

Abstract
Aspergillus fumigatus (AF) is often pathogenic in immune-deficient individuals and can cause life-threatening infections such as invasive aspergillosis. The pulmonary epithelial response to AF infection and the signaling pathways associated with it have not been completely studied. BEAS-2B cells or primary human bronchial epithelial cells were exposed to extracts of AF and challenged with IFN-β or the Toll-like receptor 3 agonist double-stranded RNA (dsRNA). Cytokine release (B-cell activating factor of the TNF family [BAFF], IFN-γ–induced protein-10 [IP-10], etc.) was assessed. AF extract was separated into low-molecular-weight (LMW) and high-molecular-weight (HMW) fractions using ultra 4 centrifugal force filters to characterize the activity. Real-time PCR was performed with a TaqMan method, and protein estimation was performed using ELISA techniques. Western blot was performed to assess phosphorylation of signal transducer and activator of transcription 1 (STAT1). IFN-β and dsRNA induced messenger RNA (mRNA) expression of BAFF (350- and 452-fold, respectively [n = 3]) and IP-10 (1,081- and 3,044-fold, respectively [n = 3]) in BEAS-2B cells. When cells were pretreated with AF extract for 1 hour and then stimulated with IFN-β or dsRNA for 6 hours, induction of BAFF and IP-10 mRNA was strongly suppressed relative to levels produced by IFN-β and dsRNA alone. When compared with control, soluble BAFF and IP-10 protein levels were maximally suppressed in dsRNA-stimulated wells treated with 1:320 wt/vol AF extract (P < 0.005). Upon molecular size fractionation, a LMW fraction of AF extract had no measurable suppressive effect on IP-10 mRNA expression. However, a HMW fraction of the AF extract significantly suppressed IP-10 expression in BEAS-2B cells that were stimulated with dsRNA or IFN-β. When BEAS-2B cells were pretreated with AF extract and then stimulated with IFN-β, reduced levels of pSTAT1 were observed, with maximum suppression at 4 and 6 hours. Our results show that AF extracts suppressed expression of inflammatory cytokines in association with inhibition of the IFN-β signaling pathway and suppression of the formation of pSTAT1.
Keywords: Aspergillus fumigatus, epithelial cells, BAFF, IP-10
Clinical Relevance
The components of Aspergillus fumigatus extract that inhibit JAK–signal transducer and activator of transcription 1 (STAT1) signaling could impair antiviral immunity and could also contribute to the skewing of adaptive immune responses toward T helper (Th) 2 observed in Aspergillus-infected individuals by undermining the Th1 responses via inhibition of the activation of STAT1. We believe that the inhibitory effect on STAT1 activation could be exploited for beneficial effects once we better understand the identity of the factor and the molecular mechanism by which STAT1 phosphorylation is inhibited.
Aspergillus fumigatus (AF) is a ubiquitous fungus that is often pathogenic in immune-deficient individuals and is a commonly isolated agent in human pulmonary infections (1). AF can cause lung diseases ranging from local inflammation of the upper airways to life-threatening infections of the lung, such as those that occur in allergic bronchopulmonary aspergillosis (ABPA) or invasive aspergillosis (2, 3).
Despite the high prevalence of AF conidia in ambient air, severe Aspergillus infections are relatively rare in healthy individuals because of mucociliary clearance and pulmonary immune responses. Available data indicate that there are efficient immune mechanisms that combat Aspergillus infection (4). Even though the innate immune functions of respiratory epithelial cells are well described in the response to bacteria and viruses, relatively little is known about the response of epithelium to fungal pathogens such as AF.
Experimental exposure to Aspergillus extracts in the lung of mice leads to a response that resembles allergic asthma, and Aspergillus is well known to induce asthma (5, 6). Allergic airway disease is characterized by eosinophilic inflammation, mucus hypersecretion, and increased airway resistance. This response results in part from antigen-specific T helper (Th) 2 cell activation characterized by the production of IL-4, IL-5, and IL-13 (7, 8). Inhaled fungi can stimulate allergic inflammation without causing active infection in an immunocompetent host. It has been demonstrated previously that direct airway exposure to AF lysates promotes a Th2-biased immune response in the lungs of mice by the accumulation of eosinophils and mucus production (9). It is unclear why exposure to fungal pathogens leads to Th2-biased immune responses.
The response to experimental lung challenge in mice is reminiscent of the immune response to AF seen in humans with ABPA and is characterized by prominent lung eosinophilia and mucus production. Allard and colleagues demonstrated that, like AF, Candida albicans antigens promote Th2 inflammation characterized by airway eosinophilia and mucus production (10). Steele and colleagues provided insight into the earliest recognition events after inhalation of AF and the importance of alveolar macrophage–associated, β-glucan–initiated, dectin-1 signaling in generating the appropriate inflammatory signals in response to AF. These authors also showed that dectin-1 is centrally involved in generating inflammatory responses to specific morphological forms of this organism in vitro and in vivo (11, 12).
Airway epithelial cells form a mucosal barrier that defends against harmful substances, including microbial and fungal pathogens, via innate production of mucins and antimicrobial substances. However, recently it has been documented that airway epithelial cells also function in the regulation of immune responses through production of cytokines and chemokines and via interactions with cells of the immune system (13–15). Epithelial cells can thus coordinate adaptive immune responses by expressing molecules that recruit dendritic cells; cells with cognate receptors, such as T cells and B cells; and innate effector cells, such as NK, NKT, ILC, and granulocytes. Epithelial cells express pathogen recognition receptors and manifest responses that can be distinct depending upon the innate stimuli that engage these receptors (13–16). It is now clear that pathogens themselves can use strategies to subvert the epithelial response and alter the alarm function of epithelium. For instance, in a recent study, Wada and colleagues showed that Alternaria inhibits Toll-like receptor (TLR) 3 expression and production of IFN-β, I-TAC, and IFN-γ–induced protein-10 (IP-10) by dendritic cells stimulated with double-stranded RNA (dsRNA) (17). Their findings demonstrate that Alternaria potently enhances Th2-type immune responses and eosinophilic inflammation in the airways. Little knowledge exists concerning how the pulmonary epithelium responds to AF infection and which receptors and signaling pathways might be involved.
This study investigated whether respiratory epithelial cells recognize AF extract and initiate an immune response. We hypothesized that AF extract would activate epithelium to produce Th2-biasing cytokines and chemokines based on the in vivo skewing to Th2 inflammation as discussed above. We found that AF did not induce Th2-biased chemokines but rather strongly inhibited a Th1-biased response in epithelium, as evidenced by suppressed expression of IP-10 and other Th1-biased cytokines induced by activation of TLR3. Suppression of Th1 biasing may de facto promote a Th2 bias by altering the balance of these responses. We present evidence to suggest that the suppression of Th1-type responses in human airway epithelial cells may result from inhibition of signaling in the JAK-STAT pathway.
Materials and Methods
Reagents
AF extract was purchased from Hollister-Stier (Spokane, WA). The extract is a commercial antigen product used to conduct skin prick testing of allergy patients in clinical settings and contains, in addition to the extract allergens, 50% (vol/vol) glycerine, 0.5% sodium chloride, and 0.275% sodium bicarbonate. dsRNA and IFN-β1a were purchased from Invivogen (San Diego, CA). The sequences of primers used for messenger RNA (mRNA) detection are IP-10, F (5-CGCTGTACCTGCATCAGCATT-3), R (5-GCTCCCCTCTGGTTTTAGGAG-3); and B-cell activating factor of the TNF family (BAFF), F (5-TCGATGTATTCAAAATATGCCTGAAA-3), R (5-TGCAATGCCAGCTGAATAGC-3), and Probe (5′-CTACCCAATAATTCC-3′). BAFF and IP-10 ELISA kits were purchased from R&D (Minneapolis, MN). Total RNA was isolated from epithelial cell cultures using Macherey-Nagel RNA II Isolation Kits (MACHEREY-NAGEL GmbH and Co. KG, Duren, Germany) following the manufacturer’s instructions. STAT1 and pSTAT1 antibodies were purchased from Cell Signaling (Danvers, MA).
Cell Culture and Treatments
The human bronchial epithelial cell line BEAS-2B was a gift from Dr. C. Harris (National Cancer Institute, Bethesda, MD) and was cultured in DMEM/F12 (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 5% heat-inactivated FBS (Invitrogen Life Technologies), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen Life Technologies). BEAS-2B cells were seeded (2 × 105 cells/well) and cultured for 48 hours in DMEM/F12 supplemented with 5% heat-inactivated FBS in a 12-well plate. Cells were kept at 37°C in a CO2 incubator and were allowed to grow for 48 to 72 hours before treatment with AF extract. At the time of 95% confluence, BEAS-2B cells were treated with AF extract diluted in medium at 1:1,280, 1:640, and 1:320 wt/vol for 1 hour and were then stimulated with 5 μg/ml dsRNA or 1,000 IU/ml IFN-β for 6 hours.
Normal human bronchial epithelial (NHBE) cells were obtained from Cambrex (East Rutherford, NJ). NHBE cells came from three independent donors and cells were maintained in serum-free bronchial epithelial growth medium (Lonza, Walkersville, MD). NHBE cells were plated in 24-well culture plates coated with collagen (Vitrogen; Cohesion Technologies, Beaufort, SC). Before stimulation, cells were cultured in bronchial epithelial growth medium without hydrocortisone for at least 2 days. NHBE cells were seeded at 3 × 104 cells per well in 12-well culture plates and cultured for 4 days. NHBE cells were treated with AF extract and stimulated as described above for BEAS-2B.
For fractionation studies, the AF extract was separated into low-molecular-weight (LMW) and high-molecular-weight (HMW) fractions using Amicon ultra 4 centrifugal force filters (UFC8 010 96; Millipore, Billerica, MA). Four milliliters of a 1:10 dilution (extract:PBS) of complete extract was loaded into a filter and centrifuged at 4,100 rpm for 15 minutes at 4°C. The flow-through was returned to 4 ml with sterile PBS and saved for further experiments as a 1:10 dilution of LMW extract. The volume of HMW extract remaining at the top of the filter was measured and restored to a total volume of 4 ml with sterile PBS and centrifuged again at 4,100 rpm for 15 minutes at 4°C. Flow through was discarded, and the remaining volume was brought to 4 ml followed by a third centrifugation with PBS at 4,100 rpm. After the final centrifugation, the concentrated HMW extract was collected from the top of the Amicon filter and reconstituted to 4 ml with sterile PBS for stimulation experiments. Preliminary experiments were conducted with different molecular weight fractions (e.g., >10, >30, >40, >50, and >100 kD).
Real-Time PCR and RT-PCR
After 6 hours of cytokine treatment, the supernatant was collected in a different tubes, and 500 μl of RNA was placed directly into the wells (Ambion, Inc., Austin, TX) for isolation of total RNA using RNeasy kits (Qiagen, Valencia, CA). cDNA was synthesized with SuperScript II reverse transcriptase (Invitrogen Life Technologies) and random primers. Real-time PCR was performed with a TaqMan method in 20-μl reactions with 10 μl of 2x TaqMan Master mix (Applied Biosystems, Carlsbad, CA), 1.6 μl of 5-μM primer sets, and 0.8 μl of 5-μM probe. Real-time PCR was performed using an Applied Biosystems 7500 System. Aliquots of cDNA equivalent to 10 ng of total RNA were used for real-time PCR. The expression levels of mRNA were normalized to the median expression of a β-actin housekeeping gene.
Protein Assays
The concentrations of BAFF and IP-10 protein in cell-free supernatants were measured with specific ELISA kits (R&D Systems). The minimum detectable limits of the kits are 31.2 pg/ml (BAFF) and 7.8 pg/ml (IP-10). After stimulation, cells were washed with PBS and lysed using M-PER (Thermo Fisher) supplemented with protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Cell lysates were centrifuged at 14,000 × g for 5 minutes. Clarified supernatants were analyzed for total protein using BCA assay (Thermo Fisher). Lysates were adjusted for protein content and used for Western blot analysis. The Bis-Tris gel electrophoresis system (Bio-Rad, Hercules, CA) was used to separate total proteins. Gel contents were transferred to a PVDF membrane using a semidry transfer system (Bio-Rad). The transferred membrane was blocked using blocking buffer (Rockland Immunochemicals, Inc., Pottstown, PA) for 1 to 2 hours. After blocking, blots were incubated with primary antibodies (pSTAT1 [1:1,000] and actin [1:10,000]) overnight. Blots were washed with 1× Tris-buffered saline with Tween 20 and incubated with infrared probe labeled secondary antibody (Licor, Lincoln, NE) for 1 hour. After washing off secondary antibodies, blots were evaluated with an Odyssey infrared scanner (Licor). Blots were stripped and used for blotting total STAT1 (1:1,000). STAT1 and actin antibodies were purchased from Cell Signaling and MP Biomedicals (Solon, OH), respectively.
Statistical Analysis
All data are presented as the mean ± SEM unless otherwise mentioned. Data were normally distributed, and differences between groups were analyzed using the paired Student’s t test. P < 0.05 was considered statistically significant.
Results
AF Extract Suppressed Expression of BAFF and IP-10 mRNA Induced in Airway Epithelial Cells by IFN-γ or TLR3 Activation
We initially screened a large number of preparations of commercial aeroallergens, seeking those that would preferentially activate Th2-biased chemokines such as eotaxins and MCP-4, but failed to find any with this property (not shown). We next conducted a series of experiments assessing inhibition of Th1-biased responses. Previous studies from our laboratory have demonstrated TLR3-induced expression of BAFF mRNA and protein in BEAS-2B cells (18). IP-10 has often been used as a marker of Th1-biased cellular responses (19). In preliminary studies (not shown), BEAS-2B cells were stimulated for 6 hours with dsRNA, a potent TLR3 ligand, and with inflammatory cytokines such as IFN-β, IFN-γ, IL-4, IL-13, IL-33, IL-25, IL-17, and TNF-α, and expression of BAFF and IP-10 was monitored. In addition to BAFF and IP-10, we also studied the expression of CCL20, TARC, TSLP, MDC, TARC-B, IFIT-1, CCL11, and CCL-26. IFN-β and dsRNA induced mRNA expression of BAFF (350-fold [n = 3; P < 0.001] and 452-fold [n = 3; P < 0.001], respectively) and IP-10 (1,081-fold [n = 3; P < 0.001] and 3,044-fold [n = 3; P < 0.001], respectively) in BEAS-2B cells (Figure 1A). BAFF and IP-10 were not up-regulated by the other cytokines (IFN-γ, IL-4, IL-13, IL-33, IL-25, IL-17, and TNF-α) tested in BEAS-2B cells (data not shown). AF extract had no effect on expression of other cytokines measured in our study. Because we observed the maximum induction of BAFF and IP-10 using dsRNA and IFN-β, we focused further studies using these two immunostimulants.
Figure 1.
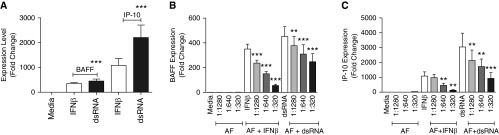
Aspergillus fumigatus (AF) extract suppressed expression of B-cell activating factor of the TNF family (BAFF) and IFN-γ–induced protein-10 (IP-10) messenger RNA (mRNA) induced in BEAS-2B airway epithelial cells by IFN-β or Toll-like receptor 3 activation. BEAS-2B cells were treated with AF extract diluted in medium at 1:1,280, 1:640, and 1:320 wt/vol for 1 hour and stimulated with 5 μg/ml double-stranded RNA (dsRNA) or 1,000 IU/ml IFN-β for 6 hours, as indicated, and then mRNA was extracted and analyzed for BAFF and IP-10 using real-time PCR. (A) IFN-β and dsRNA induced mRNA expression of BAFF and IP-10 in BEAS-2B cells: AF extract suppressed the induction of BAFF (B) and IP-10 (C) (mRNA relative to levels produced by IFN-β and dsRNA alone, expressed as fold change). The results are shown as mean ± SEM of three independent experiments. **P < 0.01 and ***P < 0.001.
AF extract by itself was not a stimulus for the expression of BAFF and IP-10. However, when BEAS-2B cells were pretreated with AF extract for 1 hour and then stimulated with IFN-β or dsRNA for 6 hours, induction of BAFF and IP-10 mRNA was strongly suppressed relative to levels produced by IFN-β and dsRNA alone. We also found that this suppression occurred in an AF extract concentration-dependent manner (Figures 1B and 1C).
AF Extract Suppressed BAFF and IP-10 Protein Secretion in BEAS-2B Cells
AF extract also suppressed the secretion of BAFF and IP-10 protein in BEAS-2B cell culture supernatants. In a previous study published by our laboratory (18), we reported that release of soluble BAFF (sBAFF) is optimal in BEAS-2B cells after 72 hours of stimulation with dsRNA. sBAFF was induced by IFN-β (66 ± 7 pg/ml at 72 h; n = 3) and by dsRNA (100 ± 17 pg/ml) at 72 hours and was not detectable in medium control stimulated cells (n = 3). To determine the effect of AF extract on sBAFF production, BEAS-2B cells were pretreated with the different concentrations of AF extract for 1 hour and then stimulated with IFN-β or dsRNA for 72 hours. Supernatants were collected and analyzed for sBAFF by ELISA. The amounts of sBAFF protein in culture supernatants were decreased in wells treated with AF extract in a concentration-dependent manner when compared with the supernatants from cells stimulated with IFN-β alone. AF extract dilutions of 1:320 wt/vol were observed to produce the maximum suppressive effect, decreasing sBAFF protein in wells stimulated with IFN-β below the detectable range of the ELISA kit. When compared with control, sBAFF protein levels were also significantly suppressed in dsRNA-stimulated wells treated with 1:640 wt/vol AF extract concentration (P = 0.05; n = 3) and 1:320 wt/vol AF extract (P < 0.001; n = 3) (Figure 2A).
Figure 2.

AF extract suppressed BAFF and IP-10 protein secretion in BEAS-2B cells. BEAS-2B cells were treated with AF extract diluted in medium at 1:1,280, 1:640, and 1:320 wt/vol for 1 hour and stimulated with 5 μg/ml dsRNA or 1,000 IU/ml IFN-β for 72 hours for BAFF or for 6 hours in studies of IP-10, as indicated. The concentrations of BAFF and IP-10 protein in cell-free supernatants were measured with specific ELISA kits. Soluble BAFF (A) and IP-10 (B) protein levels were significantly suppressed in IFN-β– and dsRNA-stimulated wells treated with AF extracts. The results are shown as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. NS, nonsignificant compared with their respective control.
IP-10 protein was induced more quickly than BAFF by IFN-β (16.6 ± 9.5 pg/ml at 6 h; n = 3) and dsRNA (25.3 ± 6.9 pg/ml at 6 h; n = 3). All dilutions of AF extract were able to inhibit IP-10 protein production in wells stimulated by dsRNA, with the AF extract concentration of 1:320 having the maximum suppression (P = 0.005; n = 3) (Figure 2B). When cells were stimulated with IFN-β, we also observed significant suppression of IP-10 protein with 1:320 wt/vol of AF extract (P = 0.005; n = 3).
To rule out the effect of diluent on gene expression, we treated the BEAS-2B cells with a similar concentration of the diluent in which AF extract was prepared. We did not find any suppression of expression of BAFF or IP-10 mRNA when BEAS-2B cells were pretreated with the diluent alone and then stimulated with IFN-β and dsRNA (see Figures E1A and E1B in the online supplement).
Inhibitory Effects of AF Extracts Resided in the HMW Fraction
To begin to characterize the nature of the compound(s) in AF extracts that produced the suppression, we separated AF extract into various LMW (<50 kD) and HMW (>50 kD) fractions by centrifugal filtration and tested both fractions following the previously described protocol. Initial experiments suggested that fractions >10 kD suppressed dsRNA and IFN-β–induced genes. Fractions with successive molecular weight (>30, >40, and >50 kD) showed the same response. In parallel, we conducted experiments with <50 kD fractions (not shown). The LMW fraction did not have any measurable suppressive effect on IP-10 mRNA expression in BEAS-2B cells (Figure 3). However, the HMW fractions of the AF extract significantly suppressed IP-10 expression in BEAS-2B cells that were stimulated with dsRNA or IFN-β, and the suppression was equivalent to the suppression by the AF extract (P < 0.001) (Figure 3). Further experiments suggested that the AF extract fraction >100 kD did not suppress the dsRNA or IFN-β–induced BAFF and IP-10 mRNA expression (not shown). Moreover, when we heated the AF extract, it no longer suppressed the dsRNA or IFN-β–induced BAFF and IP-10 in BEAS-2B cells. A tentative conclusion from these preliminary experiments is that the molecule is in the range of 50 to 100 kD and is heat labile.
Figure 3.

Inhibitory effects of AF extracts resided in the high-molecular-weight (HMW) fractions of the AF extract. For fractionation studies, the AF extract was separated into low-molecular-weight (LMW) and HMW fractions using Amicon ultra 4 centrifugal force filters. BEAS-2B cells were treated with 1:320 wt/vol concentration of AF, LMW, and HMW fraction for 1 hour and stimulated with 5 μg/ml dsRNA or 1,000 IU/ml IFN-β for 72 hours for BAFF and for 6 hours for IP-10, as indicated. The LMW fraction of the AF had no measurable suppressive effect on IP-10 mRNA expression, but the HMW fractions of the extract significantly suppressed IP-10 expression in BEAS-2B cells after stimulation with dsRNA or IFN-β. The results are shown as mean ± SEM of three independent experiments. ***P < 0.001. FE, full extract.
Fungal Extract Induced Morphologic Changes in BEAS2-B Cells
AF extract was found to induce significant morphological changes to BEAS-2B cells in culture. Although these morphological changes were not evident within 6 hours after stimulation, cell shrinkage and detachment from the culture plate were prominent at 48 and 72 hours. These morphologic changes were observed with AF extract in a concentration-dependent manner, and the maximum effects were observed at 1:320. Total number of adhered cells and viability decreased at 24, 48, and 72 hours. We assessed the effect of LMW and HMW fractions on the viability of BEAS-2B cells. The viability of BEAS-2B cells was decreased at 24 and 48 hours only by the LMW fractions, which did not have a suppressive effect on the response to dsRNA (Figure 3 and Figure E2) The HMW fraction, which strongly suppressed the cytokine response, did not have an observable effect on cell viability at any time point (Figure E2). Moreover, we did not find any detachment of cells from the surface when Beas-2B cells were treated with HMW fractions of the AF extract. These experiments suggest that the inhibitory effect of AF extract upon the cytokine response to dsRNA was mediated by a HMW molecule that does not induce toxicity or cell detachment. To further rule out global toxic or inhibitory mechanisms as responsible for the AF effect, we found that AF extract had no effect on the induction of an IL-8/CXCL8 response by IFN-β and dsRNA (Figure E3). These results also support the findings of Kauffman and colleagues, showing that AF extract itself induced IL-8 gene expression in BEAS-2B cells (20).
AF Induced Suppression of IFN-γ Signaling
dsRNA and IFN-β activate STAT1 as one of the major transcription factors that is required for the production of IP-10 (18). Because treatment with AF extract decreased the expression and secretion of IP-10 after stimulation with dsRNA and IFN-β, we hypothesized that AF extract may mediate its effect through suppression of STAT1 activation. BEAS-2B cells were stimulated with 1,000 IU/ml of IFN-β at various time points, and phosphorylation of STAT1 (pSTAT1) was measured by immunoblot analysis. We found that stimulation of BEAS-2B cells with IFN-β activated maximum pSTAT1 at 30 minutes and 1 hour. When we pretreated BEAS-2B cells with the highest concentration of AF extract and then stimulated the cells with 1,000 IU/ml of IFN-β, we observed reduced levels of pSTAT1 with maximum suppression at 4 and 6 hours (P < 0.05) (Figures 4A and 4B).
Figure 4.

Suppression of IFN-β signaling by AF extract. BEAS-2B cells were stimulated with 1,000 IU/ml of IFN-β, and at various time points phosphorylation of signal transducer and activator of transcription 1 (STAT1) (pSTAT1) was measured by Western blot analysis. (A and B) Pretreatment of BEAS-2B cells with 1:320 wt/vol AF extract followed by stimulation of the cells with IFN-β reduced the levels of pSTAT1, with maximum suppression at 4 and 6 hours. The results are shown as mean ± SEM of three independent experiments. *P < 0.05.
Confirmation of Important Findings in Primary Cells
We attempted to confirm the findings in primary NHBE cells. IFN-β and dsRNA induced IP-10 mRNA expression (872-fold [n = 3] and 3,162-fold, respectively [n = 3]) in NHBE cells (Figures 5A and 5B). When NHBE cells were pretreated with AF extract for 1 hour and then stimulated with IFN-β or dsRNA for 6 hours, induction of IP-10 mRNA was suppressed relative to the expression induced by IFN-β and dsRNA alone (P < 0.01). We also found that this suppression occurred in an AF extract concentration-dependent manner (Figures 5A and 5B). AF extract did not induce significant morphological changes to the NHBE cells during an incubation lasting up to 6 hours. After stimulation of the NHBE cells with 1,000 IU/ml of IFN-β, we observed maximal pSTAT1 expression between 30 minutes and 1 hour, and levels slowly decreased and were maintained at lower levels for up to 10 hours. Pretreatment of the NHBE cells with AF extract reduced STAT1 phosphorylation after stimulation with 1,000 IU/ml concentration of IFN-β; maximum suppression of pSTAT1 was observed at 4 and 6 hours, and the effect lasted through the 10 hours of the experiment (Figures 6A and 6B).
Figure 5.

Suppression of IP-10 mRNA expression in normal human bronchial epithelial (NHBE) primary epithelial cells by AF extract. NHBE cells were pretreated with AF extract for 1 hour and stimulated with 1,000 IU/ml of IFN-β or 5 μg/ml of dsRNA for 6 hours, as indicated, and then mRNA was extracted and analyzed for BAFF and IP-10 using real-time PCR. (A) Suppression of IP-10 mRNA relative to the expression induced by dsRNA alone. (B) Suppression of IP-10 mRNA relative to the expression induced by IFN-β alone. The results are shown as mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01.
Figure 6.

Suppression of STAT1 phosphorylation in NHBE cells. NHBE cells were stimulated with 1,000 IU/ml IFN-β, and STAT1 phosphorylation was monitored by Western blot. Peak pSTAT1 was found at 30 minutes and 1 hour. Pretreatment of the NHBE cells with AF extract reduced STAT1 phosphorylation after stimulation with IFN-β; maximum suppression of STAT1 phosphorylation was observed at 4 and 6 hours, and the effect was sustained throughout the 10-hour experiment. The results are shown as mean ± SEM of three independent experiments. *P < 0.05.
Discussion
Allergic diseases of the upper and lower airways are characterized by strong type 2 biased immune responses, in which type 2 innate lymphoid cells and Th2 cells are activated in allergic individuals and produce cytokines (e.g., IL-4, IL-5, IL-13, and others) that stimulate eosinophilic inflammation (7, 8, 21, 22). The initiating factors that promote this skewing of immune cells are not entirely known. Many fungi appear to preferentially induce type 2 cytokine responses. AF is an opportunistic fungus responsible for invasive pulmonary aspergillosis, a severe and somewhat rare disease that occurs primarily in immune compromised patients. This fungus can also cause ABPA, a highly eosinophilic form of asthma that can cause significant morbidity, and can cause AFS, another highly morbid eosinophilic disease (23). In all of these conditions, strong type 2 skewing occurs (3, 5). Epithelial cells of the respiratory tract are actively involved in initiating innate responses and can produce antifungal host defense molecules. Rather than just representing a physical barrier to prevent pathogen entry, the epithelial layer has molecular strategies to repel the organism (14, 15). Inhaled spores are trapped in the fluids and mucus lining of the epithelial surfaces of the upper respiratory tract and are removed with the help of ciliated cells (24). Finally, epithelium can also modify the host-adaptive immune response when innate immunity fails to eradicate potential pathogens. Epithelial cells can play a role in determination of the nature of the subsequent response of innate and adaptive immune cells (14, 15, 25–27). During this latter process, epithelial-derived factors can aid in selecting the type of adaptive immune response (e.g., Th1, Th2, Th17, et cetera) that is elicited.
The original goal of this study was to examine the innate response of airway epithelial cells to extracts of AF to test the hypothesis that the fungus can activate epithelial cells to release chemokines that promote type 2 responses. Initial studies with type 2 chemokines, such as CCL17 (TARC), CCL22 (MDC), and CCL11 and CCL26 (eotaxins 1 and 3), failed to find any induction (not shown). We therefore conducted a series of experiments assessing epithelial production of Th1-biased molecules induced by IFN-β and the TLR3 agonist dsRNA, including IP-10 and BAFF. We found that extracts of AF directly suppressed these markers of type 1 responses. Furthermore, we identified a potential mechanism for the suppressive effect of AF extract, demonstrating that AF extracts strongly reduced IFN-β–mediated STAT1 activation, an effect that may have the same outcome of promoting type 2 responses in immune responses. Extracts of AF did not suppress the neutrophil recruiting chemokine CXCL8 in epithelium, suggesting that they were not globally toxic and did not inhibit other important signaling pathways. AF extracts with molecular weight >100 kD did not suppress the dsRNA or IFN-β–induced BAFF and IP-10 mRNA expression, suggesting that the suppressive effect is due to a compound with molecular weight between 50 and 100 kD. This would rule out many macromolecules as the active element (e.g., HMW glucans or mannans or larger proteins).
Several other laboratories have studied the interaction of Aspergillus and its components with epithelial cells. It has been reported that metabolically inactive (UV light inactivated) resting conidia are recognized by differentiated human bronchial epithelial cells, resulting in the expression of the inflammatory mediators IFN-β and IP-10, which are known to be associated with airway hyperreactivity and airway inflammation (6). Results presented by Beisswenger and colleagues suggest that TLR3 is a receptor contributing to the activation of respiratory epithelial cells by recognizing dsRNA of conidia (24). Although we did detect a modest stimulatory effect of the AF extracts in our study (Figure E3), the more notable finding was that AF extracts profoundly suppressed induction of IP-10 and other cytokines by IFN-β and dsRNA in BEAS-2B cells and NHBE cells. Our findings are distinct from other proposed mechanisms reported previously for germinated conidia and hyphae (28, 29–32).
Results from Netea and colleagues suggest that TLR2 recognizes conidia and hyphae, whereas TLR4 detects conidia only (33). Previous studies showed that TLR3 is expressed in primary respiratory epithelial cells and that TLR3 stimulation activates the IFN-β signaling pathways (34). We used Aspergillus extract instead of hyphae and conidia in our study and found that IFN-β signaling plays an important role as a target in the mechanism of IP-10 suppression by Aspergillus extract. A recent study by Zhu and colleagues (35) explored the interaction between Alternaria and Rhinovirus, important pathogens in asthma. The authors found that fungal proteases were involved in the induction of proinflamatory cytokines and were also responsible for the enhancement of virus-induced cytokine production by Alternaria. These authors also found that Alternaria suppresses virus-induced IFN-β production by a protease-independent mechanism (35). Our studies demonstrate an AF suppression of the response to IFN-β and may help to explain some of the findings of Zhu and colleagues (35).
In the present study we report that a complete extract of AF caused morphological changes and cell detachment from the surface in a concentration-dependent manner. This cell detachment was also associated with cell death at high concentrations and with long incubations with AF extract. We did not observe morphological changes and cell detachment with a shorter (i.e., 6-h) incubation. Although the cell detachment effects of AF extract are troublesome for the studies, we do not believe that they are important to the mechanism of suppression because we were able to dissociate the suppressive property of the AF extract from the cytotoxic properties. First, the suppressive effects were observed at time points preceding the detachment or loss of viability. Second, the AF extract failed to suppress the profound release of IL-8, suggesting that the cells retained adequate viability. Third, studies with fractionation of the extract showed that the HMW fraction of the extract caused the full suppressive effect without inducing cell death or detachment (Figure E2), whereas the LMW extract caused cell death but had no suppressive effects on the expression of the response genes under evaluation. The factors within the LMW fraction that cause death and detachment likely include some of the well-known LMW toxins produced by Aspergillus, such as gliotoxin, helvolic acid, and others (36).
Extracts of AF appear to inhibit signaling activated by IFN-β. IFN-β plays an important role in the differentiation and activation of effector cells of the innate and adaptive immune system. Another function of IFN-β is the induction of a group of genes called IFN-β response genes, including that encoding IP-10, thereby linking innate and adaptive immunity (37, 38). It has been reported that IFN-β expression is also strongly induced by virus infection or dsRNA stimulation of airway epithelial cells. IFN-β is directly induced by bacterial and viral pathogen–associated molecular patterns, such as LPS, dsRNA, single-stranded RNA, and CpG motif-containing DNA via TLR-dependent or TLR-independent signaling (39–42). The receptors for IFN (IFNAR) are composed of two subunits, IFNAR1 and IFNAR2, which are associated with members of the JAK family, specifically Tyk2 and JAK1, respectively (41, 43, 44). Activation by IFN-β of the JAKs that are associated with the IFNAR results in tyrosine phosphorylation of STAT1 and STAT2. This leads to the formation of a STAT1–STAT2–IFN regulatory factor 9 complex, which is known as IFN-stimulated gene factor 3), and formation of STAT1-STAT1 homo dimers. These complexes translocate to the nucleus, bind to IFN-stimulated response elements and IFN-γ–activated sites in the promoter regions of relevant genes, and initiate gene transcription (41, 43, 44). We demonstrate in this study that AF extract has a strong suppressive effect on JAK-STAT signaling induced by IFN-β. Our findings, which were obtained in the immortalized BEAS-2B epithelial cell line and confirmed in primary human bronchial epithelial cells, suggest that a component of the HMW fraction of AF extract suppresses the phosphorylation of STAT1 or activates the dephosphorylation of STAT1 in epithelial cells. The consequences of suppression of JAK-STAT1 signaling are potentially wide reaching. Disabling this signaling pathway could lead to increased susceptibility to pathogens that are eliminated by IFN-β signaling. For example there is evidence that acute attacks of asthma are triggered or caused by rhinoviruses (45). Colonization of airways with Aspergillus could thus increase the susceptibility to infection with respiratory viruses such as rhinovirus by the mechanism that we describe here. A study by Chai and colleagues reported that Th1 responses are important in immunity to AF itself. Insofar as immunity to Aspergillus is also mediated by the IFN-β response, the ability of AF extract to disable IFN-β signaling might contribute to susceptibility to AF in individuals with ABPA, AFS, or invasive aspergillosus (46).
More germane to the present study, suppression of JAK-STAT1 signaling in epithelium could lead to diversion of the epithelial response to events that promote type 2 responses. Such events could be alterations in the activation of dendritic cells or production of chemokines or cytokines that promote Th2 immunity. Epithelium is involved in the recruitment, activation, and probably survival of Th2 cells, type 2 innate lymphoid cells, eosinophils, mast cells, and basophils (i.e., cells involved in allergic inflammation). The profound propensity for AF extract to promote Th2 responses in vivo thus could relate in part to suppression of epithelial JAK-STAT1 signaling. A shift in the balance of Th1 versus Th2 immunity could result in the observed enhanced Th2 inflammation that is associated with AF colonization and infection. In addition, the present studies, as well as previous studies, show that low concentrations of AF extracts, or fungal proteases induce morphologic changes, cell shrinking, and cell desquamation (20). By causing cell detachment, fungal proteases reduce the effectiveness of the physical barrier function of the epithelium, thereby facilitating the entry of antigens and live fungi. Loss of physical barrier is another feature of allergic diseases of the skin, sinuses, and lung (47). Thus, the combined action of AF extract to suppress counterbalancing type 1 responses and break down epithelial barrier might further promote allergic sensitization.
In conclusion, extracts of AF suppressed IP-10 mRNA and protein expression in association with inhibition of the IFN-β signaling pathway and suppression of the formation of pSTAT1. At present, the factor or factors within the HMW extracts of AF that suppress JAK-STAT1 signaling are unknown. The success of ongoing studies in identifying these factors may lead to strategies to suppress JAK-STAT1 signaling in the lungs and elsewhere. Although in the context of allergic disease the JAK-STAT1 pathway is protective (i.e., antiallergic), this pathway can cause significant morbidity and mortality when overstimulated by viruses or pathogen-associated molecular patterns, and the ability to suppress it could be valuable in cases where it causes disease. The components of AF extract that inhibits JAK-STAT1 signaling could impair antiviral immunity and could also contribute to the skewing of adaptive immune responses toward Th2 observed in Aspergillus-infected individuals by undermining the Th1 responses via inhibition of the activation of STAT1. We also believe that the inhibitory effect on STAT1 activation could be exploited for beneficial effects once we better understand the identity of the factor and the molecular mechanism by which STAT1 phosphorylation is inhibited.
Acknowledgments
Acknowledgments
The authors thank C. Harris for donating BEAS-2B cells, Derek Carter in helping with confocal microscopy to study the morphological changes in BEAS-2B and NHBE cells, Atsushi Kato for helpful discussions, and Leslie C. Grammer for providing allergens for the initial studies.
Footnotes
This study was supported by National Institutes of Health grants R37 HL068546, R01 HL078860, R01 AI072570, and U19 AI106683 (R.P.S.) and by the Ernest S. Bazley Foundation.
Author Contributions: B.B.—experiment design, conducting the experiments, data collection, analysis, writing the first draft of the manuscript, and critical revision. T.H., J.E.N., Q.S., and J.S.—data collection and conducting the experiments. D.S.G.—data collection. J.W.S.—critical revision. R.P.S.—supervising study design and experiments, critical analysis, and managing the project.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0333OC on December 4, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev. 2009;22:447–465. doi: 10.1128/CMR.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groll AH, Shah PM, Mentzel C, Schneider M, Just-Nuebling G, Huebner K. Trends in the postmortem epidemiology of invasive fungal infections at a university hospital. J Infect. 1996;33:23–32. doi: 10.1016/s0163-4453(96)92700-0. [DOI] [PubMed] [Google Scholar]
- 3.Cockrill BA, Hales CA. Allergic bronchopulmonary aspergillosis. Annu Rev Med. 1999;50:303–316. doi: 10.1146/annurev.med.50.1.303. [DOI] [PubMed] [Google Scholar]
- 4.Chazalet V, Debeaupuis JP, Sarfati J, Lortholary J, Ribaud P, Shah P, Cornet M, Vu Thien H, Gluckman E, Brücker G, et al. Molecular typing of environmental and patient isolates of Aspergillus fumigatus from various hospital settings. J Clin Microbiol. 1998;36:1494–1500. doi: 10.1128/jcm.36.6.1494-1500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogaboam CM, Carpenter KJ, Schuh JM, Buckland KF. Aspergillus and asthma: any link? Med Mycol. 2005;43:S197–S202. doi: 10.1080/13693780400025211. [DOI] [PubMed] [Google Scholar]
- 6.Medoff BD, Sauty A, Tager AM, Maclean JA, Smith RN, Mathew A, Dufour JH, Luster AD. IFN-gamma-inducible protein 10 (CXCL10) contributes to airway hyperreactivity and airway inflammation in a mouse model of asthma. J Immunol. 2002;168:5278–5286. doi: 10.4049/jimmunol.168.10.5278. [DOI] [PubMed] [Google Scholar]
- 7.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, McGarry MP, Wang H, O’Neill KR, Colbert DC, Colby TV, et al. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol. 2007;178:7879–7889. doi: 10.4049/jimmunol.178.12.7879. [DOI] [PubMed] [Google Scholar]
- 8.Chaplin DD. Overview of the human immune response. J Allergy Clin Immunol. 2006;117(Suppl):S430–S435. doi: 10.1016/j.jaci.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 9.Allard JB, Poynter ME, Marr KA, Cohn L, Rincon M, Whittaker LA. Aspergillus fumigatus generates an enhanced Th2-biased immune response in mice with defective cystic fibrosis transmembrane conductance regulator. J Immunol. 2006;177:5186–5194. doi: 10.4049/jimmunol.177.8.5186. [DOI] [PubMed] [Google Scholar]
- 10.Allard JB, Rinaldi L, Wargo MJ, Allen G, Akira S, Uematsu S, Poynter ME, Hogan DA, Rincon M, Whittaker LA. Th2 allergic immune response to inhaled fungal antigens is modulated by TLR-4-independent bacterial products. Eur J Immunol. 2009;39:776–788. doi: 10.1002/eji.200838932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricks DM, Chen K, Zheng M, Steele C, Kolls JK. Dectin immunoadhesins and pneumocystis pneumonia. Infect Immun. 2013;81:3451–3462. doi: 10.1128/IAI.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schleimer RP, Kato A, Peters A, Conley D, Kim J, Liu MC, Harris KE, Kuperman DA, Chandra R, Favoreto S, Jr, et al. Epithelium, inflammation, and immunity in the upper airways of humans: studies in chronic rhinosinusitis. Proc Am Thorac Soc. 2009;6:288–294. doi: 10.1513/pats.200808-088RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–1284. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol. 2007;19:711–720. doi: 10.1016/j.coi.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 17.Wada K, Kobayashi T, Matsuwaki Y, Moriyama H, Kita H. Alternaria inhibits double-stranded RNA-induced cytokine production through Toll-like receptor 3. Int Arch Allergy Immunol. 2013;161:75–83. doi: 10.1159/000350365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato A, Truong-Tran AQ, Scott AL, Matsumoto K, Schleimer RP. Airway epithelial cells produce B cell-activating factor of TNF family by an IFN-beta-dependent mechanism. J Immunol. 2006;177:7164–7172. doi: 10.4049/jimmunol.177.10.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 20.Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol. 2000;105:1185–1193. doi: 10.1067/mai.2000.106210. [DOI] [PubMed] [Google Scholar]
- 21.Cieslewicz G, Tomkinson A, Adler A, Duez C, Schwarze J, Takeda K, Larson KA, Lee JJ, Irvin CG, Gelfand EW. The late, but not early, asthmatic response is dependent on IL-5 and correlates with eosinophil infiltration. J Clin Invest. 1999;104:301–308. doi: 10.1172/JCI7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen HH, Ochkur SI, McGarry MP, Crosby JR, Hines EM, Borchers MT, Wang H, Biechelle TL, O’Neill KR, Ansay TL, et al. A causative relationship exists between eosinophils and the development of allergic pulmonary pathologies in the mouse. J Immunol. 2003;170:3296–3305. doi: 10.4049/jimmunol.170.6.3296. [DOI] [PubMed] [Google Scholar]
- 23.Marr KA, Carter RA, Crippa F, Wald A, Corey L. Epidemiology and outcome of mould infections in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2002;34:909–917. doi: 10.1086/339202. [DOI] [PubMed] [Google Scholar]
- 24.Beisswenger C, Hess C, Bals R. Aspergillus fumigatus conidia induce interferon-β signalling in respiratory epithelial cells. Eur Respir J. 2012;39:411–418. doi: 10.1183/09031936.00096110. [DOI] [PubMed] [Google Scholar]
- 25.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23:327–333. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 26.Message SD, Johnston SL. Host defense function of the airway epithelium in health and disease: clinical background. J Leukoc Biol. 2004;75:5–17. doi: 10.1189/jlb.0703315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaas AK, Schwartz DA. Innate immunity and the lung: defense at the interface between host and environment. Trends Cardiovasc Med. 2005;15:195–202. doi: 10.1016/j.tcm.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Balloy V, Sallenave JM, Wu Y, Touqui L, Latgé JP, Si-Tahar M, Chignard M. Aspergillus fumigatus-induced interleukin-8 synthesis by respiratory epithelial cells is controlled by the phosphatidylinositol 3-kinase, p38 MAPK, and ERK1/2 pathways and not by the toll-like receptor-MyD88 pathway. J Biol Chem. 2008;283:30513–30521. doi: 10.1074/jbc.M803149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balloy V, Si-Tahar M, Takeuchi O, Philippe B, Nahori MA, Tanguy M, Huerre M, Akira S, Latgé JP, Chignard M. Involvement of toll-like receptor 2 in experimental invasive pulmonary aspergillosis. Infect Immun. 2005;73:5420–5425. doi: 10.1128/IAI.73.9.5420-5425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubourdeau M, Athman R, Balloy V, Huerre M, Chignard M, Philpott DJ, Latgé JP, Ibrahim-Granet O. Aspergillus fumigatus induces innate immune responses in alveolar macrophages through the MAPK pathway independently of TLR2 and TLR4. J Immunol. 2006;177:3994–4001. doi: 10.4049/jimmunol.177.6.3994. [DOI] [PubMed] [Google Scholar]
- 31.Mambula SS, Sau K, Henneke P, Golenbock DT, Levitz SM. Toll-like receptor (TLR) signaling in response to Aspergillus fumigatus. J Biol Chem. 2002;277:39320–39326. doi: 10.1074/jbc.M201683200. [DOI] [PubMed] [Google Scholar]
- 32.Reed CE, Kita H. The role of protease activation of inflammation in allergic respiratory diseases. J Allergy Clin Immunol. 2004;114:997–1008, quiz 1009. doi: 10.1016/j.jaci.2004.07.060. [DOI] [PubMed] [Google Scholar]
- 33.Netea MG, Ferwerda G, van der Graaf CA, Van der Meer JW, Kullberg BJ. Recognition of fungal pathogens by toll-like receptors. Curr Pharm Des. 2006;12:4195–4201. doi: 10.2174/138161206778743538. [DOI] [PubMed] [Google Scholar]
- 34.Hess C, Herr C, Beisswenger C, Zakharkina T, Schmid RM, Bals R. Myeloid RelA regulates pulmonary host defense networks. Eur Respir J. 2010;35:343–352. doi: 10.1183/09031936.00196408. [DOI] [PubMed] [Google Scholar]
- 35.Zhu L, Lee B, Zhao F, Zhou X, Chin V, Ling SC, Chen Y. Modulation of airway epithelial antiviral immunity by fungal exposure. Am J Respir Cell Mol Biol. 2014;50:1136–1143. doi: 10.1165/rcmb.2013-0357OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abad A, Fernández-Molina JV, Bikandi J, Ramírez A, Margareto J, Sendino J, Hernando FL, Pontón J, Garaizar J, Rementeria A. What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Rev Iberoam Micol. 2010;27:155–182. doi: 10.1016/j.riam.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14:432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 38.Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 39.Kato A, Ogasawara T, Homma T, Saito H, Matsumoto K. Lipopolysaccharide-binding protein critically regulates lipopolysaccharide-induced IFN-beta signaling pathway in human monocytes. J Immunol. 2004;172:6185–6194. doi: 10.4049/jimmunol.172.10.6185. [DOI] [PubMed] [Google Scholar]
- 40.Kato A, Homma T, Batchelor J, Hashimoto N, Imai S, Wakiguchi H, Saito H, Matsumoto K. Interferon-alpha/beta receptor-mediated selective induction of a gene cluster by CpG oligodeoxynucleotide 2006. BMC Immunol. 2003;4:8. doi: 10.1186/1471-2172-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 42.Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26:469–476. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 44.Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 45.Rakes GP, Arruda E, Ingram JM, Hoover GE, Zambrano JC, Hayden FG, Platts-Mills TA, Heymann PW. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care: IgE and eosinophil analyses. Am J Respir Crit Care Med. 1999;159:785–790. doi: 10.1164/ajrccm.159.3.9801052. [DOI] [PubMed] [Google Scholar]
- 46.Chai LY, van de Veerdonk F, Marijnissen RJ, Cheng SC, Khoo AL, Hectors M, Lagrou K, Vonk AG, Maertens J, Joosten LA, et al. Anti-Aspergillus human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology. 2010;130:46–54. doi: 10.1111/j.1365-2567.2009.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hallstrand TS, Woodruff PG, Holgate ST, Knight DA. Function of the airway epithelium in asthma. J Allergy (Cairo) 2012;2012:160586. doi: 10.1155/2012/160586. [DOI] [PMC free article] [PubMed] [Google Scholar]