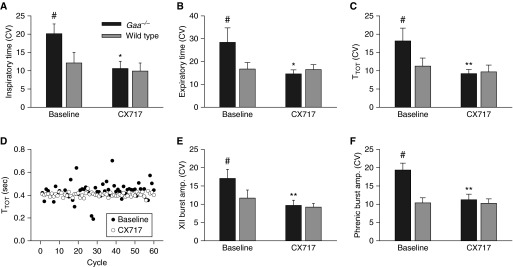
Ampakine CX717 reduces respiratory cycle variability in neurograms recorded in anesthetized Gaa−/− mice. The coefficient of variation (CV) was calculated for inspiratory time (A), expiratory time (B), and total respiratory cycle duration (TTOT) (C). At baseline, variability in these parameters was greater in Gaa−/− compared with wild-type mice. After CX717, variability was reduced and became comparable to values recorded in wild-type mice. (D) Representative data illustrating TTOT over 60 consecutive cycles. The baseline data in this image were taken during a 10-minute period recorded before CX717, and the CX717 data were taken 30 minutes after drug delivery. A reduction in inspiratory burst amplitude variability after CX717 also occurred in both the hypoglossal (E) and phrenic nerve recordings (F). *P < 0.05 when compared with baseline; **P < 0.01 when compared with baseline; #P < 0.05 when compared with wild type. Data are presented as mean (±SE).
In both strains, injection of the vehicle solution often triggered a brief increase in inspiratory burst frequency, which returned rapidly to baseline values (e.g., Figure 1). Thus, respiratory motor output had returned to baseline values within 2 minutes of the sham injection in all mice (Figure 3). In contrast, injection of 15 mg/kg CX717 produced a rapid and sustained increase in both hypoglossal and phrenic inspiratory burst amplitude in Gaa−/− mice (Figure 3). This response was unique to the Gaa−/− mice, as an equivalent dose had no impact on inspiratory burst amplitude in wild-type animals. On average, there were no persistent changes in inspiratory burst frequency after CX717 in either mouse strain (Figure 3). However, as illustrated by the representative trace in Figure 1, frequency tended to gradually decline after CX717 in Gaa−/− mice, but this did not approach statistical significance. In contrast, a statistically significant reduction in respiratory cycle variability occurred after ampakine injection in Gaa−/− mice, but not in wild-type mice. Thus, the coefficients of variation for TI, TE, and TTOT were all reduced after CX717 in Gaa−/− mice (Figure 2).
Figure 3.

Ampakine CX717 increases inspiratory burst amplitude in Gaa−/− mice. Changes in hypoglossal (XII) (A) and phrenic (B) burst amplitude are presented as a change from baseline values (%BL) after injection of vehicle or CX717. (C) CX717 did not significantly alter the frequency of the inspiratory burst. *P < 0.05 when compared with baseline; #P < 0.05 when compared with wild type. Data are presented as mean (±SE).
Ampakine CX717 Increases Ventilation in Pompe Mice
Representative examples of airflow traces recorded using whole-body plethysmography are shown in Figure 4. Ampakine CX717 had no appreciable impact when administered at 15 mg/kg, but the 30-mg/kg dose caused Gaa−/− mice to increase ventilation (Figure 5). This was reflected by increased inspiratory frequency (P = 0.038) and peak inspiratory airflow rate (P < 0.001). Respiratory volumes were also increased in Gaa−/− mice after 30 mg/kg ampakine, and similar conclusions were reached with data expressed as absolute (mls) and relative (ml/g) values (all P < 0.0.008; Figure 5). In wild-type mice, there was a tendency for increased ventilation after the 30-mg/kg ampakine dose, but this was above the threshold for statistical significance (P > 0.1), and was also considerably less than the corresponding response in Gaa−/− mice (Figure 5).
Figure 4.

Representative examples of air flow recorded using whole-body plethysmography in unanesthetized mice. Inspiration is indicated by a downward deflection in the trace. (A and B) Breathing in a Gaa−/− mouse at baseline (left) and after delivery of ampakine CX717 at 30 mg/kg (right). (C and D) The same for a wild-type mouse. The expanded traces show a few consecutive breaths from the areas indicated by the asterisks. Scaling on each trace is the same between the Gaa−/− and wild-type panels.
Figure 5.

Mean ventilatory responses to ampakine CX717 in unanesthetized mice. No ventilatory parameters were influenced by the 15-mg/kg dose of CX717. However, both respiratory timing and volumes were significantly altered after the 30-mg/kg dose in Gaa−/− mice. (A) Breathing frequency (breaths ⋅ min−1); (B) inspiratory duration (s); (C) tidal volume (ml ⋅ breath-1); (D) tidal volume (ml ⋅ breath−1/g); (E) minute ventilation (ml ⋅ min−1); (F) minute ventilation (ml ⋅ min−1/g). *P < 0.05 compared with vehicle; #P < 0.05 compared with wild-type data point. Data are presented as mean (±SE).
The 30-mg/kg ampakine dose also increased co2 in both Gaa−/− and wild-type mice. The ventilatory equivalent (i.e., e/co2), however, remained constant in wild-type mice, but was increased in Gaa−/− mice (Table 1). The mean inspiratory flow rate (i.e., tidal volume [Vt]/TI) increased by approximately 30% in Gaa−/− after the 30-mg/kg ampakine dose (Table 1).
Table 1.
Respiratory and Metabolic Parameters in Wild-Type and Gaa−/− Mice
| |
Vt/TI |
PIF |
PEF |
co2 |
|
Weight |
|---|---|---|---|---|---|---|
| (ml ⋅ breath−1/s) | (ml/s) | (ml/s) | (ml ⋅ min−1/g) | e/co2 | (g) | |
| Wild-type vehicle | 1.01 ± 0.07 | 1.80 ± 0.09 | 1.46 ± 0.10 | 0.033 ± 0.00 | 16.8 ± 0.6 | 32.1 ± 1.22 |
| Wild-type CX717 15 mg/kg | 0.99 ± 0.50 | 1.78 ± 0.08* | 1.52 ± 0.12 | 0.033 ± 0.00 | 17.0 ± 0.3 | 31.5 ± 1.47 |
| Wild-type CX717 30 mg/kg | 1.11 ± 0.05 | 1.99 ± 0.10 | 1.77 ± 0.19†,‡ | 0.039 ± 0.00†,‡ | 16.3 ± 0.4 | 31.7 ± 1.72 |
| Gaa−/− vehicle | 0.87 ± 0.06 | 1.54 ± 0.08 | 1.38 ± 0.10 | 0.038 ± 0.00† | 15.8 ± 0.3 | 25.0 ± 0.45† |
| Gaa−/− CX717 15 mg/kg | 0.81 ± 0.06‡ | 1.43 ± 0.10 | 1.30 ± 0.09‡ | 0.035 ± 0.00 | 16.1 ± 0.5 | 24.7 ± 0.41† |
| Gaa−/− CX717 30 mg/kg | 1.12 ± 0.05*,§ | 1.92 ± 0.10*,§ | 1.59 ± 0.10 | 0.042 ± 0.00*,§ | 17.3 ± 0.7*,§ | 25.5 ± 0.80† |
Definition of abbreviations: PEF, peak expiratory airflow; PIF, peak inspiratory airflow; TI, inspiratory time; co2, rate of carbon dioxide production; e, minute ventilation; Vt, inspiratory tidal volume.
Data are provided for sham treatment as well as two doses of ampakine CX717.
P < 0.05 compared with Gaa−/− mice treated with CX717 at 15 mg/kg.
P < 0.05 compared with wild-type mice after vehicle injection.
P < 0.05 compared with wild-type mice receiving CX717 at 15 mg/kg.
P < 0.05 compared with vehicle-treated Gaa−/− mice.
Brainstem Histopathology
Motoneuron pools were identified using standard anatomical landmarks, and cell bodies were confirmed via choline acetyl transferase (ChAT) immunoreactivity. Prior work established that expression of ChAT, somatostatin, and tyrosine hydroxylase is uniformly distributed in the cell soma of medullary cardiorespiratory neurons (22, 23), and this was observed in tissues from wild-type mice (Figures 6 and 7). In contrast, a clear lack of immunostaining for ChAT, somatostatin, and tyrosine hydroxylase within large areas of the soma was evident in Gaa−/− tissues (e.g., Figures 6 and 7). Indeed, virtually all motoneurons in the facial (Figure 6B), nucleus ambiguus (Figure 6D), and hypoglossal nuclei (Figure 6H) of Gaa−/− mice showed a considerably enlarged soma and profound accumulation of ChAT-immunonegative vesicles. On average, motoneurons in the facial, hypoglossal, and ambiguus nuclei were all significantly larger in Gaa−/− tissues as compared with wild type (Figure 6I). Evidence of histopathology was not ubiquitous, however, as motoneurons in the vagal nucleus had similar histologic appearance between Gaa−/− and wild-type tissues (Figures 6E and 6F).
Figure 6.
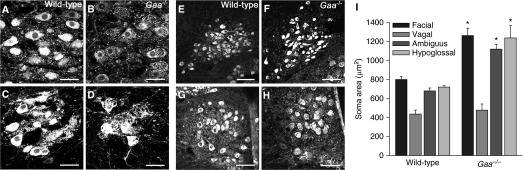
Brainstem motoneuron morphology is altered in Gaa−/− mice. Choline acetyl transferase (ChAT) immunoreactivity was used to identify motoneurons in the facial nucleus (A and B), nucleus ambiguus (C and D), the dorsal motor nucleus of the vagus (E and F), and hypoglossal nucleus (G and H). On average, cells in the facial, ambiguus, and hypoglossal nuclei were significantly larger in Gaa−/− versus wild-type tissues (I). *P < 0.05. Altered morphology is evident in these Gaa−/− motoneurons, with enlarged soma and marked accumulation of ChAT-immunonegative vesicles. In contrast, motoneurons in the vagal nucleus were apparently unaffected, with no histological evidence of a difference in size (I) or histological appearance (E–G). Scale bars indicate 50 μm (A–D) and 100 μm (E and F). Data are presented as mean (±SE).
Figure 7.

Histological appearance of medullary respiratory control neurons in wild-type and Gaa−/− mice. (A and B) Somatostatin-positive neurons within the pre-Bötzinger complex; (C and D) tyrosine hydroxylase–positive A1/C1 neurons; (E and F) serotonin-positive raphe neurons. Note in particular the presence of abnormal morphology and immunonegative vacuoles (arrows) in cells staining positive for somatostatin and tyrosine hydroxylase in Gaa−/− mice. Scale bars indicate 20 μm (A and B) and 50 μm (C–F).
We next histologically evaluated medullary neurons involved with respiratory rhythm generation and/or modulation of respiratory motor output. The preBötzinger complex (preBötC), which contains neurons that are essential for respiratory rhythmogenesis (24), was identified in the ventrolateral medulla by neurokinin 1 receptor and somatostatin immunoreactivity (25–27). Somatostatin-positive preBötC cell bodies had prominent inclusion of somatostatin-immunonegative vacuoles in Gaa−/−, but not wild-type tissues (Figures 7A and 7B). In addition, a dense network of neurokinin 1 receptor immunoreactive fibers was present in the preBötC region, with no qualitative difference between Gaa−/− and wild-type mice (data not shown). Noradrenergic neurons in the A1/C1 group of the ventral medulla displayed abnormal morphology in Gaa−/− tissues, with enlarged vesicles present throughout the soma (Figures 7C and 7D). In contrast, serotonergic raphe neurons along the rostrocaudal extension of pons and medulla in Gaa−/− mice did not display any significant altered morphology as compared with wild-type mice (Figures 7E and 7F).
Discussion
We found that an ampakine (CX717) very effectively stimulates respiratory neuromotor output and ventilation in Pompe (Gaa−/−) mice, but not in wild-type mice. This finding raises the possibility of using ampakines as an adjunctive therapy for hypoventilation in Pompe disease. In addition, brainstem histology confirmed extensive motoneuron pathology in Gaa−/− mice, and showed, for the first time, histopathology in medullary respiratory control neurons. The dramatic impact of CX717 on efferent respiratory motor output in Gaa−/− mice supports the hypothesis that respiratory neuropathology has a functional impact in Pompe disease (4), as ampakines stimulate breathing most effectively when respiratory neuromotor activity is blunted (13, 20).
Pompe Disease—Respiratory Insufficiency and the Current Treatment Approach
Respiratory insufficiency is prevalent in both infantile and late-onset Pompe disease (28–31). In early-onset patients, respiratory difficulty often becomes evident at 4–6 months of age (32), and approximately 75% of children and adolescents with Pompe disease ultimately become dependent upon mechanical ventilation (33, 34). Approximately one-third of adults with Pompe disease require mechanical ventilator support, and conditions such as aspiration and pneumonia, are prevalent (35). Obstructive sleep apnea has also been reported in Pompe disease (36, 37), and there is some evidence that central sleep apnea may also occur (37). It is also noteworthy that severe respiratory insufficiency can occur in Pompe disease without evidence for significant limb muscle weakness (31). The respiratory neuromuscular system, therefore, appears to be particularly susceptible to dysfunction in Pompe disease, and this is unique in respect to other muscular dystrophies, where locomotor impairments typically precede ventilatory insufficiency.
Biweekly intravenous infusion of recombinant GAA protein (i.e., enzyme replacement therapy) is the most widely used treatment for Pompe disease (5, 38). Although the literature clearly indicates that enzyme replacement can provide a stabilizing impact on respiratory function (9), several groups have concluded that enzyme replacement only delays the eventual requirement for mechanical ventilation (39–41). An early study from Van den Hout and colleagues (42) reported that three out of four patients initiating enzyme replacement at a young age (2.5–8 mo) nevertheless became ventilator dependent by age 4 years. Toscano and Schoser (41) longitudinally evaluated over 350 patients with late-onset Pompe after initiation of enzyme replacement. Forced vital capacity improved in 51% of patients, was unchanged in 14%, and declined in 35% (41). Another study followed adults over 12 months of enzyme replacement (43), and the wide range of responses included decreases, no change, and increases to the required ventilator support. The recombinant GAA enzyme used in enzyme replacement does not cross the blood–brain barrier (44, 45), and, thus, the limited and/or variable success of enzyme replacement, and the clear evidence of neuropathology in respiratory neurons (current study and Refs. 1, 3, 4, 7, 18), has led to the hypothesis that impaired function in respiratory neurons and networks contributes to respiratory insufficiency in Pompe disease (1). As discussed in the next section, the functional impact of ampakines in Gaa−/− mice provides further support for this hypothesis, and also raises the possibility of pharmacologically targeting the respiratory control system with ampakines.
Ampakines and Their Use in Treating Hypoventilation
Ampakines are positive modulators of the AMPA glutamate receptor subtype, and act to increase the duration of AMPA receptor gated-inward currents (20, 46). The ampakine used in this study, CX717, is metabolically stable, clinically safe, and effectively crosses the blood–brain barrier (11, 14, 47, 48). CX717 has been used in both animal models (15, 17, 20) and clinical trials to alleviate respiratory depression due to opioids (14). CX717 does not directly antagonize opioid receptors, because, in both rats and humans, CX717 counteracts respiratory depression without influencing analgesia (15–17, 20). Animal studies have confirmed that CX717 stimulates breathing by potentiating AMPA currents in respiratory neurons, including both motor and premotor neurons (15, 20). For example, ampakine CX546 stimulates respiratory frequency during opioid-induced hypoventilation—suggesting that ampakines can act on medullary rhythm-generating neurons, including the preBötC (13). On the other hand, ampakine CX717 increases both the amplitude and duration of postsynaptic AMPA currents in respiratory motoneurons (20). In our study, CX717 robustly increased inspiratory burst amplitude in the anesthetized, vagotomized Gaa−/− mouse but with little to no impact on burst frequency (e.g., Figures 1 and 2). Accordingly, the primary mechanism of activation in the anesthetized preparation is likely to be potentiation of post-synaptic AMPA currents on phrenic and hypoglossal motoneurons. Considering that the greatly increased motoneuron soma size in Pompe tissues and relevant animal models (e.g., Figure 6; see also Refs. 1, 18) is likely to decrease the electrical resistance of the cell, the “size principle” of motoneuron recruitment (49) predicts that increases in synaptic current will be required for inspiratory depolarization (1). In this scenario, ampakine-mediated potentiation of AMPA currents on respiratory motoneurons (20) would increase inspiratory motor output (e.g., Figures 1 and 2).
We also noted that the anesthetized Gaa−/− mouse showed greater variability in the respiratory cycle compared with the wild-type control (Figure 3), and this is consistent with a prior report (7). However, CX717 reduced the variability in respiratory motor output, suggesting that premotor neurons and/or respiratory rhythmogenic networks were also influenced by the drug. This is plausible, as rhythm-generating neurons in the Gaa−/− medulla showed histopathology (e.g., Figure 7), albeit not to the same extent as respiratory motoneurons. In the unanesthetized Gaa−/− mouse, the increased ventilation after CX717 treatment occurred as a result of both increased inspiratory volume and frequency. The underlying mechanism(s) are likely to include potentiation of inspiratory-related AMPA currents on respiratory motoneurons (13, 20) and pre–motor neurons associated with respiratory rhythm generation (15, 24). Another consideration, however, is that the impact of ampakines on ventilation in the conscious mouse likely reflected an interplay between increased metabolism and a direct stimulating effect on respiratory neurons. The 30-mg/kg ampakine dose increased co2; however, the ventilatory equivalent (i.e., e/co2) was not changed in the wild-type mice. In the Gaa−/− mouse, however, an increase in the e/co2 ratio suggests that at least a portion of the increased ventilation reflected neural stimulation. There was also an approximately 30% increase in Gaa−/− mean inspiratory flow rate (i.e., Vt/TI) after the 30-mg/kg ampakine dose. This is of interest, because Vt/TI increases in parallel with respiratory neuromotor drive (50, 51). Taken together, the plethsymography data (conscious mice) and nerve recordings (anesthetized mice) lead us to conclude that at least a component of increased breathing after ampakine treatment reflects a direct stimulation of respiratory-related neurons.
Commentary Regarding the Plethysmography Data
In terms of absolute respiratory volumes, Gaa−/− mice show a clear decrease as compared with the wild-type mice. However, interpretation is confounded by decreased body mass in Gaa−/− mice, because the normalized values exceed those from the wild-type mice. How can the paradox of widespread respiratory neural and muscular pathology, blunted XII and phrenic neuromotor output under anesthesia (Figure 2) (1, 7), and increased relative ventilation be explained? There are several possibilities. First, normalization of respiratory volumes to body mass may not be appropriate in degenerative disease models. Mitchell and colleagues have suggested that such an approach may in fact obviate detection of physiologic impairments in a rat amyotrophic lateral sclerosis (ALS) model (52, 53). Without clear changes in the size of the lung and chest cavity in diseased animals, but with loss of muscle mass and/or neurological problems, body mass may no longer scale with respiratory volumes in a predictable manner. Second, increased ventilation may, in fact, be the correct interpretation, reflecting an initial compensatory response to the developing neuromuscular pathology. This is something we have considered in the past, and we recently proposed a model of the progression of respiratory failure in Pompe disease in which respiratory motor output increases inappropriately during the initial stages of neuromuscular pathology (4). In the context of the current study, the primary dependent variable was the change in breathing after ampakine delivery versus the absolute level of ventilation. Importantly, the fundamental conclusion of this article—that ampakines stimulate respiratory output more robustly in Gaa−/− mice as compared with the wild-type response—is maintained regardless of the approach to the presentation of the data.
Conclusions
It is now well established that extensive neuropathology is present in advanced stages of Pompe disease, particularly in motoneurons (current data and Refs. 1, 3, 7, 18). Several independent groups have concluded that neuropathology has a significant impact on motor function (8, 49, 54), but no current therapies are aimed at improving the efferent neural regulation of the respiratory muscles (although a gene therapy clinical trial is in progress [55]). In the context of the current results, it must be emphasized that rigorously controlled in vitro and in vivo experiments indicate that CX717 robustly increases respiratory activity when respiratory neuromotor activity is suppressed, but has minimal impact under conditions of “normal” respiratory motor activity (14, 20, 47). Thus, our findings in Gaa−/− mice provide further support for the Pompe “neural hypothesis” (4, 8, 39), and raise the possibility of using ampakines as an adjunctive therapy for hypoventilation in Pompe disease. In humans, the half-life of ampakines is approximately 9 hours (S. Johnson, Cortex Pharmaceuticals Inc., personal communication), and thus, possible clinical uses for ampakines could include enabling periods of weaning from ventilator support, and potentially reducing apneic events during sleep. Future studies should also evaluate the use of ampakines as a component of neurorehabilitative strategies aimed at evoking neuroplasticity and improved independent respiratory function. Such an approach could be used in conjunction with current (e.g., enzyme replacement) (9), or future (e.g., gene therapy) (5), treatment approaches.
Acknowledgments
Acknowledgments
The authors gratefully acknowledge Cortex Pharmaceuticals for providing the ampakines used in this study.
Footnotes
This work was supported by a Parker B. Francis Fellowship and National Institutes of Health (NIH) 1 K08 HD077040-01A1 (M.K.E.), Canadian Health Institutes for Research (J.J.G.), Alberta Heritage Foundation for Medical Research (J.J.G.), and NIH 2R01HD052682-06A1 (D.D.F. and B.J.B.).
Author Contributions: M.K.E. was involved in the conception, hypotheses delineation, and study design, and was primarily responsible for acquisition, analysis, and interpretation of data and drafting the article; S.P. performed the histological analyses, as well as interpretation of and discussion on these data; S.M.T. designed the plethysmography study and assisted with data collection and manuscript preparation; A.J.C. was involved with acquisition and analysis of the plethysmography data; D.J.F. was involved with the design of the study and data interpretation; B.J.B. and J.J.G. were involved in the conception, hypotheses delineation, data interpretation, and revision the article, and can be considered as co–senior authors with D.D.F.; neurophysiology and behavioral testing were conducted in D.D.F.’s laboratory, and he was involved in the conception, hypotheses delineation, and design of the study, as well as data interpretation and writing and revising the article.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0374OC on January 8, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Jr, Mah C, Reier PJ, Byrne BJ. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease) Curr Mol Med. 2002;2:145–166. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- 3.Sidman RL, Taksir T, Fidler J, Zhao M, Dodge JC, Passini MA, Raben N, Thurberg BL, Cheng SH, Shihabuddin LS. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuller DD, ElMallah MK, Smith BK, Corti M, Lawson LA, Falk DJ, Byrne BJ. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME, Collins SW, Conlon TJ, Clement N, Cleaver BD, et al. Pompe disease gene therapy. Hum Mol Genet. 2011;20:R61–R68. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller DD, ElMallah MK, Falk DJ, Lawson L, Martin D, Smith B, Byrne BJ.The importance of targeting the central nervous system (CNS) for treatment of respiratory insufficiency in Pompe diseaseOrlando, FL: World Lysosomal Meeting; 2013 [Google Scholar]
- 7.Lee KZ, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA, Reier PJ, Byrne BJ, Fuller DD. Hypoglossal neuropathology and respiratory activity in Pompe mice. Front Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller CW, Jones HN, O’Grady G, Suarez AH, Heller JH, Kishnani PS. Language and speech function in children with infantile Pompe disease. J Pediatr Neurol. 2009;7:147–156. [Google Scholar]
- 9.van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 10.Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, LaMarca M, King C, Ward J, Sauer B, et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 11.Staubli U, Rogers G, Lynch G. Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci USA. 1994;91:777–781. doi: 10.1073/pnas.91.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arai A, Kessler M, Xiao P, Ambros-Ingerson J, Rogers G, Lynch G. A centrally active drug that modulates AMPA receptor gated currents. Brain Res. 1994;638:343–346. doi: 10.1016/0006-8993(94)90669-6. [DOI] [PubMed] [Google Scholar]
- 13.Ren J, Poon BY, Tang Y, Funk GD, Greer JJ. Ampakines alleviate respiratory depression in rats. Am J Respir Crit Care Med. 2006;174:1384–1391. doi: 10.1164/rccm.200606-778OC. [DOI] [PubMed] [Google Scholar]
- 14.Oertel BG, Felden L, Tran PV, Bradshaw MH, Angst MS, Schmidt H, Johnson S, Greer JJ, Geisslinger G, Varney MA, et al. Selective antagonism of opioid-induced ventilatory depression by an ampakine molecule in humans without loss of opioid analgesia. Clin Pharmacol Ther. 2010;87:204–211. doi: 10.1038/clpt.2009.194. [DOI] [PubMed] [Google Scholar]
- 15.Ren J, Ding X, Greer JJ. Respiratory depression in rats induced by alcohol and barbiturate and alleviation by ampakine CX717. J Appl Physiol. 2012;113:1004–1011. doi: 10.1152/japplphysiol.00752.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren J, Lenal F, Yang M, Ding X, Greer JJ. Coadministration of the AMPAKINE CX717 with propofol reduces respiratory depression and fatal apneas. Anesthesiology. 2013;118:1437–1445. doi: 10.1097/ALN.0b013e318291079c. [DOI] [PubMed] [Google Scholar]
- 17.Ren J, Ding X, Funk GD, Greer JJ. Ampakine CX717 protects against fentanyl-induced respiratory depression and lethal apnea in rats. Anesthesiology. 2009;110:1364–1370. doi: 10.1097/ALN.0b013e31819faa2a. [DOI] [PubMed] [Google Scholar]
- 18.Elmallah MK, Falk DJ, Nayak S, Federico RA, Sandhu MS, Poirier A, Byrne BJ, Fuller DD. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in Pompe mice. Mol Ther. 2014;22:702–712. doi: 10.1038/mt.2013.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falk DJ, Mah CS, Soustek MS, Lee KZ, Elmallah MK, Cloutier DA, Fuller DD, Byrne BJ. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther. 2013;21:1661–1667. doi: 10.1038/mt.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorier AR, Funk GD, Greer JJ. Opiate-induced suppression of rat hypoglossal motoneuron activity and its reversal by ampakine therapy. PLoS One. 2010;5:e8766. doi: 10.1371/journal.pone.0008766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KZ, Reier PJ, Fuller DD. Phrenic motoneuron discharge patterns during hypoxia-induced short-term potentiation in rats. J Neurophysiol. 2009;102:2184–2193. doi: 10.1152/jn.00399.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pagliardini S, Ren J, Greer JJ. Ontogeny of the pre-Bötzinger complex in perinatal rats. J Neurosci. 2003;23:9575–9584. doi: 10.1523/JNEUROSCI.23-29-09575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagliardini S, Ren J, Gray PA, Vandunk C, Gross M, Goulding M, Greer JJ. Central respiratory rhythmogenesis is abnormal in Lbx1-deficient mice. J Neurosci. 2008;28:11030–11041. doi: 10.1523/JNEUROSCI.1648-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray PA, Rekling JC, Bocchiaro CM, Feldman JL. Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBötzinger complex. Science. 1999;286:1566–1568. doi: 10.1126/science.286.5444.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stornetta RL, Rosin DL, Wang H, Sevigny CP, Weston MC, Guyenet PG. A group of glutamatergic interneurons expressing high levels of both neurokinin-1 receptors and somatostatin identifies the region of the pre-Bötzinger complex. J Comp Neurol. 2003;455:499–512. doi: 10.1002/cne.10504. [DOI] [PubMed] [Google Scholar]
- 27.Pagliardini S, Adachi T, Ren J, Funk GD, Greer JJ. Fluorescent tagging of rhythmically active respiratory neurons within the pre-Bötzinger complex of rat medullary slice preparations. J Neurosci. 2005;25:2591–2596. doi: 10.1523/JNEUROSCI.4930-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burghaus L, Liu W, Neuen-Jacob E, Gempel K, Haupt WF. Glycogenesis type II (M. Pompe): selective failure of the respiratory musculature—a rare first symptom [in German] Nervenarzt. 2006;77:181–182, 185–186. doi: 10.1007/s00115-005-2005-7. [DOI] [PubMed] [Google Scholar]
- 29.Mellies U, Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir Med. 2009;103:477–484. doi: 10.1016/j.rmed.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Mellies U, Stehling F, Dohna-Schwake C, Ragette R, Teschler H, Voit T. Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology. 2005;64:1465–1467. doi: 10.1212/01.WNL.0000158682.85052.C0. [DOI] [PubMed] [Google Scholar]
- 31.Pellegrini N, Laforet P, Orlikowski D, Pellegrini M, Caillaud C, Eymard B, Raphael JC, Lofaso F. Respiratory insufficiency and limb muscle weakness in adults with Pompe’s disease. Eur Respir J. 2005;26:1024–1031. doi: 10.1183/09031936.05.00020005. [DOI] [PubMed] [Google Scholar]
- 32.van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 33.Haley SM, Fragala MA, Skrinar AM. Pompe disease and physical disability. Dev Med Child Neurol. 2003;45:618–623. doi: 10.1017/s0012162203001129. [DOI] [PubMed] [Google Scholar]
- 34.Marsden D. Infantile onset Pompe disease: a report of physician narratives from an epidemiologic study. Genet Med. 2005;7:147–150. doi: 10.1097/01.gim.0000154301.76619.5c. [DOI] [PubMed] [Google Scholar]
- 35.Hagemans ML, Winkel LP, Hop WC, Reuser AJ, Van Doorn PA, Van der Ploeg AT. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology. 2005;64:2139–2141. doi: 10.1212/01.WNL.0000165979.46537.56. [DOI] [PubMed] [Google Scholar]
- 36.Margolis ML, Howlett P, Goldberg R, Eftychiadis A, Levine S. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest. 1994;105:947–949. doi: 10.1378/chest.105.3.947. [DOI] [PubMed] [Google Scholar]
- 37.Kansagra S, Austin S, Dearmey S, Kishnani PS, Kravitz RM. Polysomnographic findings in infantile Pompe disease. Am J Med Genet A. 2013;161A:3196–3200. doi: 10.1002/ajmg.a.36227. [DOI] [PubMed] [Google Scholar]
- 38.Beck M. Alglucosidase alfa: long term use in the treatment of patients with Pompe disease. Ther Clin Risk Manag. 2009;5:767–772. doi: 10.2147/tcrm.s5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider I, Hanisch F, Muller T, Schmidt B, Zierz S. Respiratory function in late-onset Pompe disease patients receiving long-term enzyme replacement therapy for more than 48 months. Wien Med Wochenschr. 2013;163:40–44. doi: 10.1007/s10354-012-0153-5. [DOI] [PubMed] [Google Scholar]
- 40.Byrne BJ, Kishnani PS, Case LE, Merlini L, Muller-Felber W, Prasad S, der Ploeg A. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol. 2013;260:951–959. doi: 10.1007/s00415-012-6636-x. [DOI] [PubMed] [Google Scholar]
- 42.Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 43.Orlikowski D, Pellegrini N, Prigent H, Laforet P, Carlier R, Carlier P, Eymard B, Lofaso F, Annane D. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord. 2011;21:477–482. doi: 10.1016/j.nmd.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Kikuchi T, Yang HW, Pennybacker M, Ichihara N, Mizutani M, Van Hove JL, Chen YT. Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase–deficient quail. J Clin Invest. 1998;101:827–833. doi: 10.1172/JCI1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 46.Arai AC, Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- 47.Porrino LJ, Daunais JB, Rogers GA, Hampson RE, Deadwyler SA. Facilitation of task performance and removal of the effects of sleep deprivation by an ampakine (CX717) in nonhuman primates. PLoS Biol. 2005;3:e299. doi: 10.1371/journal.pbio.0030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle J, Stanley N, James LM, Wright N, Johnsen S, Arbon EL, Dijk DJ. Acute sleep deprivation: the effects of the AMPAKINE compound CX717 on human cognitive performance, alertness and recovery sleep. J Psychopharmacol. 2012;26:1047–1057. doi: 10.1177/0269881111405353. [DOI] [PubMed] [Google Scholar]
- 49.Henneman E, Somjen G, Carpenter DO. Excitability and inhibitability of motoneurons of different sizes. J Neurophysiol. 1965;28:599–620. doi: 10.1152/jn.1965.28.3.599. [DOI] [PubMed] [Google Scholar]
- 50.Eldridge FL. Relationship between phrenic nerve activity and ventilation. Am J Physiol. 1971;221:535–543. doi: 10.1152/ajplegacy.1971.221.2.535. [DOI] [PubMed] [Google Scholar]
- 51.Grassino A, Sorli J, Lorange G, Milic-Emili J. Respiratory drive and timing in chronic obstructive pumonary disease. Chest. 1978;73:290–293. [PubMed] [Google Scholar]
- 52.Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, et al. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nichols NL, Johnson RA, Satriotomo I, Mitchell GS. Neither serotonin nor adenosine-dependent mechanisms preserve ventilatory capacity in ALS rats. Respir Physiol Neurobiol. 2014;197:19–28. doi: 10.1016/j.resp.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burrow TA, Bailey LA, Kinnett DG, Hopkin RJ. Acute progression of neuromuscular findings in infantile Pompe disease. Pediatr Neurol. 2010;42:455–458. doi: 10.1016/j.pediatrneurol.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Smith BK, Collins S, Conlon T, Mah C, Lawson LA, Martin D, Fuller DD, Cleaver B, Clement N, Phillips D, et al. Phase I/II trial of AAV1-GAA gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]