

Abstract
To explore the underlying mechanisms whereby noncoding variants affect transcriptional regulation, we identified nucleotides capable of disrupting binding of transcription factors and deactivating enhancers if mutated (dubbed candidate killer mutations or KMs) in HepG2 enhancers. On average, approximately 11% of enhancer positions are prone to KMs. A comparable number of enhancer positions are capable of creating de novo binding sites via a single-nucleotide mutation (dubbed candidate restoration mutations or RSs). Both KM and RS positions are evolutionarily conserved and tend to form clusters within an enhancer. We observed that KMs have the most deleterious effect on enhancer activity. In contrast, RSs have a smaller effect in increasing enhancer activity. Additionally, the KMs are strongly associated with liver-related Genome Wide Association Study traits compared with other HepG2 enhancer regions. By applying our framework to lymphoblastoid cell lines, we found that KMs underlie differential binding of transcription factors and differential local chromatin accessibility. The gene expression quantitative trait loci associated with the tissue-specific genes are strongly enriched in KM positions. In summary, we conclude that the KMs have the greatest impact on the level of gene expression and are likely to be the causal variants of tissue-specific gene expression and disease predisposition.
Keywords: enhancers, causal mutations, transcription factor binding sites, gene regulation
Introduction
Understanding the regulatory program is critical to understanding cellular development and disease susceptibility. However, the regulatory code is much more complex than the interpretable triplet code of protein-coding sequences and is highly lineage-specific and context-dependent (Jolma et al. 2013). The majority (88%) of Genome Wide Association Study (GWAS; Welter et al. 2014) polymorphisms are in noncoding DNA. Specifically, a noncoding variation that alters DNA-binding sites of a transcription factor (TF) and impacts transcriptional regulation might affect the pattern of gene expression and have an impact on cellular development, morphology, function, and phenotype. The fundamental mechanisms of how genetic variants disrupt TF binding and lead to downstream effects on gene expression are not yet fully understood.
Accumulating evidence implicates DNA variants within regulatory sequences in human disease and disorders (Visel, Rubin, et al. 2009; Maurano, Humbert, et al. 2012; Sakabe et al. 2012; Dickel et al. 2013; Monteiro and Freedman 2013). A growing number of genomic studies incorporated the investigation of functional properties of regulatory sequences into gene expression quantitative trait loci (eQTL; Gaffney et al. 2012) and GWAS analyses. For example, breast cancer risk-associated single nucleotide polymorphisms (SNPs) were found to be enriched in the TF-binding sites (TFBSs) of FOXA1 and ESR1, modulating the binding affinity of these TFs at distal enhancer regions. These SNPs resulted in allele-specific gene expression, exemplified by the most studied breast-cancer-associated SNP, rs4784227. This SNP within the 16q12.1 locus was experimentally verified to impact the expression of the TOX3 gene which stimulates estrogen response element-dependent transcriptional programs (Dittmer et al. 2011) and is differentially expressed in breast cancer cell lines that are metastatic to bone (Smid et al. 2006). In addition, when mapping the eQTLs and SNPs to DNase I hypersensitivity sites (DHSs; Degner et al. 2012; Maurano, Humbert, et al. 2012), approximately 50% of the eQTLs were also found to be DNase I sensitivity QTLs (dsQTLs; Degner et al. 2012), and disease-associated SNPs were found to be enriched in DHS, systematically disturbing TFBSs as well as associated with the allele-specific chromatin accessibility (Maurano, Humbert, et al. 2012). Overall, these studies indicate that sequence-encoded regulation can impact corresponding gene expression. Therefore, elucidation of sequence-encoded regulation would facilitate better understanding of the relationship between genotype and phenotype.
However, the key question is how to accurately identify causative single nucleotide variants (SNVs). Heinz et al. (2013) proposed a hierarchical collaborative model for enhancer selection and function to prioritize regulatory variants. By exploring this model in combination with transient/stable reporter assays, they found that the motif-disrupting variants of the lineage-determining TFs are the causal variants that underlie strain-specific enhancer activity. Recently, a method named “combined annotation-dependent depletion (CADD)” (Kircher et al. 2014) was developed to annotate and interpret human genetic variation. By combining diverse annotations of genetic variation into a single score, this general framework measures the likelihood of deleteriousness of all possible SNVs and could facilitate the inference of all pathogenic variants. Nevertheless, a method that could provide a straightforward way to identify most deleterious causative variants in a cell type-specific manner and infer deleterious effects of the causal variants on gene regulation is still lacking. We recently developed a computational approach to quantify the disruptive effects of SNPs in enhancers (Huang and Ovcharenko 2015), which was instrumental in devising our current study. Here, we aim to establish the complete genome-wide profile of deactivating and advantageous mutations in enhancers.
We developed an approach that extends our previous work to systematically dissect the genetic variants (all possible mutations) in enhancer regions and prioritize the genetic variants with respect to their potential deleterious effects on TF binding and functional constraints. We identified the cell type-specific motif disrupting variants that are most likely to deactivate enhancers (candidate killer mutations or KMs). We observed that KMs are likely to impact the local chromatin structure and might play an essential role in determining tissue-specific (TS) gene expression. In total, approximately 0.3% of KM positions (KMPs) carry common SNPs (∼4% of KMP clusters hold common SNPs), providing us the searching space of mutation candidates that could explain the phenotype differences among vertebrates in the future. We also demonstrate that a bimodal mutation system of regulatory elements shaped by evolutionary force relies on the co-occurrence of deleterious and restoration mutations (RSs) within enhancers.
Results
Candidate KMs in HepG2 Enhancers
The goal of our study was to identify mutations in enhancers that can disrupt binding of TFs and thus deactivate enhancers, which we refer to as candidate KMs. To identify KMs, we detected potential binding sites of TFs from TF ChIP-seq data sets and predicted mutations that alter the binding of these TFs. As it is less likely to find a functional binding site of the length k in a random sequence than in an enhancer, we identified the top enriched k-mers (k = 8) in ChromHMM HepG2 “strong enhancers” (referred to as HepG2 enhancers later on; see Materials and Methods) as the potential binding sites. We used binding significance—defined as −log10(P-value) of k-mer enrichment—to identify k-mers of interest. The greater the binding significance is, the more likely it is that k-mer is a functional binding site of an active TF. The top 522 k-mers (Bonferroni-corrected P < 10−3, 32,896 tests, supplementary table S1, Supplementary Material online) were considered significant and selected as potential binding sites, whereas 30,647 k-mers (P > 10−3 without Bonferroni correction) were considered background sites in HepG2 enhancers.
Next, to identify KMs, we computed the change in the binding significance of a k-mer caused by a mutation using a modified intragenomic replicates model (IGR [Cowper-Sal lari et al. 2012]; see Materials and Methods). In the original IGR model, the affinity of a k-mer is measured by averaging its ChIP-seq signal across the whole genome. After that, the impact on TF binding caused by a mutation was calculated as a difference in wild-type and mutated k-mer affinities (all possible k-mers overlapping a wild-type nucleotide and the mutated allele are taken into consideration and two top-scoring k-mers are used for the calculation; supplementary fig. S1, Supplementary Material online). In our model, we used k-mer binding significance instead of k-mer affinity to directly quantify the impact of mutations on TF binding (see Materials and Methods; supplementary fig. S1, Supplementary Material online). This allowed us to use this method for detection of KMs in a set of enhancers (which are enriched for binding sites of multiple TFs), whereas the original IGR model was tailored to the analysis of ChIP-seq signals of individual TFs. In all, we identified 3,756,018 enhancer positions that carry KMPs in HepG2 cell line, approximately 48% of which could cause KMs by all three possible mutations. The majority of enhancers (∼96%) have at least one position carrying KMs.
Enriched k-mers in HepG2 Enhancers Correspond to Liver TFBSs
We observed a noticeable sequence similarity among several top HepG2 enhancer k-mers, with many of them overlapping each other (supplementary fig. S2, Supplementary Material online). To eliminate the redundancy, we clustered the 522 top k-mers into 33 distinct clusters using the Markov clustering (MCL) algorithm (van Dongen and Abreu-Goodger 2012) based on the proportion of shared dimers between two k-mers (see Materials and Methods). Next, these clusters of k-mers were mapped to the TRANSFAC (Matys et al. 2006) and JASPAR (Mathelier et al. 2014) databases of TFBSs and further merged to 14 clusters using STAMP (Mahony and Benos 2007) (see Materials and Methods). Twenty-two TFBSs were matching these 14 k-mer clusters with the E-value cut-off of 5e-3. Fourteen out of these 22 TFBSs (64%) were liver-related, and the majority of k-mer clusters were associated with at least one liver-related TFBS (fig. 1A and supplementary fig. S3, Supplementary Material online). The TFBS of HNF4α was associated with the largest k-mer cluster (198 k-mers), which is concordant with the fact that HNF4α is a major TF in liver and plays a crucial role in liver development and fatty acid metabolism (Li et al. 2000; Fiegel et al. 2003; Kyrmizi et al. 2006; Martinez-Jimenez et al. 2010).
Fig. 1.
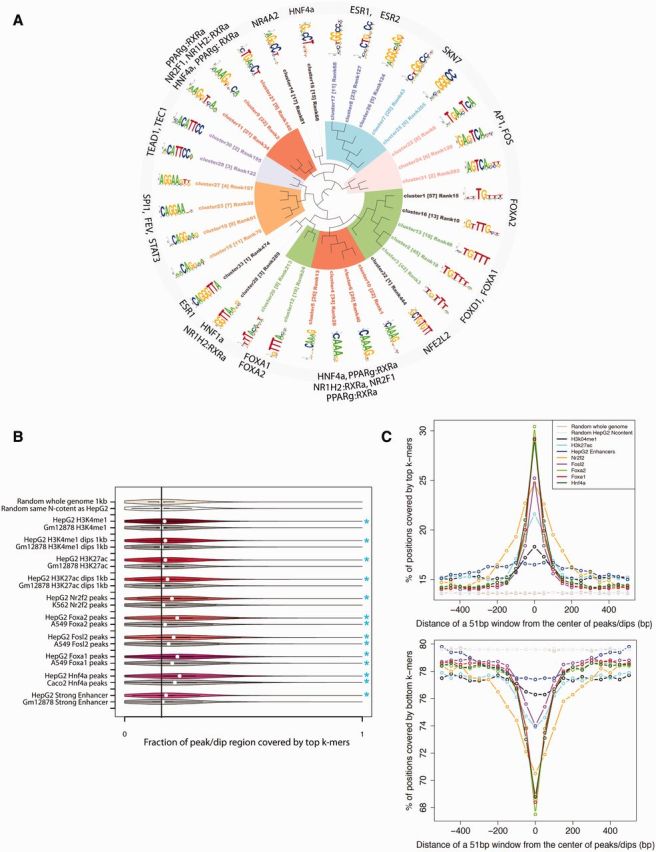
Enriched k-mers in HepG2 enhancers correspond to liver TFBSs. (A) Clusters of 522 top k-mers mapping to known TFBS. Thirty-three k-mer clusters (subclusters) were aligned and merged into 14 motif clusters. STAMP (platform for similarity, tree-building, and alignment of DNA motifs and profiles; Mahony and Benos 2007) identified 22 known TFBS in these clusters, from which 14 are liver-specific TFs. The inner-circle logos are the motifs for each k-mer subcluster, the matched known TFBSs were labeled on the outer circle. The number within the parentheses indicates the number of k-mers in each k-mer cluster. (B) Fraction of a peak region covered by the top k-mers. The top k-mers are enriched in the dip region of HepG2 histone marks as well as in HepG2 strong enhancers, but not in other cell types. The black line indicates the mean value of a random background. The significant enrichment of top k-mers in the peak regions compared with the random background is highlighted by an asterisk (Mann–Whitney test P-value < 0.001). (C) The top k-mers are biased to the center of ChIP-seq peaks of liver-specific TFs and histone marks. In contrast, the bottom 50% k-mers are depleted in the peak centers.
We speculated that if the top k-mers represent the binding sites of liver-specific TFs, they should be capable of differentiating HepG2 enhancers from a random set of sequences. To validate this assumption, we trained a support vector machine (SVM) classifier with a Gaussian kernel on the 14 clusters of the top k-mers, with each feature representing the count of a k-mer cluster in the P300 peaks located outside the HepG2 enhancers (Materials and Methods). P300 is a coactivator and its binding can accurately identify enhancers (Visel, Blow, et al. 2009). The classifier has had an overall accuracy of 0.79, measured as the area under the receiver operating characteristic curve (supplementary fig. S4, Supplementary Material online), indicating that the top k-mers could be used for distinguishing HepG2 enhancers from random sequences.
As a final validation step, we reasoned that if a k-mer is a binding site of a particular TF, that k-mer should be enriched in the ChIP-seq peaks of the TF compared with random sequences. We collected ChIP-seq peaks of 56 TFs available for the HepG2 and other cell lines from the ENCODE project (Bernstein et al. 2012). We also generated random sequences across the whole genome in two different ways: 1) randomly sampling 1-kb sequences; 2) randomly sampling sequences with the same length and repeat content as the HepG2 enhancer sequences. First, we demonstrate that the top k-mers are enriched in HepG2 enhancers and enhancer-associated histone marks (H3K27ac and H3K4me1) compared with random sequences (fig. 1B; Mann–Whitney test, P < 0.0001). We observe a higher top k-mer coverage at the dips of the two histone marks than at the histone marks themselves (as dips of H3K27ac, H3K4me1, and H3K4me2 are often correlated with TF binding [Ernst et al. 2011]). Histone mark enrichment is not observed in other cell lines (Gm12878), further indicating that the top k-mers are likely to be HepG2-specific TFBSs. As for the coverage of top k-mers in the ChIP-seq peaks of 56 TFs, 19 TFs (18 liver-specific TFs and P300) shows a significantly higher top k-mer coverage than expected (fig. 1B and supplementary fig. S5, Supplementary Material online; Mann–Whitney test, P < 0.0001). Notably, the top k-mers were enriched in ChIP-seq peaks of HNF4α, FOXA1, and FOSL2 (a subunit of AP-1) in both HepG2 and at least one other cell line to a very similar extent, suggesting that these k-mers correspond to binding sites of TFs that may be ubiquitously active. In contrast, the top k-mers of the remaining 16 TFs (the 19 TFs excluding HNF4α, FOXA1, and FOSL2), are either enriched in ChIP-seq peaks in both cell lines but with a much higher level of enrichment in HepG2 cell line, such as FOXA2, NR2F2 (fig. 1B), or enriched in HepG2 ChIP-seq peaks only, such as RXRα, P300, SP1 etc. (supplementary fig. S5, Supplementary Material online). According to the clustering result of top k-mers (fig. 1A), the top 522 k-mers mainly capture the TFBSs of six TFs: HNF4α (198 k-mers), PPARγ (170 k-mers), PPARα (165 k-mers), NR1H2 (163 k-mers), NR2F1 (160 k-mers), and FOXA1 (146 k-mers). Combining all the results above, we speculated that the high abundance of top k-mers in ChIP-seq peaks of all other 16 TFs only in the HepG2 cell line could primarily be caused either by their direct or indirect interaction with at least one of the six TFs (HNF4α, PPARγ, PPARα, NR1H2, NR2F1, and FOXA1). For example, in figure 1B, the top k-mers are enriched in both cell types for HNF4α, FOXA1, and FOSL2, but are much less enriched in other cell line for FOXA2 and NR2F2, suggesting that the top k-mers are enriched in FOXA2 due to interactions between FOXA2 and FOXA1 or HNF4α to a large extent. Further investigation of an overlap between all the ChIP-seq peaks of 56 TFs indicates that certain TF pairs such as FOXA1-Nr2f2, FOXA1-FOXA2, FOXA1-HNF4α, HNF4α-HNF4γ, HNF4α-RXRA, and HNF4γ-RXRA tend to bind closer to each other as compared with a random expectation; Nr2f2 has a strong tendency to bind close to many active liver TFs, as does FOXA1 (supplementary fig. S6, Supplementary Material online; Materials and Methods). Both Nr2f2 and FOXA1 tend to bind close to each other as well, suggesting a possible interaction between these two TFs. The ChIP-seq peak regions of the remaining 37 TFs (56 TFs excluding the 19 TFs with top k-mer enriched in the ChIP-seq peaks in at least HepG2 cell line) were not enriched with the top k-mers at least in HepG2 cell line (supplementary fig. S7, Supplementary Material online).
Considering that the ChIP-seq technology does not have a single base-pair resolution, the top k-mers should be concentrated in the center of peaks if they are the binding sites of the TF of the ChIP-seq data set or the binding sites of another TF that is interacting with the TF of the ChIP-seq data set. To validate this assumption, we picked a [−500-bp, 500-bp] interval around the ChIP-seq peak center for the 56 TFs available both in HepG2 and other cell lines, and separated this interval into 21 contiguous 51-bp windows (each two adjacent 51-bp windows had a 1-bp overlap with each other). Further validating our results, the closer a 51-bp window is located to the center of the ChIP-seq peak, the more the window is covered by the top k-mers for P300 and 22 liver-specific TFs (six extra TFs with ChIP-seq data only available in the HepG2 cell lines are also included into this analysis in addition to the 19 TFs mentioned above, three of which were liver-specific TFs: HNF4γ, Mbd4, and Mybl2; fig. 1C and supplementary fig. S8, Supplementary Material online). The top k-mers are also enriched in the centers of HepG2 enhancers and dips of enhancer-associated histone modification marks (H3K4me1 and H3K27ac). On the contrary, the bottom 50% k-mers are depleted in the centers of ChIP-seq peaks (fig. 1C and supplementary fig. S9, Supplementary Material online), which is accordant with the assumption that the bottom k-mers are unlikely to be TFBSs of liver-specific TFs. Neither the top nor bottom k-mers have a location bias in the random control sequences (fig. 1C; supplementary figs. S8 and S9, Supplementary Material online).
Bimodal Mutation Profile of Enhancers
For each position in a potential binding site, all three possible mutations could produce different levels of binding significance change (fig. 2A). The mutations that change a significant k-mer to a background k-mer could imply a possible binding site loss and the mutations in the opposite direction suggest a potential binding site gain. Since either losing or gaining a binding site might lead to a phenotype change, we were interested in both candidate KMs that could lead to a binding site loss and candidate RSs that could lead to a binding site gain. The positions with KMs are called KMPs, and the positions with RSs are called RS positions (RSPs). We paid extra attention to the positions where all three possible mutations would change a significant k-mer to a background k-mer (defined as fragile KMPs [fKMPs], dubbed fKMPs) and the positions where any mutation would change the background k-mer to a significant one (defined as fragile RSPs [fRSPs], dubbed fRSPs). We defined the positions where only one or two mutations were KMs as “stable KMPs” (sKMPs). Analogously, positions with only one or two mutations as RS mutations were defined as “stable RSPs” (sRSPs). Figure 2A shows an example of eight KMPs located with a binding site of FOXA2 and overlapping with a ChIP-seq peak of FOXA2. The positions 1, 3, 4, 5, and 7 are fKMPs; yet, the positions 2, 6, and 8 are sKMPs. One of the fKMPs (chr6-139829666) corresponds to the SNP that was coincided with the HDL cholesterol GWAS trait (SNP ID rs605066) targeted to gene Cited2 (Teslovich et al. 2010; Willer et al. 2013). This position also corresponds to a strongly conserved nucleotide C in the binding site of FOXA2 (fig. 2A) which has been identified to be a major TF mediating HDL cholesterol level via regulation of apolipoprotein M in mouse (Wolfrum et al. 2008) and human (Hu et al. 2012). Therefore, we speculate that this fKMP is carrying a SNP causing a change in the HDL cholesterol level. In addition, this SNP residing in the fKMP is also a proxy of the tag SNP rs592423, associated with the trait of adiponectin levels (Dastani et al. 2012) and another tag SNP rs632057, coincides with trait of mean corpuscular hemoglobin (Kamatani et al. 2010) (fig. 2A). The target gene of these two SNPs is also Cited2 (Kamatani et al. 2010; Dastani et al. 2012). Considering that the chromatin states of the two tag SNPs are weak transcription and polycomb repressed state based on the ChromHMM segmentation (fig. 2A), the SNP rs605066 is also likely to be the causative SNP for the traits of adipopectin levels and mean corpuscular hemoglobin via modulating the FOXA2 regulation of the gene Cited2.
Fig. 2.
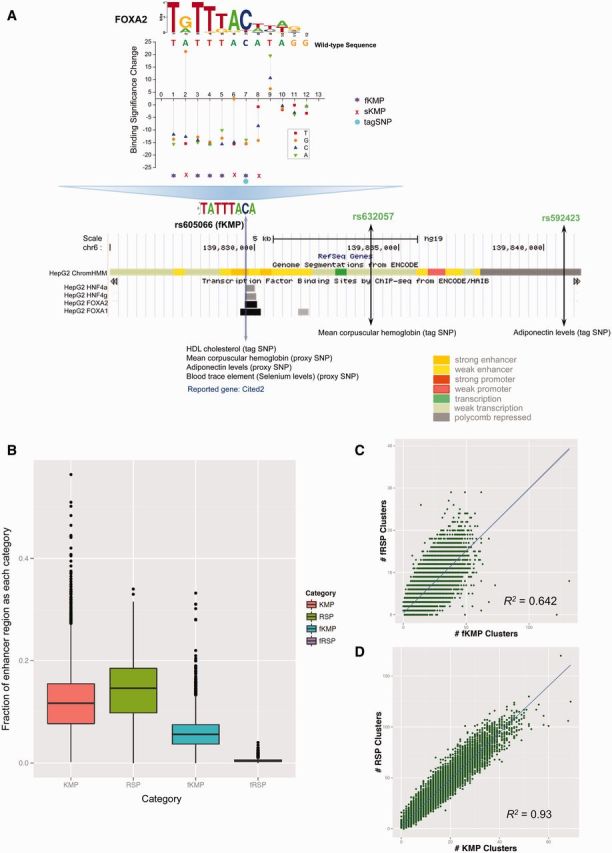
Distributions of KMPs/RSPs in enhancers. (A) Examples of a SNP associated with trait of HDL cholesterol in fKMPs. rs605066 is likely to be a causal SNP for the trait of HDL cholesterol. This position associates with the strongly conserved nucleotide C of the FOXA2 binding site and it does overlap with a FOXA2 ChIP-seq peak. This SNP is also in the LD blocks of two SNPs with liver-related traits (mean corpuscular hemoglobin and adponectin levels). (B) The boxplot demonstrates enhancer content of KMP, RSP, fKMP, and fRSP. (C) The number of fKMP clusters per enhancer is positively correlated with the number of fRSP clusters per enhancer (R2 = 0.642; PCC = 0.8). (D) The number of KMP clusters per enhancer is positively correlated with the number of RSP clusters per enhancer (R2 = 0.93; PCC = 0.96).
In total, 96.7% of HepG2 enhancers have at least one fKMP in them, which is partially a reflection of the top k-mer abundance in HepG2 enhancers (see supplementary Material, Supplementary Material online, for details). On average, 5.7% of HepG2 enhancer positions are fKMPs. Similarly, 87.2% of HepG2 enhancers have at least one fRSP in them. However, only 0.49% of HepG2 enhancer positions are fRSPs (fig. 2B and table 1), indicating that fRSPs are much more sparsely distributed in enhancer regions compared with fKMPs. This greater than 10-fold enrichment of fKMPs over fRSPs in HepG2 enhancers is not unexpected, given that the abundance of active TFBSs in HepG2 enhancers is the source of KMPs destroying these sites (only ∼1% of k-mers of HepG2 enhancers are significant k-mers), while fRSPs correspond to de novo creation of cell-specific TFBSs in the background (∼93% of k-mers of HepG2 enhancers are background k-mers). The amount of sKMPs is similar to fKMPs; however, there are many more sRSPs than fRSPs in HepG2 enhancers (fig. 2B and table 1; supplementary fig. S10, Supplementary Material online). By clustering two fKMPs located within 8 bp from each other and applying the same clustering procedure to fRSPs, we observed that fKMPs tend to form “hot spots” in enhancer regions whereas fRSPs tend to form “singletons” (supplementary fig. S11, Supplementary Material online), which is due to fKMPs being approximately 10-times more abundant than fRSPs. Interestingly, the number of fKMP clusters and fRSP clusters is positively correlated for HepG2 enhancers, as are the KMP and RSP clusters (fig. 2C and D; supplementary fig. S12, Supplementary Material online). In addition, fRSPs are located much closer to fKMP clusters than expected (supplementary fig. S13A, Supplementary Material online). Over 60% of fRSPs are within 10 bp of an fKMP cluster (supplementary fig. S13B, Supplementary Material online). The distance of fRSPs to fKMP clusters is significantly smaller than expected (Mann–Whitney test, P < 2.23e-308, Materials and Methods). Similar location bias is also observed in KMPs clusters and RSPs clusters (supplementary fig. S14, Supplementary Material online). The interdependency between KMPs and RSPs further implies the “restoration” function of RSPs in compensation for the deactivating effects of KMPs on enhancer activity. Our results indicate presence of hot spots of TFBS creation and deactivation in the sequence of HepG2 enhancers.
Table 1.
Statistics of KMPs and RSPs.
| fragile | stable | Total KMP/RSP | |
|---|---|---|---|
| KMP | 1,818,923 | 1,937,095 | 3,756,018 |
| RSP | 139,180 | 4,347,169 | 4,486,349 |
We next studied the clusters of TFBSs overlapping KMPs and examined whether the binding sites of certain TF pairs tended to be lost together (Materials and Methods). To achieve this goal, enrichment of SNPs that are candidate KMs (called KM SNPs) co-occurring in TFBS clusters were analyzed here. We found that many TF pairs, including HNF4A-FOXA1, FOXA1-RXRA, HNF4A-HNF1A, PPARG-RXRA, HDAC2-PPARG, and HNF4A-RXRA, featured more KM SNPs occurring in both TFBSs simultaneously than expected by chance (fig. 3). Most (73.9%) of these KM SNP pairs belong to the same linkage disequilibrium (LD) block, suggesting that a pair of TFBSs is lost in a KM mutant.
Fig. 3.
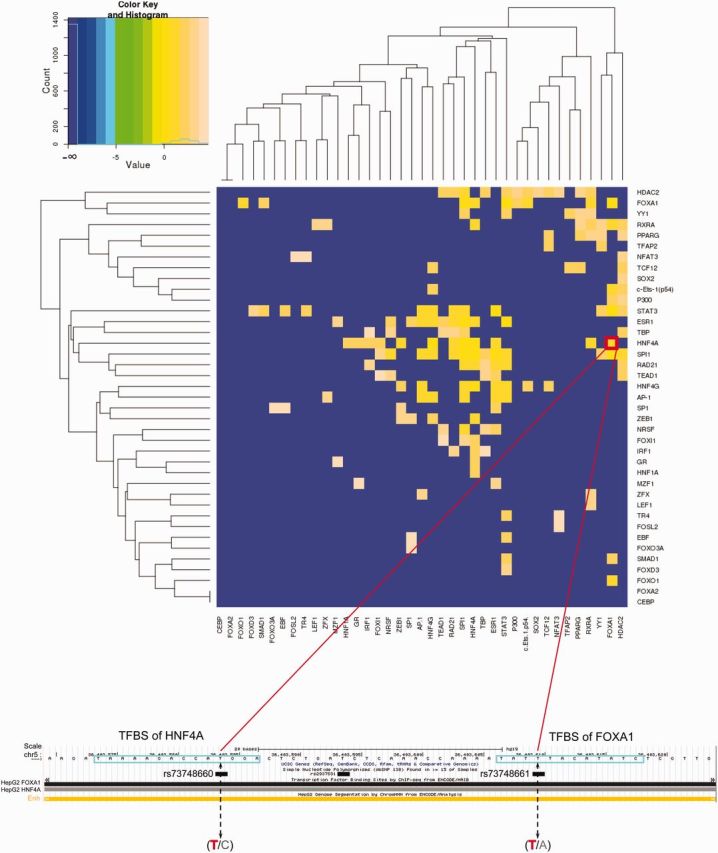
KM SNPs tend to coexist in TFBS clusters. Values in the heat map represent the log (ratio of the fraction of TF pairs both having a KM SNP to the fraction of random TF pairs both having a KM SNP), that is, the log-ratio of proportion of TFBS clusters overlaying KM SNPs associated with certain TF pairs to the product of that overlaying KM SNPs associated with each TF separately (Materials and Methods). The red rectangle focuses on the coexistence of binding sites of HNF4A-FOXA1. The nucleotide colored in red within the parenthesis is the reference allele; the one colored in gray is the alternative allele (KM allele).
Functional Constraints of KMPs and RSPs
Because mutations at either KMPs or RSPs may cause a binding site loss or gain and may lead to phenotype modulations, KMPs and RSPs might bear more functional constraints and be under stronger pressure of purifying selection than other enhancer regions. We therefore first compared the conservation levels of KMPs and RSPs with other positions in an enhancer region. phyloP (Cooper et al. 2005) was applied to measure the conservation level at a single nucleotide resolution. The fKMPs are the most conserved positions over the enhancer region, followed by the sKMPs. The KMPs are significantly more conserved than the RSPs (Mann–Whitney test P < 2.23e-308), which are in turn more conserved than regular enhancer regions (supplementary fig. S15A, Supplementary Material online). To compare the proportion of conserved KMPs and RSPs to the proportion of conserved enhancer nucleotides (excluding KMPs and RSPs), all the positions were separated into two categories (supplementary fig. S15B, Supplementary Material online): 1) conserved (phyloP ≥ 1), in which the fKMPs and sKMPs are strongly enriched (Fisher’s exact test, P < 2.23e-308), followed by fRSPs and sRSPs (Fisher’s exact test, P < 1.78e-110); 2) nonconserved (phyloP ≤ -1), in which the KMPs are depleted (Fisher’s exact test, P < 2.23e-308) whereas RSPs are not. Kircher et al. (2014) developed a scoring framework named CADD that measures the deleteriousness caused by any possible human SNV (Kircher et al. 2014). We implemented the phred-like (Ewing and Green 1998) CADD score (scaled C score) to evaluate the functional constraint on each position (KMP, RSP, and other enhancer position) by averaging the scaled C score of all three possible mutations at that particular position. Similar results are obtained with the CADD framework (fig. 4A and B).
Fig. 4.

The CADD score of KMPs and RSPs is higher than those of enhancer positions. (A) Box plot of CADD scores of fKMP, sKMP, fRSP, sRSP, and enhancer positions, respectively. fKMPs are the most conserved category, followed by sKMPs, fRSPs, and sRSPs. The P-values were calculated using the Mann–Whitney test. (B) KMPs and RSPs are both enriched in the conserved category (scaled C-score ≥ 6), and depleted in the nonconserved category (scaled C-score ≤ 3). The mean values in the legend are the average scaled C-scores for the corresponding categories. Both P-values for enrichment and depletion were calculated using the Fisher’s exact test. An asterisk represents significant enrichment compared with control (enhancers), P < 2.23e-308. A triangle represents significant depletion compared with control (enhancer), P < 2.23e-308. (C) The average C-score of KMP clusters is positively correlated with the average C-score of the nearest RSP cluster. (R2 = 0.79; PCC = 0.89).
Moreover, since a greater binding significance of a particular k-mer indicates a greater likelihood of that k-mer being a functional TFBS, we can quantify the deleteriousness of a KM/RS as a change in binding significance of the k-mer. The smallest binding significance change of the three possible mutations () was used to evaluate the deleterious effect of mutations on the fKMPs/fRSPs (see Materials and Methods for details). A larger value of indicates a greater deleterious effect of mutations on the fKMP/fRSP.
We again used the conservation level (phyloP score) of a single nucleotide to further validate the assumption that the deleterious effect would be a good estimator for the functional constraints on a position, considering that evolutionary constraints often indicate functional constraints. Indeed, there is a positive correlation between conservation level and deleterious effect of fKMP (supplementary fig. S16A, Supplementary Material online). fKMPs, where the mutations would cause larger drops in binding significance, show stronger evolutionary constraints. In contrast, no such clear relationship is observed for fRSPs (supplementary fig. S16B, Supplementary Material online), which might be caused by the relative small number of fRSPs and the small magnitude of the deleterious effect on fRSPs. Moreover, the C-score also has a positive correlation with the deleterious effect of fKMPs (supplementary fig. S17A, Supplementary Material online), whereas the correlation disappears again when dealing with RSPs (supplementary fig. S17B, Supplementary Material online).
To further examine the coordination between KMPs and RSPs, RSP clusters were assigned to their most proximal KMP clusters, then the assigned RSP clusters and the KMP clusters were denoted as correlated pairs. We observed that SNPs are more likely to exist at the correlated pairs simultaneously compared with random pairs of KMP clusters and RSP clusters (fold enrichment = 1.28, binomial test P-value = 3.42e-12; Materials and Methods), with approximately 52.7% of the coexisting SNP pairs located in a single LD block. The correlated KMP clusters and RSP clusters seem to have similar levels of functional constraints (average C-scores) (fig. 4C). The fRSPs were also assigned to the fKMPs by the same principle. The associated fKMP-fRSP cluster pairs also tend to host similar levels of functional constraints (supplementary fig. S15C, Supplementary Material online) and are more likely to carry SNPs simultaneously than random pairs (fold enrichment = 1.33, binomial P-value = 0.11) (Materials and Methods). The similar levels of functional constraints and co-occurrence of SNPs at the correlated KMP clusters and RSP clusters suggest that if a binding site is lost due to single-nucleotide mutation, a new binding site nearby is likely to be gained by another single-nucleotide mutation. In this way, the organism might maintain its function simply through reordering the binding sites in enhancer regions.
KMs Have the Most Deleterious Effect on Enhancer Activity
We used the results of a massively parallel reporter assay (MPRA) experiment (Kheradpour et al. 2013) to directly evaluate the disruptive effects of our predictions. The original study tested 2,104 145-bp enhancer sequences containing manipulated target motifs of HepG2-specific TFs (HNF1, HNF4, FOXA) and K562-specific TFs (GATA, NFE2L2). There were five single-nucleotide mutations on each enhancer sequence including max 1-bp decrease, least 1-bp change, max 1-bp increase, and two separate random 1-bp changes on the 145-bp enhancers. These motif manipulations were conducted in the assay because they reduce, improve, make the smallest change and make random changes, respectively, to the PWM match score. To take full advantage of the MPRA data set and to strengthen the statistical significance of our results, we also applied our framework in K562 cell line to predict KMPs/RSPs. Considering that top K562-enriched k-mers chiefly represent potential binding sites of GATA and NFE2L2, whereas the top k-mers in HepG2 mainly consist of potential binding sites of HNF4α and FOXA1, we only selected the corresponding enhancer sequences containing motif instances of GATA, NFE2L2, HNF4, and FOXA.
After mapping the 580 engineered enhancer variants to our predictions (Materials and Methods), we grouped them into four categories: 153 KMs at fKMPs, 71 KMs at sKMPs, 38 RSs (3 RSs at fRSPs), and 151 controls (mutations that keep a k-mer within the insignificant set). A total of 167 variants in the engineered enhancers did not belong to the above three categories of our predictions (the details of their effect on enhancer activity are provided in supplementary materials, Supplementary Material online).
We first compared the effect sizes (magnitude of enhancer activity change measured as a drop of the normalized expression value) of mutations in each category with the control and investigated whether there was a correlation between the effect size of a mutation and the binding significance change caused by the mutation. Because the scale of the binding significance score depends on the size of the null distribution of enhancers, to facilitate quantifying the change of significance values (−log10 of P-value) without a large standard error, we defined a phred-like (Ewing and Green 1998) significance score (scaled significance score, Materials and Methods) ranging from 0 to 45, on the basis of the rank of each k-mer, to interpret the decrease of the binding significance caused by a mutation. As shown in figure 5A, the enhancers with KMs at fKMPs feature the most pronounced decrease in enhancer activity (Mann–Whitney test P = 3.765e-05 as compared with the control set), followed by KMs at sKMPs (Mann–Whitney test P = 0.0082). In contrast, the RS mutations do not show a significant increase in enhancer activity (Mann–Whitney test P = 0.127). The smaller effect size of RSs compared with KMs might be partly due to the smaller magnitude of binding significance changes caused by the RSs compared with those caused by KMs (fig. 5A). On the other hand, this result suggests enhancers might cease their activity by losing a specifically positioned active binding site which might be a part of a complex enhancer structure, as opposed to gaining a randomly positioned binding site which might not be sufficient to create a functional enhancer alone.
Fig. 5.
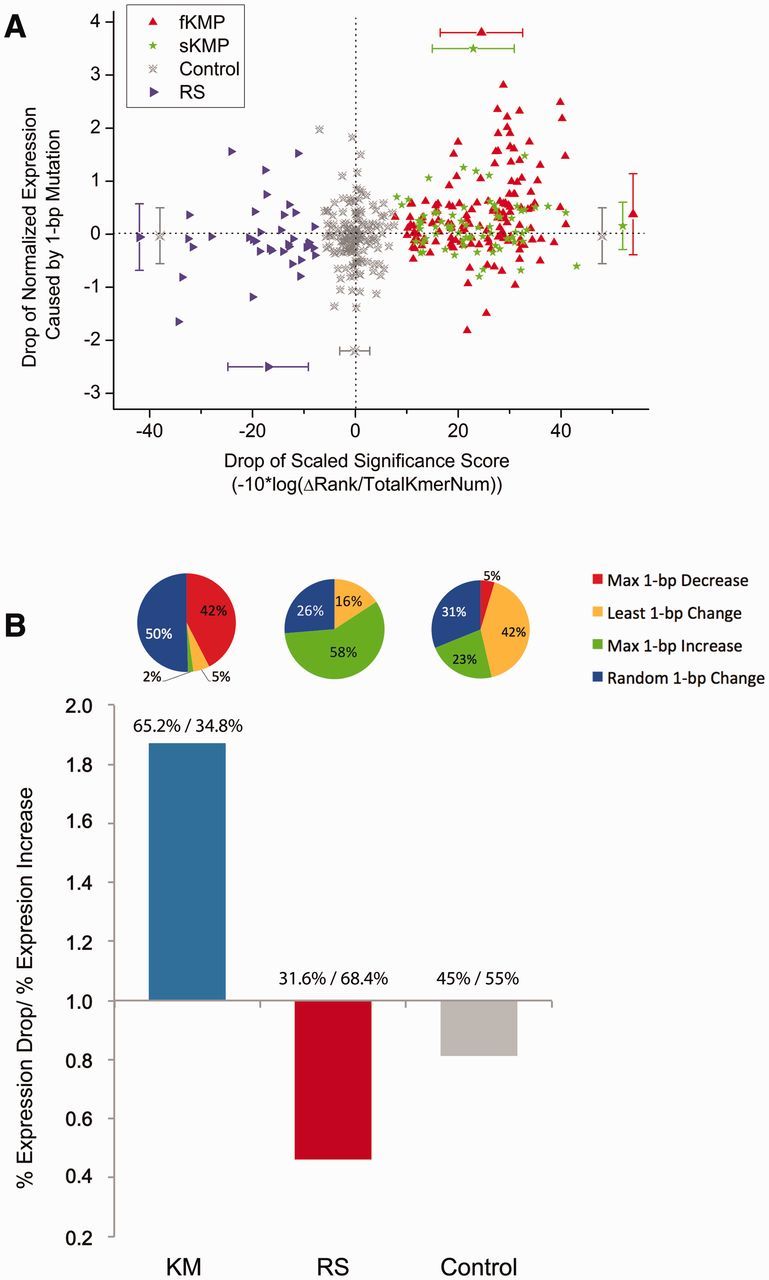
Candidate KMs have the most deleterious effect on enhancer activity. (A) Relationship between the drop of the scaled significance score and the drop of the normalized expression level of a 145-bp enhancer. The y axis is the normalized expression level drop caused by a 1-bp mutation, and x axis is the drop of the scaled significance of the mutated k-mer relative to the original k-mer. Error bars indicate the mean and the standard deviation of the corresponding axis in each category. (B) Tendency of decreased expression of each predicted mutation category. The y axis is the ratio of proportion of mutations that decrease expression to that of mutations that increase expression. The fraction above each bar indicates the ratio. The numerator is the percentage of mutations leading to a decrease in expression, the denominator is the percentage of mutations that increase expression. KM: fKMPs and sKMPs. RS: fRSPs and sRSPs. The pie chart above each bar indicates the distribution of the PWM manipulations in our predicted mutation categories.
To understand the relationship between the effect size and the deleterious effect of mutations at fKMPs (as defined previously) we binned fKMPs into four percentile intervals of deleterious effect: [0, 5%], [5%, 25%], [25%, 50%], [50%, 100%]. Then we mapped the 153 experimentally characterized KMs at fKMPs to the four intervals. As expected, the fKMPs with greater deleterious effects tend to have larger effect sizes in deactivating enhancers (supplementary fig. S18, Supplementary Material online), which is accordant with the previous conclusion that the fKMPs with larger deleterious effect might bear more functional constraints and, therefore, tend to have a greater disruptive effect on enhancer activity once mutated.
Secondly, we examined the tendency of mutations to diminish the enhancer activity in all four categories of engineered enhancer variants by comparing the proportion of mutations that decreased the expression levels of enhancers. As expected, the KMs (at both fKMPs and sKMPs) have the largest portion (65.2%) of mutations that reduce enhancer activity. In contrast, the RSs have the smallest portion of mutations that reduce enhancer activity (31.6%) (fig. 5B; supplementary table S2, Supplementary Material online).
In summary, these results show that KMs and RSs are associated with decrease and increase in enhancer activity, respectively. Across all the SNVs, the KMs have the largest proportion of mutations that deactivate enhancers. The effect size in enhancer activity disruption by KMs is larger than the one in increasing enhancer activity of RS mutations.
HepG2 KMs Are Enriched in Liver-Related GWAS Traits
To test whether HepG2 KMs are correlated with liver disorders and traits, we used the NHGRI GWAS Catalog (Welter et al. 2014) to assess the association between KMs and liver-related diseases (see Materials and Methods). Once overlapping our predictions with the GWAS LD blocks, the KM SNPs are strongly associated with the liver-related traits such as HDL-cholesterol, triglycerides, adiponectin levels, liver enzyme levels, blood trace elements (see levels), type I diabetes, and several other liver-related traits when compared with either all SNPs and enhancer SNPs (fig. 6; supplementary tables S3 and S4, Supplementary Material online). As an example, the KM SNP rs10422861 located within the ChIP-seq peaks of FOXA1, FOXA2, HNF4α, and HNF4γ resides in the intronic region of the gene PEPD. The KMP SNP is in strong LD with a tag SNP rs3786897 of the trait Type II diabetes (Cho et al. 2012), which is also an eQTL linked to gene PEPD (Schadt et al. 2008). Although the tag SNP rs3786897 is also located in the same enhancer region, but it does not overlap with other HepG2 ChIP-seq peaks. This KM SNP is also a proxy of another tag SNP rs731839 associated with traits of triglycerides, HDL cholesterol, and adiponectin levels, which is also linked to the gene PEPD (Willer et al. 2013; supplementary fig. S19, Supplementary Material online). All these results suggest that the KM SNP rs10422861 is the causative SNP modulating the regulation of the gene PEPD, and has a strong correlation with type II diabetes, HDL cholesterol, triglycerides, and adiponectin levels (supplementary fig. S19, Supplementary Material online). In another example, the KM SNP rs6037083, located within the ChIP-seq peaks of FOXA1/FOXA2/AP1, is in strong LD with a tag SNP rs7267979, associated with the trait of liver enzyme levels, and targeted to gene ABHD12 (Chambers et al. 2011). This tag SNP is also an eQTL linked to the expression level of gene ABHD12 (Schadt et al. 2008), which is located in the intronic region of ABHD12, with an expression state based on the ChromHMM prediction. Similarly, it is highly likely that the KM SNP rs6037083 is the causal SNP of the trait liver enzyme levels by regulating the expression of gene ABHD12. Another two examples are also listed in supplementary figure S19, Supplementary Material online, both of which also suggest the strong association between KMs and liver diseases/phenotypes.
Fig. 6.
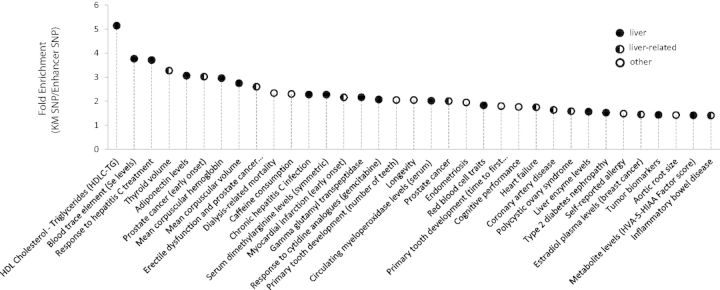
Enrichment of GWAS traits in KM SNPs relative to enhancer SNPs. The y axis is the ratio of fold enrichment of KM SNPs as compared with random expectation to the fold enrichment of enhancer SNPs as compared with random expectation. Only the top 35 enriched GWAS traits are presented here. Full results are presented in supplementary table S3, Supplementary Material online.
KMs Play an Important Role in Differential TF Binding and Affect Local Chromatin Accessibility
KMs are likely to disrupt the enhancer activity through altering binding fitness of major TFs. Therefore if KMs are a crucial factor for differential TF binding, KMs should be enriched in the differential TF-binding regions. We studied the enrichment of KMs in variable TF-binding regions from two mother–father–daughter trios (supplementary table S5, Supplementary Material online) in the 1000 Genomes Project (Abecasis et al. 2012). TF-binding data were available for lymphoblastoid cell lines (LCLs). We redid our analysis on LCL and identified the most overrepresented k-mers, which were subsequently mapped to TFs whose motif variants have the most deleterious impact on enhancer activity in LCLs (Materials and Methods). In total, 498 significant k-mers were identified representing the binding sites of major TFs in LCLs including PU.1 and IRF. PU.1 (also known as SPI1) is an essential TF that plays key roles in differentiation and proliferation of B-lymphocytes (Lloberas et al. 1999). Therefore, we picked PU.1 as an example to study the effect of KMs on differential TF binding. Next, we identified PU.1 KMs in both the maternal and paternal genomes (Kasowski et al. 2013; Materials and Methods) using 313 significant k-mers and 31,370 background k-mers.
The PU.1 ChIP-seq peaks were first separated into three categories based on the comparison between any two individuals: 1) individual-specific peaks (peaks that exist in only one individual), 2) differential peaks (peaks that exist in both individuals but have differential binding signals), and 3) similar peaks (peaks that exist in both individuals and have similar binding signals). We applied Homer (Heinz et al. 2010) to systematically identify PU.1 peaks belonging to these three categories (Materials and Methods). Compared with the similar ChIP-seq peaks, the KMs are significantly enriched in both individual-specific and differential ChIP-seq peaks (P-value ≤ 1.02e-06; fig. 7A and B; supplementary fig. S20, Supplementary Material online).
Fig. 7.
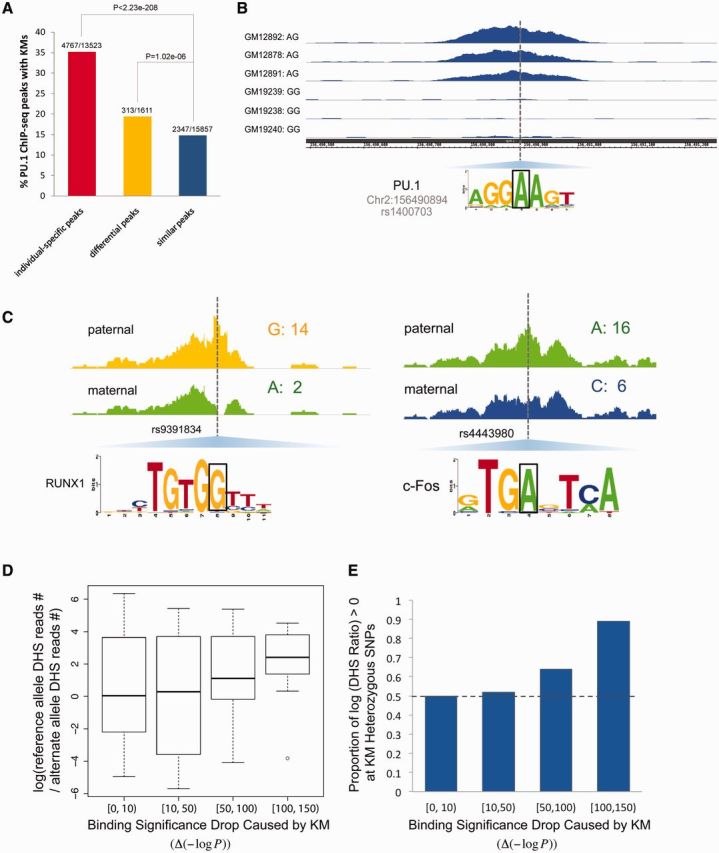
Candidate KMs affect TF binding and modulate local chromatin accessibility. (A) KMs/RSs are enriched in the differential and individual-specific ChIP-seq peaks of PU.1. P-values were calculated based on the Fisher’s exact test. The fraction above each bar indicates the percentage of peaks in the corresponding category containing KMs. The numerator is the number of peaks with KMs, the denominator is the number of peaks (B). One example showing that KM/RS is the causal variant that causes different PU.1 binding. The regions shown here are 1.2 kb long. (C) Examples of allele-specific DNase I sensitivity in GM12878 for KMs that disrupt TF binding (801-bp windows centered on a heterozygous KM SNP). In total, 299 heterozygous SNPs with at least 11 DNase-seq reads are KMs. The number after each nucleotide indicates the number of reads associated with that nucleotide. (D) Correlation between local accessibility and binding significance. The y axis is the log odds ratio of DNase I read coverage of the reference allele to that of the alternative KM allele. The x axis is the decrease in binding significance of the original reference allele caused by a KM allele. (E) Proportion of positive log odds ratio of DHS coverage (reference allele/KM allele) versus binding significance drop caused by a KM allele.
Although we do not know whether chromatin modification is the cause or the consequence of the change in TF binding, one mechanism that may be important is that the functional variants that alter a TF recognition sequence also frequently alter the local chromatin accessibility (Maurano, Wang, et al. 2012; Kasowski et al. 2013). Therefore, we speculated that the KMs that disrupt TF binding should be enriched in the dsQTLs, which are regions of genomic variants typically affecting chromatin accessibility in a range of about 200–300 bp (Degner et al. 2012). There were 32,728 heterozygous KM SNPs in the maternal and paternal genomes of the two trios (Materials and Methods), 1,082 of which were dsQTLs. As for the 826,362 enhancer SNPs, 10,864 were dsQTLs. Hence, consistent with our expectation, the heterozygous KM SNPs were strongly enriched in dsQTLs relative to enhancer SNPs with a 2.51-fold enrichment (Hypergeometric test P-value = 1e-164). The enhancer SNPs were also strongly enriched in dsQTLs compared with the random SNP sets with a 2.56-fold enrichment (binomial test P-value < 2.23e-308) (supplementary fig. S21A, Supplementary Material online). We further evaluated the imbalance in the fraction of DNase-seq reads obtained from each allele in heterozygous KM SNPs to manifest the affect of KMs on the local chromatin state. We detected 299 heterozygous KM SNPs in GM12878 with sufficient DNase-seq reads coverage (at least 11 reads covered each heterozygous KM SNP) within the high-confidence DHSs (at a false discover rate [FDR] of 5%; Materials and Methods). It seemed that the KM SNPs are associated with the local chromatin states, with the higher binding significance allele (reference allele) exhibiting higher accessibility (fig. 7C). KM alleles that are more deleterious are less likely to exhibit open chromatin states, and have more DNase-seq reads associated with the reference allele on the heterozygous KM loci (fig. 7D and E). This imbalance in the reads obtained from each allele is indicative of the negative effects of the KMs on local chromatin accessibility. Figure 7C shows two examples of heterozygous KM SNPs that would disrupt TF binding and affect local chromatin accessibility. The SNP rs9391834 (G/A) associated with a putative binding site of RUNX1 located in the intronic region of gene HLA-B, which is the top susceptibility gene for psoriasis (Tiilikainen et al. 1980; Nair et al. 2006). In addition, a previous work indicated that dysregulation of two target genes (SLC9A3R1 or NAT9) by RUNX1 is a susceptibility factor for psoriasis based on the study on cohorts of psoriasis patients from the United States (Helms et al. 2003). Therefore, it is likely that the KM allele A of rs9391834 might affect the expression of psoriasis-associated gene HLA-B through disrupting the binding of RUNX1 and modulating the local nucleosome occupancy around its binding site. As another example, the KM SNP rs4443980 (A/C) was associated with the putative binding site of c-Fos (component of AP1), which may participate in B cell differentiation (Corcoran 2005; Ohkubo et al. 2005). By altering the binding affinity of the TF c-Fos and the local chromatin structure, the KM allele C might be the causative allele associated with affecting B cell differentiation and the autoimmune system.
eQTLs are genomic regions that are correlated with a change in the level of gene expression (Rockman and Kruglyak 2006). An extensive number of studies have characterized the level and patterns of regulatory variation and eQTLs over the last decade (Morley et al. 2004; Cheung et al. 2005; Stranger et al. 2005, 2012). eQTLs have been studied in diverse tissues and cell lines (Myers et al. 2007; Emilsson et al. 2008; Schadt et al. 2008; Grundberg et al. 2009; Stranger et al. 2012). More importantly, it has been discovered that as many as 55% of eQTLs are also dsQTLs (abbreviated as eQTL-dsQTLs) (Degner et al. 2012), leading us to the possible underlying mechanism by which eQTLs affect gene expressions: when the alternative alleles at a particular heterozygous SNP site cause allele-specific TF binding or different nucleosome occupancy at the TF-binding regions, this in turn might cause allele-specific differences in the rates of transcription (Degner et al. 2012), thus further lead to allele-specific gene expression levels. Interestingly, the heterozygous KM SNPs are 2.62-fold enriched in eQTL-dsQTLs compared with enhancer SNPs (supplementary fig. S21B, Supplementary Material online; hypergeometric test P-value = 1.3e-39). The enrichment of KM SNPs in eQTL-dsQTLs indicates that the KMs are likely to affect the gene expression level through modulating affinity of TF binding as well as changing the local chromatin accessibility.
KMs at fKMPs Significantly Affect Cell Type-Specific Gene Expression
Next, considering the lack of a genome-wide map of chromatin structure that links enhancers to their targeted genes in LCL, we investigated the potential effects of the KMs on the TS gene expression by studying the enrichment of TS eQTLs at the fKMPs. We utilized a data set of eQTLs in LCLs with associated gene expression data of the CEU populations from the HapMap3 project (Stranger et al. 2012) to uncover the potential effects of KMs on gene expression. Since the eQTLs and the associated genes are only available for two individuals (GM12891 and GM12892) of the two trios in our study, we included only the data of these two individuals into our analysis (supplementary table S6, Supplementary Material online). Unlike housekeeping genes that are ubiquitously expressed and perform basic cellular functions, the TS genes are highly expressed only in a few tissues. Due to the limited knowledge of the genomic features and mechanism responsible for expression of TS genes, we selected the 2,000 most highly expressed genes in LCL based on the genome-wide expression profile of the Human U133A/GNF1H Gene Atlas (Su et al. 2004) and considered them as the potential TS genes in LCL. By mapping the common SNPs (MAF ≥ 0.01) from the 1000 Genome Project and the eQTLs associated with the 2,000 TS genes of LCL to the fKMPs and enhancers, we found that the TS-gene-associated eQTLs are 2.6-fold enriched in enhancer SNPs relative to sets of matched random SNPs (binomial test, P = 6.9e-131), indicating that the causal variants for cell type-specific gene expression tend to be located in enhancer regions. Meanwhile, the eQTLs associated with the TS genes in LCL are 3.6-fold enriched in fKMP SNPs compared with enhancer SNPs (Hypergeometric test, P = 3.4e-17). The enrichment of eQTLs in SNPs located at fKMPs is even greater when the 1,000 most highly expressed genes are considered (fold enrichment = 3.9, hypergeometric test, P = 1.78e-10) (supplementary fig. S22 and table S7, Supplementary Material online). The results indicate that fKMPs are more enriched in eQTLs associated with TS genes as compared with random enhancer SNPs, suggesting an essential role of fKMPs in regulation of TS genes.
Discussion
We developed a framework to identify essential positions in enhancer sequences, which are likely to have either a deleterious (KM) or advantageous (RS) effect on the function of the enhancers if mutated. KMPs and RSPs that we identified in HepG2 enhancers are strongly associated with the functional binding sites of several major liver-specific TFs including HNF4α, FOXA1, PPARα/PPARγ, AP-1 (FOSL2), NR1H2, and NR2F1. Our framework is capable of identifying binding sites of essential TFs within any set of ChIP-seq enhancers, where mutations are likely to impact enhancer activity and affect the precise pattern of TS gene expression and cause a phenotype change.
We observed that the vast majority of HepG2 enhancers contain at least one KMP, and both KMPs and RSPs are more conserved than other enhancer regions during the course of evolution. Fragile fKMPs and RSPs show even greater functional constraint. The stronger pressure of purifying selection acting on these positions confirms that mutations at these positions are likely to result in a functional outcome. KMPs are more conserved than RSPs, and they both tend to be located near each other, forming mutational hot spots in enhancer sequences. We also found that the conservation level between the nearest KMP-RSP cluster pairs is strongly correlated, with SNPs coexisting in cluster pairs, suggesting binding site reshuffling. The significant correlation between these two types of mutations suggests a bimodal mutation system in the regulatory genome, which is shaped by evolutionary forces: an active binding site can be gained rapidly in the proximity of “fragile” enhancer regions hosting deleterious nucleotides. An organism might utilize this bimodal cis-regulatory mechanism to maintain enhancer activity during evolution.
We also observed that KM SNPs are likely to co-occur in the clusters of TFBSs overlapping KMP hot spots, suggesting that the major TFs with deleterious motif variants tend to lose their binding sites together.
A vital part of a genomic association study is the identification of causal genetic variants. Based on our findings, eQTLs that are linked to TS genes are significantly enriched in fKMPs, indicating that fKMPs should be the primary candidates in the search for causal noncoding variants. The causal role of fKMP mutations was further supported by their impact on the level of gene expression based on our analysis of the massively reporter assay (Kheradpour et al. 2013). Although the in vivo scenario of the regulatory circuit might be determined by multiple combinatorial factors rather than a single enhancer, we can still propose a solid conclusion that mutations at fKMPs are most likely to deactivate enhancers and have the largest effect size on reducing enhancer activity as compared with other genetic variants in the enhancer region. In addition, KMs at the fKMPs with higher deleterious effect tend to have a greater impact on the level of gene expression. Due to the most deleterious variants at fKMPs, enhancers would easily cease their activity. However, it would not be so easy to create a functional enhancer by simply gaining a randomly positioned binding site, although the RS mutations at RSPs do have the largest portion of mutations increasing the enhancer activity. Additionally, the functional analysis of GWAS SNPs indicates that the KM SNPs are strongly associated with TS traits/diseases, suggesting the causative role of KMs in underlying TS phenotype and disorders.
Our previous work (Huang and Ovcharenko 2015) reported enrichment of KM SNPs at the allele-specific TFBSs, but the underlying mechanism for the allele specificity in chromatin structure had not been fully studied. This motivated the analysis of the association between KMs and open chromatin accessibility in our study. We observed that KMs and RSs are likely to be the causal variants that underlie local chromatin modifications and differential binding of TFs. In summary, by altering the local chromatin accessibility of the TFBSs and the binding affinity of the TFs, KMs, and RSs are likely to affect the transcription rate and therefore decrease and increase enhancer activity, respectively. This conclusion was further supported by the enrichment of dsQTL-eQTL in the heterozygous KM/RS SNP sites.
Unlike CADD (Kircher et al. 2014), which annotates all possible genetic variants based on a single score (C-score) generated by integrating diverse annotations into a single measure, our pipeline prioritizes genetic variants in enhancer regions and focuses on identifying causal variants which have the most deleterious effects on the cis-regulatory role of enhancers given a specific tissue/cell type. These variants are more likely to be the pathogenic ones and could determine the TS gene expressions. By determining the KMPs/RSPs that are strongly correlated with loss and gain of cis-regulatory elements of the major TS TFs, the proposed approach is likely to identify driver mutations that underlie the TS diseases and phenotype divergence. However, our framework does have several limitations. First, although the MPRA data facilitate the validation of the deleterious effects of KMs/RSs, to further limit false positives in our predictions, large-scale gold-standard experimental data on the noncoding regions are in a great need. Second, the overall precision of KMs in predicting differential/individual-specific binding is 68%, whereas the sensitivity of KMs in predicting differential/individual-specific binding is low (35% for individual-specific binding, 19% for differential binding; fig. 7A). This may be caused by the complex scenarios of TF binding, which cannot be completely explained by a simple disruption of a canonical binding site. It is known that a large portion of TFs that bind to noncanonical motifs or interact with a partner which binds to either a canonical or noncanonical motif (Wang et al. 2012). Either scenario cannot be detected by our framework since our approach targets deleterious mutations in canonical motifs. Future work could expand our framework to include the local mutation rate, nucleotide divergence, and genomic data across different species in order to identify pathogenic and causal variants which underlie phenotype divergence during evolution. We hope our framework will provide the research community a valuable source for the study of the phenotype divergence across species during evolution in near future.
Materials and Methods
Data Availability
We used the GRCh37 (hg19) assembly of the human genome, which we downloaded from the UCSC Genome Browser (Kent et al. 2002).
Putative strong enhancers no longer than 3-kb predicted by ChromHMM (Ernst and Kellis 2012) were used as the training set for the enrichment analysis of k-mers. HepG2 and LCLs (GM12878) ChromHMM strong enhancers and ChIP-seq peaks (narrowPeak format) of 62 TFs were downloaded from the USCS Genome Browser (Kent et al. 2002; http://genome.ucsc.edu/, last accessed May 23, 2015). HNF4α ChIP-seq data in differentiated Caco-2 cells were downloaded from NCBI Gene Expression Omnibus (GEO) (accession number: GSM575229).
For the analysis in LCLs, our study targeted individual genomes of two father–mother–daughter trios (supplementary table S5, Supplementary Material online). The first trio was of Utah residents of European ancestry (CEU), and the second was Yoruban from Ibadan Nigerian ancestry (YRI). Genetic variation data were downloaded from the 1000 Genomes Project (Abecasis et al. 2012) and dbSNP 138 (Sherry et al. 2001). Genomic coordinates of SNPs were mapped from hg18 to hg19 using the UCSC liftOver tool (Hinrichs et al. 2006). The personal genomes of each individual of the two trios were obtained by overlaying the maternal and paternal SNPs haplotypes onto the hg19 genome (Kasowski et al. 2013). As for the differential binding of PU.1 in LCLs, we used the ChIP-seq data of PU.1 from the study in which the ChIP-seq reads were aligned against the maternal and paternal genomes of the corresponding individual (Kasowski et al. 2013). The DNase I data used in the analysis of local chromatin accessibility in GM12878 were downloaded from GEO with accession number GSE29692. This DNase I data includes reads mapped to hotspot DHS at an FDR threshold of 5% (Maurano, Humbert, et al. 2012). For the prediction of KMs/RSs of maternal and paternal genomes, fKMPs/fRSPs in LCL ChromHMM strong enhancers and the analysis of enrichment of eQTLs in fKMPs/fRSPs, we used the ChromHMM strong enhancers of the two individuals from the study conducted by Kasowski et al. (2013). The eQTL data with linkage to associated genes for CEU populations were obtained from a study (Stranger et al. 2012), and the gene expression profile of 79 tissues was downloaded from BioGPS (Wu et al. 2009, 2013) (accession number: GSE1133).
k-mer Analysis of Enhancer Sequences
We generated a set of controls for each ChromHMM strong enhancer sequence. Controls were randomly sampled from the whole genome with the same GC-content, repeat-content, and length as the corresponding enhancer sequence. Twenty-four control sequences were extracted for each enhancer. In cases when not enough controls with our strict criteria (ΔGC-content ≤ 0.005, Δrepeat-content ≤ 0.01) could be identified, we created additional controls by reshuffling enhancer sequences.
We use k-mers to identify potential binding sites in enhancers. We determined the optimal length of k-mers by considering the trade-off between sensitivity and specificity. The sensitivity of k-mers could be evaluated by coverage of known TF binding motifs in the TFBS database such as TRANSFAC (Matys et al. 2006) and JASPAR (Mathelier et al. 2014). We observed that the informative regions of 78% of known TF-binding motifs are best modeled by 8-mers (supplementary fig. S23, Supplementary Material online). In a previous study (Cowper-Sal lari et al. 2012), 8-mers have been used to successfully identify causative SNPs in breast cancer cell lines Gorkin et al. (2013) also used 6-mers to build a classifier for enhancer prediction in melanocytes. To test the specificity of 8-mers, we compared the enrichment of the regulatory domains of liver-specific genes in KMPs identified by 6-mers, 8-mers, and 10-mers, respectively. We observed that KMPs identified using 8-mers have the highest enrichment in the regulatory domains of liver-specific genes (the most highly expressed genes in liver relative to other tissues, supplementary material and fig. S24, Supplementary Material online). Therefore, for each of the possible 32,896 k-mers (k = 8), we used the Fisher’s exact test to evaluate enrichment of k-mers in the HepG2 ChromHMM strong enhancer set and identified the top 522 k-mers significantly enriched in enhancers (P ≤ 1e-3 after Bonferroni correction; supplementary table S1, Supplementary Material online) as potentially functional k-mers and 30,647 insignificant k-mers (P > 1e-3 without Bonferroni correction) as background k-mers. Finally, the significance of the P-value (–log10(P-value)) was used to estimate the functional constraint of a k-mer. The putative strong enhancers predicted by ChromHMM (Ernst and Kellis 2012) were chosen as the positive set because these DNA segments correspond to histone marks H3K4me1 and H3K27ac correlated with transcriptionally active chromatin and high expression level of associated genes (Zentner et al. 2011). For the k-mers enriched in HepG2 enhancers with high significance (small P-value), there is only a small chance of finding them in a random sequence. Therefore, the top k-mers are likely to represent binding sites of active TFs in the HepG2 cell line. In other words, the statistical measure (–log10(P-value)) could be a good estimation for the binding fitness of the k-mer.
To remove redundancy among the top k-mers and identify associated motifs, we clustered the top k-mers in two steps: the first step was to cluster the k-mers without alignment, and the second step was to align k-mer clusters and map the aligned k-mer clusters to known TFBSs. To calculate the similarity between any two k-mers, each k-mer was treated as a node in the graph, there would be an edge connecting two nodes if the two k-mers share at least five dimers without alignment (two dimers in both k-mers would be considered the same if and only if they have the same letter contents and are located in the same position in the two k-mers), that is, the similarity score (formula 2) between the two corresponding k-mers need to be no smaller than 5/(8−1). We next applied the Markov Cluster Algorithm (MCL) algorithm (Dongen 2000; van Dongen and Abreu-Goodger 2012) to find clusters on the graph with each node representing a k-mer. The motif profiles generated by k-mer MCL clusters were further aligned and merged and matched to the known TFBS database, including JASPAR (Mathelier et al. 2014) and TRANSFAC (Matys et al. 2006), using the web-based tool STAMP(Mahony and Benos 2007).
| (2) |
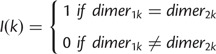 |
(3) |
where
dimerik is the di-mer starting from the kth position of the ith k-mer
We also built an SVM classifier on the 14 clusters of the top k-mers to validate their ability to discriminate enhancers from controls. Since the 522 k-mers were picked due to their enrichment in ChromHmm HepG2 enhancers, we used the 21,944 P300 peaks located outside ChromHmm HepG2 enhancers for the 5-fold cross validation test. For each P300 ChIP-seq peak, we randomly sampled 24 × control sequences genome-wide with the same length and GC- and repeat content. We used a Gaussian kernel SVM with a vectorized representation of sequences, with each feature representing a k-mer cluster, considering that the number of features (14 top k-mer clusters) is significantly smaller than the number of data points (21,241 strong enhancers). We applied the package libsvm (Lin 2011) to build the classifier.
To study the potential effect of candidate KMs on differential binding of the TF PU.1 in LCLs, we applied the same pipeline to PU.1 ChIP-seq regions in the two trios to identify its potential binding sites, resulting in 313 significant k-mers and 31,370 insignificant k-mers. In the LCLs dsQTL and eQTL analysis, for the prediction of top k-mers in LCLs, the same pipeline was performed for GM12878 (one individual in LCL cell lines) ChromHMM strong enhancers and 498 significant k-mers and 30,741 insignificant k-mers have been identified.
Candidate KMs and Candidate RMs
Once we identified top k-mers in the positive training set, we applied a modified IGR model (Cowper-Sal lari et al. 2012) to predict mutations with potential phenotypic effects in HepG2, K562, and LCL ChromHMM strong enhancers.
IGR analyzes k-mer composition change caused by a mutation to estimate a change in TF-binding affinity. For both the wild type and derived nucleotides, there were eight 8-mers associated with each nucleotide, respectively. The highest scoring k-mer (maxima k-mer) was extracted from the set of k k-mers overlapping the position of the mutation. TF-binding affinity was estimated as a genome-wide average TF ChIP-seq signal for each of the two k-mers and Student’s t-test was used to estimate the affinity change between these k-mers (see [Cowper-Sal lari et al. 2012] for details). Same as IGR, our approach was constructed based on the model that if a canonical binding motif is altered by a mutation, there should be no alternative binding motif in the immediate proximity of the mutation in either orientation. In our study, we utilized the same approach for selection of maxima k-mers and comparison between maxima k-mers for each allele given the genomic context of the mutation position as in the IGR approach, except that the score of the k-mer is its binding significance (−log10P). Instead of studying the binding affinity change of a particular TF caused by a SNP, we were interested in identifying the most deleterious motif-disrupting variants that are associated with multiple TFs and could deactivate enhancers. This precluded us from applying the IGR approach directly. Therefore, we needed to recognize the potential binding sites of the most essential liver-specific TFs in enhancer regions. The putative strong enhancers predicted by ChromHMM (Ernst and Kellis 2012) were chosen as the positive set to identify the mostly enriched TFBSs (k-mers). Since higher binding significance indicates stronger functional constraints, we used the change of binding significance ΔSig (formula (4), P1 is the P-value of the original k-mer, and P2 is the P-value of the mutated k-mer) of the k-mers to evaluate the change of binding fitness caused by the SNV (supplementary fig. S1, Supplementary Material online).
| (4) |
More importantly, we considered all three possible nucleotide substitutions at an enhancer position to quantify binding significance change. At the positions where the original associated maxima k-mer belonged to the 522 significant k-mers, the nucleotide substitution that changed a significant k-mer to an insignificant one was considered a candidate KM, due to which a binding site might be lost in the enhancer region. At the positions where the original associated maxima k-mer belongs to the 30,647 insignificant k-mers, the nucleotide substitution that changes an insignificant k-mer to a significant one was considered a RS, leading potentially to a gained TFBS. Because either losing or gaining a binding site could lead to phenotype change, we were particularly interested in the positions that are more easily to lose or gain a binding site due to a single-nucleotide mutation. Specifically, the positions where all three mutations would cause a binding site loss (change a significant k-mer to an insignificant one) were defined as fKMPs; the positions where all three mutations would cause a binding site gain (change an insignificant k-mer to a significant one) were defined as fRSPs. To differentiate the fKMPs from all the other positions where only one or two mutations are KMs, we defined positions with KMs caused by no more than two mutations as sKMPs. Similarly, the positions with RSs caused by no more than two mutations were defined as sRSPs. fKMPs and sKMPs together are called KMPs. fRSPs and sRSPs together are termed as RSPs.
Since different single-nucleotide substitutions on a position have different levels of binding significance modulations on the original k-mer, the minimum absolute modulation of binding significance (formula 5) among the modulations caused by all three possible single-nucleotide substitutions on the fKMP/fRSP were used to evaluate the level of disruption on TF binding once a mutation occurred at that particular position. To further validate the correlation between of a fKMP/fRSP and the functional constraints on the position, we first sorted the fKMP/fRSP by their (defined as deleterious effect) decreasingly and partitioned the sorted fKMPs/fRSPs into 20 bins. Within each bin, we checked the proportion of positions with high phyloP score (≥ 2).
| (5) |
To identify the fKMPs/fRSPs and KMs/RSs in LCL cell lines, using the top 498 significant k-mers enriched in GM12878 ChromHMM strong enhancers as well as the 30,741 insignificant k-mers, we applied the modified IGR approach with the same parameters to both the paternal and maternal genomes in each individual of the two trios. To study the association of KMs and differential binding of PU.1 in LCL cell lines, the same pipeline was applied to the PU.1 ChIP-seq peaks by considering both maternal and paternal genome of each individual of the two trios. When counting KMs in the individual-specific/differential/similar PU.1-binding regions between two individuals, one KM would be counted if the reference allele was also an RS for the k-mer associated with the killing allele.
Analysis of Correlation between RSP and KMP Clusters
Any two KMPs located within an 8-bp window were clustered together, and a KMP was joined with its nearest cluster if its minimum distance to the cluster was not larger than 8 bp. Furthermore, any two KMP clusters with their minimum distance not larger than 8 bp were merged into one cluster. We applied the same clustering procedure to RSPs. The distance of a RSP cluster to a KMP cluster is defined as the minimum distance between the two clusters. To study the statistical significance of the distance of a given RSP cluster to its nearest KMP cluster, for the KMP cluster we randomly picked a set of non-KMP positions (with the same number of positions and the same relative distances as the RSP cluster) from the enhancer 1,000 times. The distances between the 1,000 random sets of positions and the KMP clusters formed an expected empirical background. As shown in supplementary figure S14, Supplementary Material online, the distances from RSP clusters to the nearest KMP cluster are significantly smaller than the null distribution, with P-value < 2.23e-308 using Mann–Whitney test. As for the coexistence of common SNPs in the coordinated KMP and RSP clusters, we applied the binomial test to examine enrichment: for the coexistence of SNPs in the assigned pair of KMP and RSP clusters, the probability of finding it in a randomly picked KMP-RSP cluster pair was calculated as the product of frequency of the SNPs in KMP clusters and RSP clusters separately.
Enrichment of Closely Bound TF Pairs
We considered that the ChIP-seq peaks of two TFs overlap each other if the distance between the centers of the two peaks is no greater than 50-bp. The ratio of their overlapping is estimated by ratio = where A, B represent the ChIP-seq peaks for TFA and TFB, respectively, |A| represents the size of the ChIP-seq peaks of TFA. is the size of overlapping peaks; is the total amount of ChIP-seq peaks of TFA and TFB. We speculated that if two TFs bound close to each other, there was likely to be functionally cooperation between these two TFs, either by direct or indirect interactions. To evaluate the significance of the interaction between A and B, we established a null distribution by randomly generated 1,000 independent pairs of TF ChIP-seq peaks, each similar in size as the tested TFs. The enrichment of the interacting TF pairs was then evaluated by ratioreal/rationull.
Enrichment of KM SNPs in the Clusters of TFBSs
We first extended the sequence of the overlapping top k-mer clusters (associated with a KMP cluster) with 6-bp in both directions and applied FIMO (Bailey et al. 2009) with TRANSFAC (Matys et al. 2006) and JASPAR (Mathelier et al. 2014) matrices of TF binding specificities to search for known TFBSs overlapping the KMP clusters. The overlapping TFBSs were ranked by their widths covering the top k-mer clusters, and only the top one TFBS was kept. The TF would be considered if at least 2,000 of its binding sites were overlapping KMP clusters. The neighboring TFBSs no more than 30-bp between them were further clustered. To determine the enrichment of the co-occurrence of common SNPs in the TFBS pairs within a TFBS cluster, we applied the same procedure of the enrichment analysis of co-occurrence of SNPs as in KMP-RSP clusters.
Validation of Disruptive Effects of Our Predictions Using MPRA
We only selected the engineered enhancer sequences with HNF4 and FOXA motif instances from the MPRA experiment (Kheradpour et al. 2013). Because there are five single-nucleotide mutations on each enhancer sequence including the max 1-bp decrease, least 1-bp change, max 1-bp increase of the motif match score, and two separate random 1-bp changes of the 145-bp enhancers, and there are 30 enhancers for the targeted TFs: HNF4, FOXA, and GATA, 26 enhancers for NFE2L2. In total, 116 enhancers with 580 engineered variants were selected in our study and superimposed onto our predictions.
Scaled Significance Score
Instead of using the raw score of binding significance (S = −log10P) to evaluate the biological significance of a k-mer and to estimate the modulation of binding significance caused by SNVs, we used the rank of k-mers as the variant to define a normalized significance score with a comparable unit, that is, the phred-scaled significance score (scaled-significance = −10 × log10(rank_S/N), where rank_S is the rank of the binding significance score and N the total number of k-mers). For example, a scaled binding significance of 10 referred to the top 10% of all 32,896 k-mers. With scaled binding significance score it would be easier to infer the significance of the probability of picking a k-mer(s) at that score or greater when selecting randomly from the control set.
Enrichment Analysis of GWAS Traits
The NHGRI GWAS Catalog was downloaded in February 2015 (Welter et al. 2014). To study the enrichment of a set of SNPs coinciding with a certain trait, we generated a null distribution composed of 1,000× random SNP sets with the same size as the tested SNP set. The P-value of the association between the set of the SNPs and the studied trait was estimated using binomial distribution. The enrichment of KM SNPs coinciding with a trait relative to enhancer SNPs was evaluated as the ratio of the enrichment of KM SNPs on this trait relative to the null distribution to that of enhancer SNPs on this trait relative to the null distribution. In all, 665 traits with at least three tag SNPs were kept for the association study. The tag SNPs coinciding with the 665 GWAS traits were further expanded by LD (r2 > 0.8, minimum distance of 500-bp). In total, 219 KM SNPs coincided with liver or liver-related traits (supplementary table S4, Supplementary Material online).
Peak Calling
We used BEDTools (Quinlan and Hall 2010) to transfer mapped bam files (a binary version of a tab-delimited text file that contains sequence alignment data; Li et al. 2009) of the ChIP-seq data to bed files (tab-delimited text file that defines a feature track; Kent et al. 2002). Then we applied Homer (Heinz et al. 2010) to identify peak regions of TF ChIP-seq data sets and dip regions of histone mark ChIP-seq data sets, using “-style factor” and“-size 1000 -nfr” parameters, respectively.
To study the effect on differential TF binding of candidate KMs of the two trios in LCLs, we first separated the ChIP-seq peaks into three categories: common peaks (ChIP-seq peaks having similar binding signals between two individuals), differential peaks (ChIP-seq peaks existing in both individuals but having differential binding signals), and individual-specific peaks (ChIP-seq peaks that are present in one individual while missing in another). To identify individual-specific and common-peaks of PU.1 shared between any two individuals of the two trios, we applied Homer again to merge any two peaks (in two individuals) with at most 100-bp between the two peak centers using the parameter “-d 100” for the command “mergePeaks.” We also applied Homer to identify differential ChIP-seq peaks of PU.1 between any two individuals using the command “getDifferentialPeaks,” which by default identifies peaks that have more than a four-fold difference of tag counts between two experiments with a cumulative Poisson P-value ≤ 0.0001.
If a peak had an individual-specific/differential binding signal between two individuals with the same KM(s) in at least one pair-wise comparison, it could be considered as one individual-specific/differential peak caused by certain KMs. The peaks which were not individual-specific/differential peaks between any two individuals were considered as similar peaks. The similar peaks are also binned to two categories: 1) similar peaks without KMs for any pair-wise comparison; 2) similar peaks with KMs (same as individual-specific/differential peaks, similarly bound peaks sharing the same KMs across different pair-wise comparisons are considered as one peak).
Allele-Specific Local Chromatin Structure
To study the allelic imbalance in chromatin accessibility caused by a KM/RS, we compared the fraction of reads obtained from each allele. We first called the KMs/RSs in both the paternal and maternal genomes of GM12878: one position in the paternal genome would be considered to carry a KM if the maternal allele at the position is a KM for the paternal genome, meanwhile the paternal allele at the position is a RS for the maternal genome, and vice versa. The DNase I data includes reads mapped to hotspot DHS at an FDR threshold of 5% (Maurano, Humbert, et al. 2012). At 8,766 KM/RS sites, the DNase-seq reads were extracted using SAMtools (Li et al. 2009). Because in the original aligned data (Maurano, Humbert, et al. 2012), the DNase I reads were mapped to GRCh37/hg19 human reference sequence, to correct for mapping bias caused by mismatches, reads containing the maternal or paternal reference allele were only counted if they have at most one mismatches. Sites with less than 11 reads were filtered for library or mapping noise and lack of statistical power, leaving 299 sites for further analysis.
dsQTL Analysis in LCLs
In total, 5,450 dsQTLs coincided with common SNPs (MAF ≥ 0.01) from the 1000 Genomes Project and dbSNP. We collected all proxy SNPs of the dsQTL SNPs based on LD analysis by using SNAP (Johnson et al. 2008) to expand the LCL dsQTL SNP set. The r2 threshold of 0.8, D′ ≥ 0.9, and the maximal distance between two proxy SNP of 500 bp were applied. To study the enrichment of dsQTL in enhancer SNPs compared with all common SNPs, the empirical control set of random SNPs had to be generated in order to alleviate the ascertainment bias. For each enhancer SNP, 500 SNPs located at approximately the same distance to the nearest TSS were chosen at random. Then the binomial test b(x;n,p) was applied to calculate the enrichment of enhancer SNPs coinciding with dsQTLs, setting the first parameter x to the number of enhancer SNPs coinciding with dsQTLs, the second parameter n to the number of enhancer SNPs, and the third parameter p to the proportion of random matched SNPs carrying dsQTLs (P = 0.00513). For the enrichment of dsQTLs in KM/RS SNPs compared with enhancer SNPs, we counted the number of dsQTLs matched with both categories and studied the enrichment using the hypergeometric test.
eQTL Analysis in LCLs
We first applied the same modified IGR approach to the strong enhancers of GM12891 and GM12892 to identify the KMPs/RSPs. The predicted ChromHMM strong (active) enhancers were generated by another study (Kasowski et al. 2013).
There were originally 67,758 eQTL-gene links in LCLs from CEU populations based on a previous study (Stranger et al. 2012). To expand the LCL eQTL SNP set, we identified all proxy SNPs of the eQTL SNPs based on an LD analysis using SNAP (Johnson et al. 2008) with the stringent criteria: r2 threshold of 0.8, D′ ≥ 0.9, and maximal distance between two proxy SNPs of 500-bp. In the analysis of the enrichment of eQTLs associated with highly expressed genes in enhancer common SNPs relative to the matched random SNP sets described earlier, binomial test b(x; n,p) was applied to calculate the enrichment of enhancer SNPs coinciding with eQTLs linked to highly expressed genes, setting the first parameter x to the number of enhancer SNPs coinciding with eQTLs linked to highly expressed genes, the second parameter n to the number of enhancer SNPs, and the third parameter p to the proportion of random matched SNPs that carry eQTLs linked to the same set of genes. For the enrichment of eQTLs in fKMP/fRSP SNPs compared with enhancer SNPs, we counted the number of eQTLs matched with both categories and studied the enrichment also using hypergeometric test.
Supplementary Material
Supplementary text, figures S1–S34, and tables S1–S7 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. The authors are grateful to Di Huang, Dorothy L. Buchhagen, Gesine Cauer, Konnor La, and Parsa Hosseini for critical reading of the manuscript.
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37:W202-W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, et al. 2011. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 43(11):1131-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-C, Lin C-J. 2011. LIBSVM : a library for support vector machines. ACM Transactions on Intelligent Systems and Technology. 2:27:1–27:27. Available at: http://www.csie.ntu.edu.tw/~cjlin/libsvm. [Google Scholar]
- Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437:1365-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, et al. 2011. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 44(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15:901-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran L. 2005. Molecular analysis of B lymphocyte development and activation. Berlin Heidelberg: Springer. [Google Scholar]
- Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, Moore JH, Lupien M. 2012. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 44:1191-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, Henneman P, Heid IM, Kizer JR, Lyytikäinen LP, et al. 2012. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 8:e1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482:390-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel DE, Visel A, Pennacchio LA. 2013. Functional anatomy of distant-acting mammalian enhancers. Philos Trans R Soc Lond B Biol Sci. 368:20120359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer S, Kovacs Z, Yuan SH, Siszler G, Kögl M, Summer H, Geerts A, Golz S, Shioda T, Methner A. 2011. TOX3 is a neuronal survival factor that induces transcription depending on the presence of CITED1 or phosphorylated CREB in the transcriptionally active complex. J Cell Sci. 124:252-260. [DOI] [PubMed] [Google Scholar]
- Dongen SV. 2000. Graph clustering by flow simulation [Ph.D thesis]. University of Utrecht. [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452:423-428. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 9:215-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186-194. [PubMed] [Google Scholar]
- Fiegel HC, Lioznov MV, Cortes-Dericks L, Lange C, Kluth D, Fehse B, Zander AR. 2003. Liver-specific gene expression in cultured human hematopoietic stem cells. Stem Cells 21:98-104. [DOI] [PubMed] [Google Scholar]
- Gaffney DJ, Veyrieras JB, Degner JF, Pique-Regi R, Pai AA, Crawford GE, Stephens M, Gilad Y, Pritchard JK. 2012. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 13:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. 2013. Discovery and refinement of loci associated with lipid levels. Nat Genet. 45:1274-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Lee D, Reed X, Fletez-Brant C, Bessling SL, Loftus SK, Beer MA, Pavan WJ, McCallion AS. 2012. Integration of ChIP-seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res. 22(11):2290–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Kwan T, Ge B, Lam KC, Koka V, Kindmark A, Mallmin H, Dias J, Verlaan DJ, Ouimet M, et al. 2009. Population genomics in a disease targeted primary cell model. Genome Res. 19:1942-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 38:576-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, Glass CK. 2013. Effect of natural genetic variation on enhancer selection and function. Nature 503:487-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms C, Cao L, Krueger JG, Wijsman EM, Chamian F, Gordon D, Heffernan M, Daw JA, Robarge J, Ott J, et al. 2003. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat Genet. 35:349-356. [DOI] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34:D590-D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YW, Zheng L, Wang Q, Zhong TY, Yu X, Bao J, Cao NN, Li B, Si-Tu B. 2012. Vascular endothelial growth factor downregulates apolipoprotein M expression by inhibiting Foxa2 in a Nur77-dependent manner. Rejuvenation Res. 15:423-434. [DOI] [PubMed] [Google Scholar]
- Huang D, Ovcharenko I. 2015. Identifying causal regulatory SNPs in ChIP-seq enhancers. Nucleic Acids Res. 43:225-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. 2008. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24:2938-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G. 2013. DNA-binding specificities of human transcription factors. Cell 152:327-339. [DOI] [PubMed] [Google Scholar]
- Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N. 2010. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 42:210-215. [DOI] [PubMed] [Google Scholar]
- Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek, et al. 2013. Extensive variation in chromatin states across humans. Science 342:750-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res. 12:996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. 2013. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 23:800-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46:310-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. 2006. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev. 20:2293-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ning G, Duncan SA. 2000. Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev. 14:464-474. [PMC free article] [PubMed] [Google Scholar]
- Lloberas J, Soler C, Celada A. 1999. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunol Today. 20:184-189. [DOI] [PubMed] [Google Scholar]
- Mahony S, Benos PV. 2007. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 35:W253-W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. 2010. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol. 30:565-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou, Ienasescu H, et al. 2014. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42:D142-D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34:D108-D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Wang H, Kutyavin T, Stamatoyannopoulos JA. 2012. Widespread site-dependent buffering of human regulatory polymorphism. PLoS Genet. 8:e1002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AN, Freedman ML. 2013. Lessons from postgenome-wide association studies: functional analysis of cancer predisposition loci. J Intern Med. 274:414-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG. 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430:743-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, et al. 2007. A survey of genetic human cortical gene expression. Nat Genet. 39:1494-1499. [DOI] [PubMed] [Google Scholar]
- Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, Jenisch S, Weichenthal M, Abecasis GR, Lim HW, Christophers E, et al. 2006. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 78:827-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y, Arima M, Arguni E, Okada S, Yamashita K, Asari S, Obata S, Sakamoto A, Hatano M, O-Wang J, et al. 2005. A role for c-fos/activator protein 1 in B lymphocyte terminal differentiation. J Immunol. 174:7703-7710. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L. 2006. Genetics of global gene expression. Nat Rev Genet. 7:862-872. [DOI] [PubMed] [Google Scholar]
- Sakabe NJ, Savic D, Nobrega MA. 2012. Transcriptional enhancers in development and disease. Genome Biol. 13:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. 2008. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29:308-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smid M, Wang Y, Klijn JG, Sieuwerts AM, Zhang Y, Atkins D, Martens JW, Foekens JA. 2006. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 24:2261-2267. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, Hunt S, Kahl B, Antonarakis SE, Tavaré S, et al. 2005. Genome-wide associations of gene expression variation in humans. PLoS Genet. 1:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. 2012. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8:e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 101:6062-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. 2010. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiilikainen A, Lassus A, Karvonen J, Vartiainen P, Julin M. 1980. Psoriasis and HLA-Cw6. Br J Dermatol. 102:179-184. [DOI] [PubMed] [Google Scholar]
- van Dongen S, Abreu-Goodger C. 2012. Using MCL to extract clusters from networks. Methods Mol Biol. 804:281-295. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457:854-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, Pennacchio LA. 2009. Genomic views of distant-acting enhancers. Nature 461:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Yet al. 2009. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22(9):1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. 2014. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42:D1001-D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Howell JJ, Ndungo E, Stoffel M. 2008. Foxa2 activity increases plasma high density lipoprotein levels by regulating apolipoprotein M. J Biol Chem. 283:16940-16949. [DOI] [PubMed] [Google Scholar]
- Wu C, Macleod I, Su AI. 2013. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41:D561-D565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW , III, et al. 2009. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10:R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21(8):1273–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We used the GRCh37 (hg19) assembly of the human genome, which we downloaded from the UCSC Genome Browser (Kent et al. 2002).
Putative strong enhancers no longer than 3-kb predicted by ChromHMM (Ernst and Kellis 2012) were used as the training set for the enrichment analysis of k-mers. HepG2 and LCLs (GM12878) ChromHMM strong enhancers and ChIP-seq peaks (narrowPeak format) of 62 TFs were downloaded from the USCS Genome Browser (Kent et al. 2002; http://genome.ucsc.edu/, last accessed May 23, 2015). HNF4α ChIP-seq data in differentiated Caco-2 cells were downloaded from NCBI Gene Expression Omnibus (GEO) (accession number: GSM575229).
For the analysis in LCLs, our study targeted individual genomes of two father–mother–daughter trios (supplementary table S5, Supplementary Material online). The first trio was of Utah residents of European ancestry (CEU), and the second was Yoruban from Ibadan Nigerian ancestry (YRI). Genetic variation data were downloaded from the 1000 Genomes Project (Abecasis et al. 2012) and dbSNP 138 (Sherry et al. 2001). Genomic coordinates of SNPs were mapped from hg18 to hg19 using the UCSC liftOver tool (Hinrichs et al. 2006). The personal genomes of each individual of the two trios were obtained by overlaying the maternal and paternal SNPs haplotypes onto the hg19 genome (Kasowski et al. 2013). As for the differential binding of PU.1 in LCLs, we used the ChIP-seq data of PU.1 from the study in which the ChIP-seq reads were aligned against the maternal and paternal genomes of the corresponding individual (Kasowski et al. 2013). The DNase I data used in the analysis of local chromatin accessibility in GM12878 were downloaded from GEO with accession number GSE29692. This DNase I data includes reads mapped to hotspot DHS at an FDR threshold of 5% (Maurano, Humbert, et al. 2012). For the prediction of KMs/RSs of maternal and paternal genomes, fKMPs/fRSPs in LCL ChromHMM strong enhancers and the analysis of enrichment of eQTLs in fKMPs/fRSPs, we used the ChromHMM strong enhancers of the two individuals from the study conducted by Kasowski et al. (2013). The eQTL data with linkage to associated genes for CEU populations were obtained from a study (Stranger et al. 2012), and the gene expression profile of 79 tissues was downloaded from BioGPS (Wu et al. 2009, 2013) (accession number: GSE1133).