

Abstract
Lipid-laden macrophages, or “foam cells,” are observed in the lungs of patients with fibrotic lung disease, but their contribution to disease pathogenesis remains unexplored. Here, we demonstrate that fibrosis induced by bleomycin, silica dust, or thoracic radiation promotes early and sustained accumulation of foam cells in the lung. In the bleomycin model, we show that foam cells arise from neighboring alveolar epithelial type II cells, which respond to injury by dumping lipids into the distal airspaces of the lungs. We demonstrate that oxidized phospholipids accumulate within alveolar macrophages (AMs) after bleomycin injury and that murine and human AMs treated with oxidized phosphatidylcholine (oxPc) become polarized along an M2 phenotype and display enhanced production of transforming growth factor-β1. The direct instillation of oxPc into the mouse lung induces foam cell formation and triggers a severe fibrotic reaction. Further, we show that reducing pulmonary lipid clearance by targeted deletion of the lipid efflux transporter ATP-binding cassette subfamily G member 1 increases foam cell formation and worsens lung fibrosis after bleomycin. Conversely, we found that treatment with granulocyte-macrophage colony-stimulating factor attenuates fibrotic responses, at least in part through its ability to decrease AM lipid accumulation. In summary, this work describes a novel mechanism leading to foam cell formation in the mouse lung and suggests that strategies aimed at blocking foam cell formation might be effective for treating fibrotic lung disorders.
Keywords: pulmonary fibrosis, foam cells, alveolar macrophages, type II pneumocytes, oxidized phospholipids
Clinical Relevance
This study is the first to suggest that foam cells play a causal role in the development of pulmonary fibrosis and that strategies aimed at blocking their formation might be effective in limiting the onset and/or progression of disease.
Fibrotic lung diseases are a heterogeneous group of disorders that, when progressive, can cause respiratory failure from increased deposition of connective tissue in the distal airspaces of the lung. Although the underlying mechanisms leading to fibrotic responses in the lung remain poorly understood, recent studies indicate that injury to, or dysfunction of, the alveolar epithelium plays a causal role in the disease. This epithelial injury hypothesis is supported by multiple lines of evidence, including human genetic studies, which link mutations in epithelial-specific genes to the development of idiopathic and familial forms of pulmonary fibrosis (1, 2). However, opponents of this theory argue that injury to the epithelium does not easily explain the myriad of other pathological features common to the fibrotic lung, such as alveolar macrophage (AM) M2 polarization and oxidant/antioxidant imbalances (3–5).
An important function of lung epithelium is the de novo synthesis of lipids performed exclusively by alveolar type II epithelial (ATII) cells. This function is required for producing the lipid-rich surfactant that bathes the underlying respiratory epithelium, reducing surface tension and protecting against environmental challenges (6, 7). Maintenance of surfactant lipid homeostasis is required for normal lung function; perturbations in the quantity or quality of surfactant lipids are associated with the development of many lung disorders, including pulmonary fibrosis (8–10).
The precise regulation of surfactant lipids requires a balance between synthesis and clearance of lipids (8, 11). Lipid clearance is ordinarily mediated by ATII cells and, to a lesser extent, AMs (12, 13). Lipid-laden AMs, also called “foam cells,” are common pathological features in animals models of lung fibrosis (14, 15) and in histological specimens from patients with various chronic fibrotic lung diseases (16, 17). Although foam cells are thought to be a consequence, rather than cause, of fibrotic lung disease, their very presence signifies a disruption in surfactant lipid homeostasis. In this study, we sought to elucidate the relationship between foam cells and epithelial injury and to determine whether foam cells play a pathogenic role in fibrotic responses in the lung.
Materials and Methods
Wild-type C57BL/6 mice (6–8 wk old) were purchased from Charles River Laboratories (Bar Harbor, ME). Abcg1−/− mice on C57BL/6 background were generated and maintained as described previously (18). Profibrotic injuries used in these studies included bleomycin, silica, and radiation. Delivery of bleomycin (0.075 U) (Enzo Laboratories, Farmingdale, NY) and silica (20 mg of sterile silica crystal, median diameter, 1–5 μm) (Sigma-Aldrich, St. Louis, MO) was performed using the tongue-pull maneuver in isoflurane-anesthetized mice as described previously (19). Radiation-induced lung injury was induced by exposure to a one-time dose of 16 Gy delivered to the thorax while shielding the rest of the body. For granulocyte-macrophage colony-stimulating factor (GM-CSF) studies, mice received 10 μg of GM-CSF protein (R&D Systems Inc., Minneapolis, MN) or vehicle in a total volume of 100 μl of sterile PBS by daily intraperitoneal injection beginning on Day 3 after bleomycin intratracheal instillation. The bleomycin- and silica-treated mice were killed on Days 1, 3, 7, 14, and 21 after profibrotic injury. In the radiation-induced lung fibrosis model, mice were killed on Weeks 1, 4, and 8 after radiation exposure. Before performing studies, animal protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the Thomas Jefferson University and the National Institute of Environmental Health Sciences. All mice were maintained in a pathogen-free environment certified by the American Association for Accreditation of Laboratory Animal Care.
Mouse Bronchoalveolar Lavage Recovery and Fractionation
Bronchoalveolar lavage (BAL), total cell counts, and differential cell counts were performed as previously described (20). Details are provided in the online supplement.
Oil Red O Staining
Frozen lung tissue and cell pellets from BAL fluid were incubated for 15 minutes at room temperature in Oil Red O solution. After staining, slides were immersed in 85% propylene glycol, rinsed twice in distilled water, and counterstained with Harris hematoxylin.
Isolation of Primary ATII Cells
ATII cells were isolated as described previously (21). Further details are provided in the online supplement.
Lipid Extraction and Analysis
Total lipids were extracted from BAL fluid, macrophages, lung tissue, and conditioned media using a modified method of Bligh and Dyer in chloroform/methanol (2:1) (22). Further details regarding the extraction methods and separation protocols used in our studies are provided in the online supplement.
ATP and Lactic Acid Measurement
Extracellular ATP and lactic acid in BAL fluid was quantified using Colorimetry kits (Bio Vision, Mountain View, CA) according to the manufacturer’s instructions.
Measurement of Oxidized Phospholipids
The level of oxidized phosphatidylcholine (oxPc) in BAL fluid was measured by ELISA using E06 antibody (Avanti Polar Lipids, Alabaster, AL) as described (23).
OxPc Intratracheal Administration
1-Palmitoyl-2(5′-oxi-valeroyl)-sn-glycerol-3-phosphocholine (POVPC) (Avanti Polar Lipids) was administered by intratracheal instillation at a dose of 200 μg/mouse. Mice were killed on Days 5 and 10 after oxPc installation.
Lung Collagen Measurements
Whole lung collagen was determined using a Sircol Collagen Assay kit (Biocolor Ltd., Belfast, Northern Ireland, UK) according to the manufacturer’s instructions.
Lung Histology
Details are provided in the online supplement.
Immunohistochemistry
Details are provided in the online supplement.
Electron Microscopy
Details are described in the online supplement.
RNA Isolation and Analysis
The following gene transcripts were evaluated in this study: sterol regulatory element-binding protein (Srebp), carbohydrate response element binding protein (Chrebp), acetyl CoA carboxylase (Acc), fatty acid synthase (Fasn), 3-hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr), liver X receptor (Lxr)-a, proliferator-activated receptor (Ppar)-γ, ATP-binding cassette subfamily G member 1 (Abcg1), ATP-binding cassette, subfamily A, member 1 (Abca1), cluster of differentiation 36 (Cd36), scavenger receptor A (Sra)-1, transforming growth factor (Tgf)-β1, Tnf-α, Il-1-α, arginase (Arg1), Cd86, Cd163, Ym1, and chemokine (c-c motif) receptor 7 (Ccr7). The housekeeping genes glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and hypoxanthine phosphoribosyltransferase 1 (Hprt1) were used for normalization. Specific details regarding quantitative RT-PCR and the olignucleotide sequences used in these studies are provided in the online supplement.
Western Blot Analysis
Western blot analysis was performed for 5′ adenosine monophosphate-activated protein (AMPK), phosphorylated AMP-activated protein kinase (pAMPK), ACC, phosphorylated ACC (pACC), FASN, HMGCR, SREBP-1, ABCG1, ABCA1, and GAPDH; detailed protocols are described in the online supplement.
In Vitro Exposure to Bleomycin
Details are provided in the online supplement.
In Vitro Production of Foam Cells
Details are provided in the online supplement.
Liposome Preparation
Liposomes were prepared as described previously (24). Details are provided in the online supplement.
Measurement of Liposome Uptake by ATII Cells
The uptake of liposomes was performed as described previously (25). Details are provided in the online supplement.
Measured of TGF-β1 by ELISA
TGF-β1 was measured using a commercially available DuoSet ELISA development kit (R&D Systems Inc.). Details are provided in the online supplement.
Statistical Analysis
Statistics were performed using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA). Two-group comparisons were analyzed by unpaired Student’s t test, and multiple-group comparisons were performed by one-way ANOVA followed by Tukey’s post hoc analysis. Statistical significance was achieved when P < 0.05 at the 95% confidence interval.
Results
Foam Cell Formation Precedes Fibrotic Injury
To understand the relationship of foam cell formation to lung fibrosis, we isolated cells from BAL fluid at several time points after the initiation of a profibrotic injury. Bleomycin injections cause a predictable fibrotic response in the lung, with initial induction of TGF-β1 occurring around Day 3 and with peak collagen deposition taking place at 14 to 21 days after drug exposure. This response is also characterized by early and sustained AM infiltration into the terminal air spaces (see Figure E1 in the online supplement), along with a host of other immune responses within the distal lung. Relevant to this time course, foam cells (Oil Red O staining) were initially detected at 3 days after bleomycin exposure (Figure 1A). As expected, this time point coincided with induction of TGF-β1 protein and Col1α1 messenger RNA (mRNA) expression but preceded histological and biochemical onset of fibrosis in the lung (Figures 1B–1E). The number of foam cells and the amount of lipids within macrophages increased at subsequent time points after injury. At 14 days after bleomycin administration, when pulmonary TGF-β1 expression and collagen deposition were maximally detected, AMs were significantly increased in size, with approximately one-fifth (22 ± 2% SE) of the cells staining for neutral lipids (Oil Red O staining) and all cells demonstrating increased staining for phospholipids (Figure 1F). Further analysis demonstrated a 4-fold increase in intracellular cholesterol and a 2.5-fold increase in phospholipids in freshly isolated AMs at this time point (Figure 1G). Moreover, electron microscopy of affected lungs demonstrated progressive cytoplasmic phospholipid accumulation (i.e., lamellar bodies) in AMs from 3 to 14 days (Figure 1H).
Figure 1.
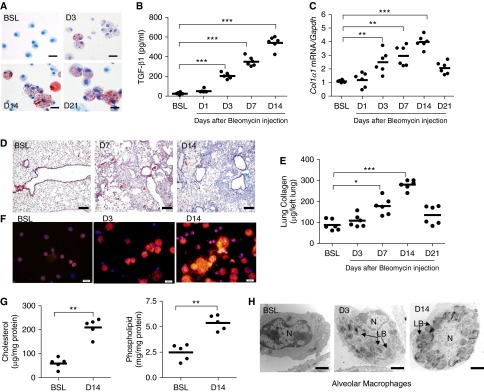
Bleomycin induces foam cell formation in the lung. (A) Oil Red O staining of alveolar macrophages (AMs) cytospun onto glass slides. AMs were isolated from bronchoalveolar lavage (BAL) fluid at baseline (BSL) and at Day (D) 3, D14, and D21 after bleomycin (n = 5, each group). Scale bar, 20 μm. (B) Transforming growth factor (TGF)-β1 in BAL fluid at BSL and at D1, D3, D7, and D14 after bleomycin (n = 6, each group). (C) Quantitative messenger RNA (mRNA) expression for Col1α1 gene in whole lung at BSL and at D1, D3, D7, D14, and D21 after bleomycin administration. Gene expression is normalized to glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (n = 6, each group). (D) Trichrome staining of lung at BSL and D7 and D14 after bleomycin administration. Increases in collagen (blue) are evident in lung at D7 and D14 after bleomycin (n = 3) but not at D3 after injury (data not shown). The same magnification was used in all images (scale bar, 75 μm). (E) Quantification of lung collagen (Sircol assay) at BSL and at D3, D7, D14, and D21 after bleomycin administration (n = 6). (F) Nile red staining of AMs after cells were cytospun onto glass slides. AMs were isolated at BSL and at D3 and D14 after bleomycin (n = 5). Scale bar, 20 μm. (G) Cholesterol and phospholipid content in freshly isolated AMs from BSL and D14 bleomycin-injured mice (n = 5). (H) Representative electromyographic images of AMs at BSL and at D3 and D14 after bleomycin. Marked intracellular phospholipid accumulation (black onion-skin structures consistent with lamellar bodies [LB]) is evident in AMs at D3 and D14 after bleomycin injury (scale bar, 2 μm). The nucleus is labeled N. Data are expressed as mean ± SE. In B, C, and E, the statistical significance was assessed with one-way ANOVA; in G, Student’s t test was used. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the BSL group.
To determine the generalizability of our observation that a profibrotic insult promotes foam cell formation, we tested alternative models of fibrotic pulmonary insult. We observed foam cell formation in the lung after silica exposure (Figure E2A) and after radiation injury (Figure E2B); in both cases, the initial detection of foam cells preceded (radiation) or coincided with (silica) the biochemical and the histological onset of fibrosis in these model systems (Figures E2C–E2E and data not shown). Foam cell formation was associated with changes in macrophage gene expression characteristic of increased intracellular lipid accumulation; when compared with macrophages from uninjured lungs, we observed increased transcript levels for high-affinity lipid receptors, including Cd36, Sra-1, and Lxr-α, and for the lipid export proteins ATP binding cassette transporters A1 (Abca1) and G1 (Abcg1) at 3 and 14 days after bleomycin administration (Figures E3A and E3B). Increased ABCA1 and ABCG1 protein expression was also observed at 14 days after bleomycin (Figure E3C), and 14-day silica treatment also increased Cd36, Sra-1, and Lxr-α mRNA expression (Figure E3D). These findings indicate that foam cell formation after bleomycin or silica administration precedes overt fibrosis in several models of pulmonary fibrosis.
Profibrotic Injury Promotes Extracellular Surfactant Lipid Accumulation
Macrophages are capable of de novo lipid production and sequestration of extracellular lipids (21, 22). We first tested whether foam cell formation occurred in the setting of increased extracellular lipids by performing quantitative lipid analysis of BAL fluid from control and injured mice. On Day 3 after bleomycin or silica administration, we observed that cholesterol, triglyceride, and phospholipid levels were markedly increased in cell-free BAL fluid (Figures 2A–2C; Figure E4), paralleling the time points at which foam cells were initially detected in these model systems. All three lipid fractions remained elevated in BAL fluid at 14 days after insult, indicating that the increases in surfactant lipid concentrations were sustained after the initial injury. The peak in BAL fluid lipid concentration after bleomycin occurred at 14 days (Figures 2A–2C), which corresponded to the time point when TGF-β1 protein and collagen mRNA and protein expression were maximally expressed in the fibrotic lung (Figures 1B, 1C, and 1E). Similar results were observed in the radiation injury model of pulmonary fibrosis (Figures E5A–E5D). Collectively, these findings indicate that profibrotic injury triggers surfactant lipid accumulation, which in turn promotes AM foam cell formation in the injured lung.
Figure 2.
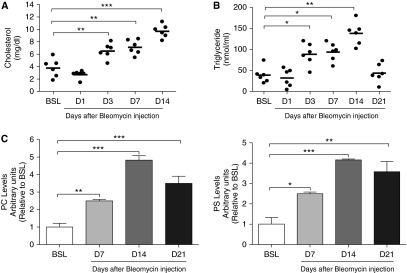
Profibrotic injury promotes surfactant lipid accumulation in BAL fluid after bleomycin. (A) Total cholesterol in BAL fluid at BSL and at D1, D3, D7, and D14 after bleomycin (n = 6). (B) Triglyceride concentration in BAL fluid at BSL and at D1, D3, D7, and D14 days after bleomycin (n = 6). (C) Thin-layer chromotography performed for phospholipids on BAL fluid from mice (n = 6) at BSL and at D7, D14, and D21 after bleomycin. Phosphatidylcholine (PC) and phosphatidylserine (PS) are increased at D7, D14, and D21 (P < 0.01) after bleomycin (based on densitometry measurements). Data are expressed as mean ± SE. The statistical significance was assessed with a one-way ANOVA test. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the BSL group.
Bleomycin Induces Metabolic Stress in ATII Cells
To evaluate whether increased lipid production in pulmonary cells in general, and in ATII cells in particular, was responsible for the marked changes in BAL lipid levels, we performed gene expression profiling on whole lung lysates. Because similar lipid abnormalities were observed in each of the three profibrotic models of lung injury, the remainder of our investigation was focused on the bleomycin model. Despite the observed increase in extracellular lipids (above), we found that transcript levels for the component of the lipid synthesizing machinery, including Srebp-1c, Chrebp, Fasn, and Hmgcr, were decreased in the lung at Day 1 and remained below baseline expression levels at 3, 7, and 14 days after injury (Figure 3A). Down-regulation of these transcripts was verified in purified populations of ATII cells isolated from Day 7 bleomycin-injured lungs (Figure E6A). Western blot analyses of whole lung lysates confirmed down-regulation of SREBP-1c, FASN, and HMGCR proteins at 7 days after bleomycin injury (Figures E6B–E6D), suggesting that lipid synthesis is inhibited by bleomycin.
Figure 3.
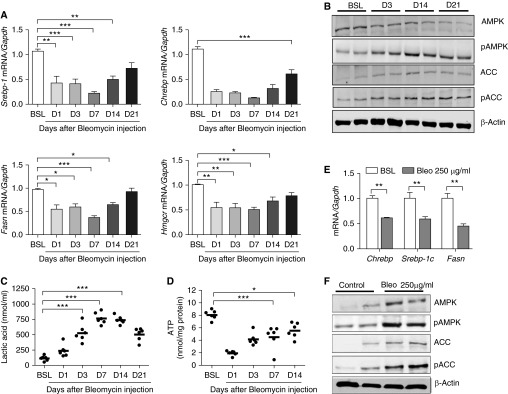
Bleomycin induces metabolic stress in alveolar type II epithelial (ATII) cells. (A) Quantitative mRNA expression for sterol-response element binding protein (Srebp-1), carbohydrate response element binding protein (Chrebp), fatty acid synthase (Fasn), and 3-hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr) at BSL and at various time points after bleomycin (Bleo; n = 6). (B) Western blot analysis for total adenosine monophosphate–activated protein kinase (AMPK) and phosphorylated AMPK (pAMPK) (Thr172) and acetyl CoA carboxylase (ACC) (Ser79) in whole lung homogenates at BSL and at D3, D14, and D21 after bleomycin. The image is representative of two independent experiments. (C) Lactic acid in BAL fluid at BSL and at D1, D3, D7, D14, and D21 after bleomycin (n = 6). (D) Tissue ATP concentration in whole lung at BSL and at D1, D3, D7, and D14 after bleomycin (n = 6). (E) Quantitative mRNA expression for Chrebp, Srebp-1, and Fasn in MLE-12 cells cultured in the presence or absence of bleomycin (n = 3 independent experiments). (F) Western blot analysis for total and pAMPK and ACC in MLE-12 cells cultured in the presence or absence of bleomycin. The image is representative of at least two independent experiments. Data are expressed as mean ± SE. In A, C, and D, the statistical significance was assessed with one-way ANOVA; Student’s t test was used in E. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the BSL group. pACC = phosphorylated acetyl CoA carboxylase.
Because down-regulation of lipid synthesis is a classic starvation response (26), we assessed the metabolic status of the lung by measuring levels of pAMPK at various time points after bleomycin injury. AMPK is the master energy “switch,” which, when activated by phosphorylation at Thr172, serves to inhibit anabolic pathways (such as lipid synthesis) and to activate catabolic pathways (such as fatty acid oxidation) (27). Consistent with our gene expression profiling results, bleomycin induced early and sustained phosphorylation of AMPK and phosphorylation of its downstream target, ACC, in the lung (Figure 3B). These metabolic changes were associated with a shift toward aerobic glycolysis represented by increased lactic acid production (Figure 3C). Consistent with these observations, tissue ATP levels, which provide a measure of energy stores, were significantly decreased in whole lung tissues at 3, 7, and 14 days after bleomycin administration (Figure 3D). At later time points, when fibrotic responses were decreasing (e.g., TGF-β1 and collagen levels were falling), lactic acid and tissue ATP levels began to normalize (Figures 3C and 3D), suggesting that resolution of fibrotic responses is associated with the recovery of metabolic homeostasis.
We next assessed the ability of bleomycin to directly induce a starvation response in alveolar epithelial cells using the ATII model cell line MLE-12 (28, 29). After exposure to bleomycin for 24 hours, metabolic changes in MLE-12 cells recapitulated those observed in whole lungs, including down-regulation of factors necessary for lipid synthesis (Chrebp, Srebp-1c, and Fasn) (Figure 3E), phosphorylation of AMPK and ACC (Figure 3F), and increased production of lactic acid (Figure E7). Taken together, these findings indicate that bleomycin induces metabolic stress and inhibits surfactant lipid synthesis in ATII cells.
Bleomycin Promotes Release and Impairs the Reuptake of Surfactant Lipids from ATII Cells
Because bleomycin inhibited lipid synthesis in ATII cells, we tested whether the injury-induced accumulation of lipids in BAL fluid resulted from release of intracellular lipids from the pulmonary epithelium. Using Oil Red O staining, we quantified epithelial cell lipid levels and observed a reduction in the number of lipid droplets in the ATII cells on Day 3 after bleomycin exposure (data not shown). Moreover, quantitative analysis of tissue lipid concentrations showed that cholesterol, triglyceride, and phospholipid levels were markedly decreased after injury (Figures 4A and 4B), and this corresponded to time points when lipids were increased in BAL fluid. We observed decreased staining for the lamellar body–associated protein ABCA3, which suggested that the reduction in tissue lipid concentrations resulted from depletion of intracellular lipid pools from the ATII cells (Figure 4C). These findings were corroborated by electron microscopy of the lung, which showed that the average size of the lamellar bodies in ATII cells was markedly decreased at Days 3 and 14 after bleomycin administration (Figure 4D).
Figure 4.
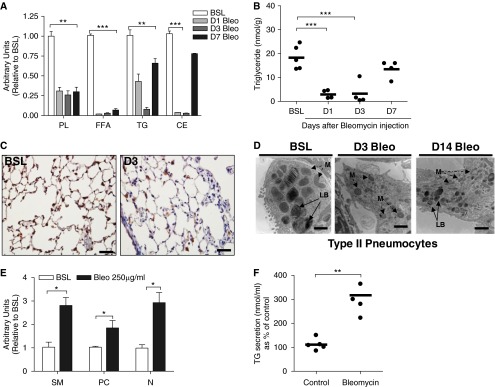
Bleomycin injury induces surfactant release from ATII cells. (A) Thin-layer chromatography for tissue lipids (phospholipids [PL], free fatty acids [FFA], triglycerides [TG], and cholesterol esters [CE]) in whole lung at BSL and at D1, D3, and D7 after bleomycin. By densitometry, PL, FFA, TG, CE are decreased at D1, D3, and D7 after bleomycin (n = 5, each group). (B) Triglyceride concentration in whole lung at BSL and at D1, D3, and D7 after bleomycin (n = 4, each group). (C) Immunohistochemical staining for ABCA3. Staining for ABCA3 (brown) is decreased on D3 after bleomycin. The same magnification was used in all images detection with a 40× objective lens. Scale bar, 100 μm. (D) Representative electron microscopic images of ATII cells at BSL and at D3 and D14 after bleomycin. Marked reduction in the size of LBs is observed in ATII cells after bleomycin. Scale bar, 2 μm. M, mitochondria. (E) Bleomycin promotes release of phospholipids from MLE-12 cells. By densitometry, PC, sphingomyelin (SM), and neutral lipids (N) are increased after bleomycin treatment (P < 0.05). The image is representative of two independent experiments. (F) Bleomycin promotes release of triglycerides from MLE-12 cells (n = 5 independent experiments). Data are expressed as mean ± SE. In A through D, the statistical significance was assessed with one-way ANOVA; a Student’s t test was used in E and F. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the BSL group.
We next tested whether bleomycin directly stimulates lipid release from ATII-type cells. Bleomycin treatment of MLE-12 cells resulted in marked enhancement of phospholipid and triglyceride levels in conditioned media (Figures 4E and 4F). We speculate that the observed release of surfactant lipids from ATII cells is mediated in part by extracellular ATP because we detected marked accumulation of this well-known surfactant secretagogue in the extracellular fluid after injury (Figure E8A).
Because ATII cells continuously recycle surfactant lipids, we also examined whether impaired lipid reuptake might contribute to extracellular lipid accumulation in injured lungs. We assessed the ability of MLE-12 cells to internalize lipids in the presence or absence of bleomycin. As shown in Figure E8B, treatment with bleomycin markedly reduced surfactant lipid uptake in MLE-12 cells. This impairment was also observed when MLE-12 cells were treated with 2-deoxy-d-glucose, an inhibitor of glycolysis, indicating that metabolic stress, even in the absence of profibrotic injury, impairs surfactant lipid regulation (Figure E8C). Collectively, our data indicate that in the injured lung, surfactant lipid accumulation in BAL fluid results from increased lipid release paired with reduced reuptake of lipids in ATII cells. Our gene expression profiling of injured lungs and MLE-12 cells suggests reduced capacity of injured ATII cells to synthesize lipids de novo.
Foam Cell Formation Induces the Production of TGF-β1
In order for alveolar foam cells to be implicated in the fibrotic response to pulmonary insult, we reasoned that foam cells would exhibit altered immune responses when compared with healthy AMs. We first examined whether lipid uptake by AMs is sufficient to alter macrophage immune phenotype. We found that transcript levels for Tgf-β1 were markedly increased in freshly isolated AMs from Day 14 bleomycin-injured lungs compared with baseline AMs (Figure 5A). Moreover, expression of the M2 marker Ym1 (Figure 5B) and the lipid receptor/transporter genes (Cd36, Sra-1, Lxr-α, Abca1, and Abcg1; see Figures E3A and E3B) were increased in macrophages from the bleomycin-injured lung. No differences in expression of the M1 marker Tnf-α were observed in macrophages from control and bleomycin-injured lungs, further suggesting a shift toward an M2 or “reparative” macrophage phenotype (25, 26) that favors collagen deposition and fibrosis.
Figure 5.
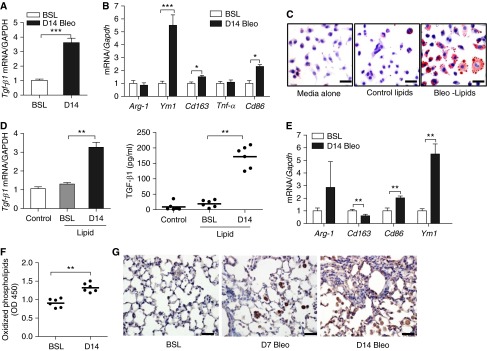
Foam cell formation promotes macrophage production of TGF-β1. (A) Quantitative mRNA expression for Tgf-β1 in AMs isolated from lung at BSL and at 14 days after bleomycin (n = 6). (B) Quantitative mRNA expression for Arg-1, Cd163, Cd86, and Ym1 in AMs isolated from BAL fluid at BSL and at 14 days after bleomycin (n = 6). (C) Oil Red O staining of MH-S cells after culture in media alone or in media supplemented with surfactant lipid extracts from the lungs of mice at BSL or at 14 days after bleomycin. Scale bar, 20 μm. (D) Measurement of Tgf-β1 transcript (left) and protein (right) in MH-S cells. Cells were cultured in media alone or in media supplemented with lipid extracts from uninjured or Day 14 bleomycin-injured lungs (n = 3 independent experiments). (E) Quantitative mRNA expression for Arg-1, Cd163, Cd86, and Ym1 in MH-S cells. Cells were cultured in media alone or in media supplemented with lipid extracts from uninjured or from Day 14 bleomycin-injured lungs (n = 3 independent experiments). (F) ELISA for oxidized phosphatidylcholine (oxPc) in BAL fluid at BSL and at 14 days after bleomycin (n = 5). (G) Immunohistochemical staining for oxPc (brown stain) in lung at BSL and at 7 and 14 days after bleomycin. Staining for oxPc is increased in AMs after bleomycin. The same magnification was used in all images detection with a 40× objective lens. Data are expressed as mean ± SE. Scale bar, 20 μm. In A through F, the statistical significance was assessed with Student’s t test; one-way ANOVA was used in B and E. OD, optical density. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the BSL group or control.
To further assess the effects of lipid uptake on macrophage phenotype, we cultured murine AM MH-S cells with lipid extracts isolated from BAL fluid of either control or Day 14 bleomycin-exposed mice. The decision to isolate lipids on Day 14 after injury was based on the fact that surfactant lipids, foam cells, and TGF-β1 levels were most abundant at this time point. As shown in Figure 5C, lipid extracts from control and injured mice induced foam cell formation; however, the number and size of foam cells were noticeably increased when MH-S cells were cultured with lipids from Day 14 bleomycin-injured lung. Consistent with our in vivo findings, lipid extracts from bleomycin-injured lungs also induced expression of Tgf-β1 (Figure 5D) and Ym1 (Figure 5E) when compared with equivalent quantities of lipids from control mice (Figure E9A). This observation suggests that the type of lipid, rather than the quantity of lipids, is a determinant of foam cell phenotype and that bleomycin-exposed BAL fluid is a potent inducer of TGF-β1 production.
Oxidized Phosphatidylcholine Promotes M2 Polarization and Induces Lung Fibrosis in Mice
Having observed that the lipid profile of injured lungs is altered and that these changes in lipid composition modulate AM functions, we sought to characterize the response of AMs to diverse lipid subtypes. Because reactive oxygen species are thought to play a role in the pathogenesis of fibrotic lung diseases (30), we postulated that reactive oxygen species–mediated lipid peroxidation might explain the differential effects of lipids from control and bleomycin-injured mice on TGF-β1 production in AMs. We detected modest but significant increases in oxPc in the BAL fluid of bleomycin-treated mice (Figure 5F). In contrast, immunohistochemical staining of lung demonstrated marked intracellular accumulation of oxPc in AMs at early and late time points after bleomycin injury (Figure 5G). These findings suggest that oxPc may play a role in driving macrophage fibrotic responses. To further test this hypothesis, MH-S cells were cultured in the presence of oxPc or nonoxidized phosphatidylcholine (Pc). Treatment with oxPc, but not native Pc, resulted in marked enhancement of Tgf-β1 mRNA and protein expression (Figure E9B). Moreover, oxPc induced expression of Ym1, Cd36, Cd163, and Cd86 transcripts, indicating polarization of macrophages to an M2 macrophage phenotype (Figure E9C). Culture of MH-S cells with oxidized low-density lipoprotein did not induce TGF-β1 production (data not shown), suggesting specificity for oxPc in promoting profibrotic responses. Consistent with findings with murine AMs, oxPc induced TGF-β1 production and promoted M2 polarization by increasing transcript levels for Ym1 and Cd163 in primary human AMs (Figures E9D and E9E). Finally, to confirm the sufficiency of phospholipid oxidation to induce lung fibrosis, we instilled oxPc directly into the tracheal lumen of mice. We found that oxPc promoted foam cell formation and readily induced lung fibrosis, as demonstrated by increased Tgf-β1, Col1α1 transcript levels, and collagen content in the lung at 5 and 10 days after instillation (Figures 6A–6E). As expected based on the abundance of Pc in the lung, instillation of native Pc did not induce lung fibrosis (data not shown).
Figure 6.
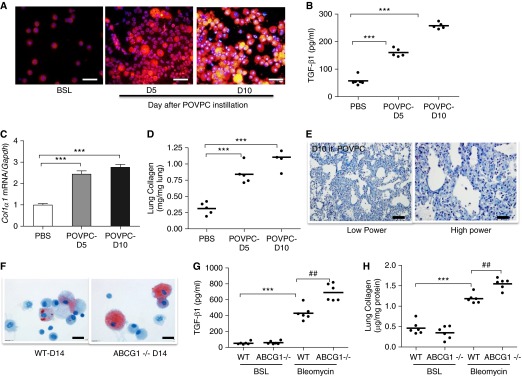
Oxidized phospholipids induce lung fibrosis in mice. (A) Nile red staining of AMs after cells were cytospun onto glass slides. AMs were isolated at BSL and at 5 and 10 days after oxidized phospholipid (POVPC) instillation (n = 5). Scale bar, 20 μm. (B) TGF-β1 in BAL fluid at BSL and at 5 and 10 days after POVPC instillation (n = 5). (C) Quantitative mRNA expression for Col1α1 in whole lung at BSL and at 5 and 10 days after POVPC instillation. Gene expression is normalized to Gapdh. (D) Quantification of lung collagen (Sircol assay) at BSL and at 5 and 10 days after POVPC. (E) Trichrome stain of the lung at BSL (data not shown) and at 10 days after POVPC. Collagen is markedly increased after POVPC administration. Low power (scale bar, 100 μM); high power (scale bar, 20 μm). (F) Oil Red O staining of AMs from wild-type (WT) and Abcg1−/− mice at 14 days after bleomycin. The number and size of foam cells is increased in Abcg1−/− mice when compared with WT mice (n = 6). Scale bar, 20 μm. (G) TGF-β1 in BAL fluid from WT and Abcg1−/− mice at 14 days after bleomycin. TGF-β1 concentration is increased in BAL fluid from Abcg1−/− mice when compared with age-matched WT mice (n = 6). (H) Quantification of lung collagen (Sircol assay) in WT and Abcg1−/− mice at 14 days after bleomycin. Collagen content is increased in lung from Abcg1−/− mice when compared with age-matched WT mice (n = 6). Data are expressed as mean ± SE. The statistical significance was assessed with one-way ANOVA. ***P < 0.001 versus the BSL or PBS groups; ##P < 0.01 versus the WT-bleomycin group.
Targeted Deletion of Abcg1 Increases Foam Cell Formation and Worsens Lung Fibrosis
Having established that oxPc is sufficient to induce foam cell formation from AMs and to promote fibrosis in mice, we tested whether impaired macrophage lipid clearance could further exacerbate the fibrotic response by inducing lung fibrosis with bleomcyin in mice that are globally deficient in the lipid efflux transporter Abcg1. Compared with control mice, the number (17 ± 2% wild-type versus 35 ± 2% Abcg1−/− mice) and size of foam cells was increased in lungs from Abcg1−/− mice at 14 days after bleomycin administration (Figure 6F). Moreover, bleomycin treatment of the Abcg1−/− mice was associated with exaggerated pulmonary fibrotic responses as determined by increases in TGF-β1 production, collagen content, and Col1α1 transcript levels (Figures 6G and 6H and data not shown). Together, these findings suggest that differences in the ability to clear lipids from the lung may influence the onset and/or progression of fibrotic responses in the lung.
Suppressing Lipid Release from ATII Cells Decreases Foam Cell Formation and Attenuates Bleomycin-Induced Lung Fibrosis
Because GM-CSF is known to regulate surfactant lipid homeostasis, at least in part, by its ability to suppress lipid release from ATII cells (31, 32), we tested whether this cytokine might be effective in reducing lung fibrosis by limiting toxic lipid accumulation. We detected a sustained down-regulation in GM-CSF in lung at 1, 3, 7, 14, and 21 days after bleomycin administration, suggesting that its deficiency might play a role in surfactant accumulation (Figure 7A). We administered GM-CSF (10 μg) or vehicle to mice by daily intraperitoneal injection after bleomycin administration. Consistent with its role in modulating surfactant lipid metabolism, we found that treatment with GM-CSF reduced Oil Red O staining in AMs and significantly decreased cholesterol, triglyceride, and free fatty acid in BAL fluid (Figure 7B–7E). This reduction in lipid accumulation was also associated with decreased staining for oxidized phospholipids in the lung. Moreover, we found that GM-CSF decreased fibrotic responses in the lung as quantified by decreased Col1α1 transcripts, TGF-β1 levels, and collagen content in the lung after bleomycin treatment (Figures 7F–7J).
Figure 7.
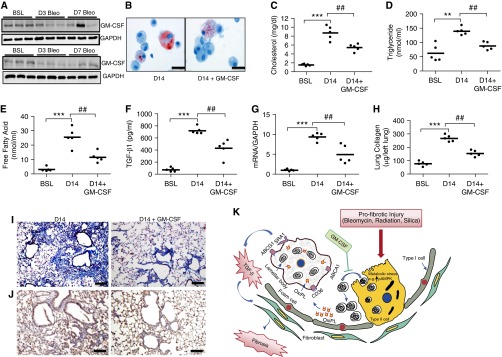
Treatment with granulocyte-macrophage colony-stimulating factor (GM-CSF) attenuates lung lipid abnormalities and abrogates the fibrotic response to bleomycin injury. (A) Western blot analysis for GM-CSF at BSL and at D3, D7, D14, and D21 after bleomycin (n = 6). The image is representative of at least two independent experiments. (B) Oil Red O staining of AMs cytospun onto glass slides. Scale bar, 20 μm. AMs were isolated from BAL fluid at D14 after bleomycin in mice treated with vehicle or GM-CSF for 14 consecutive days (n = 5, each group). (C–E) Cholesterol, triglyceride, and free fatty acid concentration in BAL fluid from bleomycin-injured mice treated with daily with vehicle or GM-CSF (10 μg, intraperitoneally) (n = 5). (F) TGF-β1 in BAL fluid at D14 after bleomycin in mice treated with vehicle or with daily GM-CSF (10 μg, intraperitoneally) (n = 5). (G) Quantitative mRNA expression for Col1α1 in lung at BSL and at D14 after bleomycin in mice treated with vehicle or with daily GM-CSF (10 μg, intraperitoneally) (n = 6). (H) Quantification of lung collagen (Sircol assay) at BSL and at D14 after bleomycin in mice treated with vehicle or with daily GM-CSF (intraperitoneally; n = 5). (I) Trichrome staining of the lung at BSL and at D14 after bleomycin in mice treated with vehicle or with daily GM-CSF (10 μg, intraperitoneally). The same magnification was used for acquiring all images. Scale bar, 100 μm. (J) Immunohistochemical staining for oxPc (brown stain) in lung at D14 after bleomycin in mice treated with daily vehicle or GM-CSF (10 μg, intraperitoneally). The same magnification was used for acquiring all images. Scale bar, 100 μm. (K) Schematic illustration of the proposed sequence of events leading to lung fibrosis after bleomycin. Profibrotic injury induces metabolic stress in lung epithelium, leading to accumulation of abnormal lipids in the distal airspaces of the lung. Uptake of oxPc by macrophages leads to foam cell formation and promotes TGF-β1 production. Treatment with GM-CSF attenuates metabolic stress, extracellular lipid accumulation, foam cell formation, and the severity of lung fibrosis after bleomycin. Data in this figure are expressed as mean ± SE. The statistical significance was assessed with one-way ANOVA. **P < 0.01 and ***P < 0.001, bleomycin versus the BSL group; ##P < 0.01, bleomycin alone versus the bleomycin plus GM-CSF group. ABCA1, ATP-binding cassette, subfamily A, member 1; ABCG1, ATP-binding cassette subfamily G member 1; CD36, cluster of differentiation 36; OxiPL, oxidized phospholipid; SRA1, scavenger receptor A1.
To further elucidate the mechanisms by which GM-CSF attenuates lung fibrosis, we examined the effects of GM-CSF on metabolism and lipid regulation in lungs tissue and ATII cells. Pretreatment with GM-CSF significantly reduced bleomycin-induced metabolic stress in whole lungs and blocked AMPK phosphorylation (Figure E10A), largely reversed the down-regulation of lipogenic genes (Chrebp, Srebp-1c, and Fasn) (Figure E10B) in response to bleomycin, and suppressed the production of lactic acid (Figure E10C). Further, extracellular lipid accumulation was reduced with GM-CSF treatment (Figure E10D), providing a possible explanation for why extracellular lipids and foam cells were decreased in BAL fluid in GM-CSF–treated mice. These findings suggest that GM-CSF attenuates lung fibrosis, at least in part, through its ability to reduce metabolic stress in ATII cells and to block toxic lipid accumulation, which alters macrophage functions (Figure 7K).
Discussion
There are two competing theories regarding the pathogenesis of lung fibrosis; one proposes that epithelial cell injury is the inciting event (33), and the other maintains that inflammation drives the development of the disease (34). In this study, we provide evidence supporting both models: our work identifies a paracrine lipid signaling axis linking epithelial injury to macrophage activation, collectively leading to pulmonary fibrosis.
Our work indicates that diverse epithelial insults can culminate in the disruption of alveolar epithelial cell metabolism and surfactant lipid homeostasis. Specifically, we show that profibrotic injury induces early and sustained metabolic changes to ATII cell metabolism and that these changes involve the establishment of an energy-deprived state characterized by activation of AMPK, inhibition of lipid synthesis, and the release of stored lipids into the distal airspaces of the lung (35). Ensuing accumulation of oxidized lipids in the extracellular space is sufficient to promote development of macrophage foam cells that engender a fibrotic cascade.
We postulate that these metabolic changes are in part an adaptive response, enabling ATII cells to reduce their energy demands while responding to injury. Increased activation of AMPK was recently noted in the lungs of patients with idiopathic pulmonary fibrosis, suggesting a common pathway that links a metabolic stress response to pulmonary fibrosis in mice and humans (36). Our work also identifies a relationship between recovery from metabolic stress and the resolution of fibrotic responses. This relationship implies that monitoring metabolic changes in the lung may be useful for assessing disease activity in patients with lung fibrosis. This hypothesis is supported by recent studies showing that lactic acid and extracellular ATP levels are increased in BAL fluid from patients with idiopathic pulmonary fibrosis and that higher levels are found during acute exacerbations of the disease (37).
Another important manifestation of ATII cell injury identified in this study was the accumulation of surfactant lipids, including oxidized phospholipids, in the BAL fluid. This was found to be a critical element in inducing foam cell formation, which, in turn, promotes the fibrotic response. We found that extracellular lipid accumulation initially results from the depletion of intracellular lipid stores as both the number and size of lamellar bodies in ATII cells were markedly decreased at early time points after injury. We also found that profibrotic injury impaired the reuptake of surfactant lipids by ATII cells, explaining how surfactant lipid levels remained elevated despite sustained decreases in expression of the lipid synthetic machinery. Although not yet confirmed, we postulate that lipid accumulation serves as an important stimulus for macrophage influx into the lung, a necessary response if excess extracellular lipids are to be cleared from the airspaces and if fibrosis is to be promoted to protect an injured epithelial barrier. This hypothesis might explain why macrophages accumulate in the human fibrotic lung and continue to migrate into the murine lung even at very late time points after bleomycin injury (Figure E1).
Pulmonary macrophages are known to be a major source of TGF-β1 in fibrotic lungs, and their depletion in mice has been shown to attenuate the development of lung fibrosis (38–41). That said, there has been ongoing debate as to whether macrophage activation is the cause or the consequence of fibrotic disease. In this study, we demonstrate that macrophage activation and induction of TGF-β1 is not only a consequence of epithelial injury but is also a response that reflects a change in macrophage phenotype to an M2-like (foam cell) phenotype. Macrophage functions are altered after fibrotic insult, with up-regulation of lipid receptors and consequent lipid accumulation, presumably all part of an adaptive response to a fibrotic insult. These findings might explain the general lack of efficacy of anti-inflammatory therapies in treating fibrotic lung conditions; suppressing immune cell functions might actually impede an adaptive response to injury.
Our investigation of macrophage foam cells in the lung has been informed by important studies in the field of atherosclerosis (42, 43). Taking advantage of prior understanding regarding mediators of macrophage lineages, we were able to demonstrate that deletion of a known macrophage lipid transporter, Abcg1, exacerbates lung fibrosis (presumably by limiting the ability of foam cells to efflux intracellular lipids). These findings suggest that pharmacological approaches aimed at enhancing lipid efflux or preventing foam cell formation may prove effective in preventing or treating lung fibrosis. Consistent with this hypothesis, it was recently reported that mice deficient in the lipid uptake receptor Cd36 produce lower levels of profibrotic cytokines and are protected from developing pulmonary fibrosis after treatment with bleomycin (44). Although not yet proven, we postulate that reduced foam cell formation and suppressed TGF-β1 production may account for the improved pulmonary phenotype in these mice. The fine line between appropriate inflammatory response to insult and an exuberant response that leads to permanent fibrosis remains to be fully established.
Although oxidative stress has been a recognized feature of fibrotic lung diseases, our work provides insight into how oxidative injury contributes to the pathogenesis of these disorders (45). We determined that oxidized phospholipids accumulate in the insulted murine lung and that oxPc serves as an important driver of profibrotic macrophages in rodents and humans. Although the role of oxPc in human fibrotic lung disease has not been adequately studied, it is well recognized that oxidized phospholipids accumulate in the lungs of patients with various fibrotic lung conditions (46). Collectively, these findings suggest that blocking oxPc uptake by macrophages may have a therapeutic role in limiting fibrotic response in the human lung.
Although our work did not test a wide array of oxidized lipid species for their ability to generate a fibrotic response, the inability of oxidized low-density lipoprotein to induce a reparative macrophage phenotype suggests that not all oxidized lipids can promote fibrosis. Further, the observation that nonoxidized phospholipids fail to induce macrophage TGF-β1 production provides a possible explanation for why patients with pulmonary alveolar proteinosis, a condition associated with massive phospholipid accumulation in the airspaces, do not develop lung fibrosis; this condition is not associated with ATII injury or with perturbations in oxidant/antioxidant balance (47, 48).
We believe that our findings suggest an important role for GM-CSF in lung fibrosis. The significant decrease in GM-CSF levels in the lung after bleomycin treatment and the inhibition of fibrosis after intraperitoneal administration of GM-CSF argue that this cytokine is likely to be protective against fibrosis. Our work indicates that the protective effects of GM-CSF are mediated in part by limiting ATII cell injury, lipid accumulation, and alveolar foam cell formation. We recognize, however, that GM-CSF is a pleiotropic hormone that has been shown to attenuate fibrotic responses through alternative mechanisms, such as augmenting production of antifibrotic factors and enhancing wound epithelialization (49, 50). In addition, GM-CSF has been shown to enhance macrophage lipid clearance. However, whether GM-CSF therapy will protect against pulmonary fibrosis in humans remains to be studied.
In summary, our work describes a mechanism whereby diverse triggers of epithelial injury cause metabolic perturbations that lead to the release of lipids into the airspaces, thereby stimulating the development of macrophage foam cells, which then in turn promote pulmonary fibrosis. Our work provides a rationale for pursuing new pharmacological approaches aimed at restoring metabolic health and reducing foam cell formation in the lungs of patients with chronic fibrotic lung disease.
Footnotes
This work was supported by National Institutes of Health grant R01HL105490 and by Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences grant Z01 ES102005.
Author Contributions: F.R. and R.S. conceived of the study. M.K., K.W., R.B.P., M.B.F., and C.B.K. assisted with the design of individual experiments. F.R., D.S., M.D., and W.S. performed the in vitro experiments. B.L., F.R., D.S., M.D., W.S., J.M., and M.B.F. performed the in vivo experiments. F.R. and R.S. wrote the paper. C.B.K., K.W., and M.B.F. assisted with editing the manuscript. R.S. supervised the entire project.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0343OC on November 19, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, van Es HW, van den Bosch JM, Grutters JC. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182:1419–1425. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- 2.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walters DM, Cho HY, Kleeberger SR. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid Redox Signal. 2008;10:321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 4.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol. 2014;50:825–837. doi: 10.1165/rcmb.2013-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingenito EP, Mora R, Mark L. Pivotal role of anionic phospholipids in determining dynamic behavior of lung surfactant. Am J Respir Crit Care Med. 2000;161:831–838. doi: 10.1164/ajrccm.161.3.9903048. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt R, Meier U, Yabut-Perez M, Walmrath D, Grimminger F, Seeger W, Günther A. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med. 2001;163:95–100. doi: 10.1164/ajrccm.163.1.9903029. [DOI] [PubMed] [Google Scholar]
- 8.Sunaga H, Matsui H, Ueno M, Maeno T, Iso T, Syamsunarno MR, Anjo S, Matsuzaka T, Shimano H, Yokoyama T, et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat Commun. 2013;4:2563. doi: 10.1038/ncomms3563. [DOI] [PubMed] [Google Scholar]
- 9.Young LR, Gulleman PM, Bridges JP, Weaver TE, Deutsch GH, Blackwell TS, McCormack FX. The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am J Respir Crit Care Med. 2012;186:1014–1024. doi: 10.1164/rccm.201207-1206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pantelidis P, Veeraraghavan S, du Bois RM. Surfactant gene polymorphisms and interstitial lung diseases. Respir Res. 2002;3:14. doi: 10.1186/rr163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goss V, Hunt AN, Postle AD. Regulation of lung surfactant phospholipid synthesis and metabolism. Biochim Biophys Acta. 2013;1831:448–458. doi: 10.1016/j.bbalip.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Forbes A, Pickell M, Foroughian M, Yao LJ, Lewis J, Veldhuizen R. Alveolar macrophage depletion is associated with increased surfactant pool sizes in adult rats. J Appl Physiol (1985) 2007;103:637–645. doi: 10.1152/japplphysiol.00995.2006. [DOI] [PubMed] [Google Scholar]
- 13.Bates SR, Dodia C, Tao JQ, Fisher AB. Surfactant protein-A plays an important role in lung surfactant clearance: evidence using the surfactant protein-A gene-targeted mouse. Am J Physiol Lung Cell Mol Physiol. 2008;294:L325–L333. doi: 10.1152/ajplung.00341.2007. [DOI] [PubMed] [Google Scholar]
- 14.Yasuda K, Sato A, Nishimura K, Chida K, Hayakawa H. Phospholipid analysis of alveolar macrophages and bronchoalveolar lavage fluid following bleomycin administration to rabbits. Lung. 1994;172:91–102. doi: 10.1007/BF00185080. [DOI] [PubMed] [Google Scholar]
- 15.Azuma A, Li YJ, Abe S, Usuki J, Matsuda K, Henmi S, Miyauchi Y, Ueda K, Izawa A, Sone S, et al. Interferon-beta inhibits bleomycin-induced lung fibrosis by decreasing transforming growth factor-beta and thrombospondin. Am J Respir Cell Mol Biol. 2005;32:93–98. doi: 10.1165/rcmb.2003-0374OC. [DOI] [PubMed] [Google Scholar]
- 16.Basset-Léobon C, Lacoste-Collin L, Aziza J, Bes JC, Jozan S, Courtade-Saïdi M. Cut-off values and significance of Oil Red O-positive cells in bronchoalveolar lavage fluid. Cytopathology. 2010;21:245–250. doi: 10.1111/j.1365-2303.2009.00677.x. [DOI] [PubMed] [Google Scholar]
- 17.Hayes D, Jr, Kirkby S, S McCoy K, Mansour HM, Khosravi M, Strawbridge H, Tobias JD. Reduction of lipid-laden macrophage index after laparoscopic Nissen fundoplication in cystic fibrosis patients after lung transplantation. Clin Transplant. 2013;27:121–125. doi: 10.1111/ctr.12020. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy MA, Barrera GC, Nakamura K, Baldán A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Konter JM, Parker JL, Baez E, Li SZ, Ranscht B, Denzel M, Little FF, Nakamura K, Ouchi N, Fine A, et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol. 2012;188:854–863. doi: 10.4049/jimmunol.1100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Govender P, Romero F, Shah D, Paez J, Ding SY, Liu L, Gower A, Baez E, Aly SS, Pilch P, et al. Cavin1; a regulator of lung function and macrophage phenotype. PLoS One. 2013;8:e62045. doi: 10.1371/journal.pone.0062045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messier EM, Mason RJ, Kosmider B. Efficient and rapid isolation and purification of mouse alveolar type II epithelial cells. Exp Lung Res. 2012;38:363–373. doi: 10.3109/01902148.2012.713077. [DOI] [PubMed] [Google Scholar]
- 22.Iverson SJ, Lang SL, Cooper MH. Comparison of the Bligh and Dyer and Folch methods for total lipid determination in a broad range of marine tissue. Lipids. 2001;36:1283–1287. doi: 10.1007/s11745-001-0843-0. [DOI] [PubMed] [Google Scholar]
- 23.Hörkkö S, Miller E, Dudl E, Reaven P, Curtiss LK, Zvaifler NJ, Terkeltaub R, Pierangeli SS, Branch DW, Palinski W, et al. Antiphospholipid antibodies are directed against epitopes of oxidized phospholipids: recognition of cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. J Clin Invest. 1996;98:815–825. doi: 10.1172/JCI118854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poelma DL, Zimmermann LJ, Scholten HH, Lachmann B, van Iwaarden JF. In vivo and in vitro uptake of surfactant lipids by alveolar type II cells and macrophages. Am J Physiol Lung Cell Mol Physiol. 2002;283:L648–L654. doi: 10.1152/ajplung.00478.2001. [DOI] [PubMed] [Google Scholar]
- 25.Poelma DL, Ju MR, Bakker SC, Zimmermann LJ, Lachmann BF, van Iwaarden JF. A common pathway for the uptake of surfactant lipids by alveolar cells. Am J Respir Cell Mol Biol. 2004;30:751–758. doi: 10.1165/rcmb.2003-0127OC. [DOI] [PubMed] [Google Scholar]
- 26.Carling D, Mayer FV, Sanders MJ, Gamblin SJ. AMP-activated protein kinase: nature’s energy sensor. Nat Chem Biol. 2011;7:512–518. doi: 10.1038/nchembio.610. [DOI] [PubMed] [Google Scholar]
- 27.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiménez-Alonso J, Jaimez L, Barrios L, Pérez-Jiménez F, Costán G, Jiménez-Perepérez JA. Salivary gamma-glutamyl transferase activity in internal diseases. Arch Intern Med. 1984;144:1804–1806. doi: 10.1001/archinte.1984.00350210126022. [DOI] [PubMed] [Google Scholar]
- 29.Mahto SK, Tenenbaum-Katan J, Greenblum A, Rothen-Rutishauser B, Sznitman J. Microfluidic shear stress-regulated surfactant secretion in alveolar epithelial type II cells in vitro. Am J Physiol Lung Cell Mol Physiol. 2014;306:L672–L683. doi: 10.1152/ajplung.00106.2013. [DOI] [PubMed] [Google Scholar]
- 30.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest. 1996;97:649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikegami M, Jobe AH, Huffman Reed JA, Whitsett JA. Surfactant metabolic consequences of overexpression of GM-CSF in the epithelium of GM-CSF-deficient mice. Am J Physiol. 1997;273:L709–L714. doi: 10.1152/ajplung.1997.273.4.L709. [DOI] [PubMed] [Google Scholar]
- 33.Uhal BD, Nguyen H. The Witschi Hypothesis revisited after 35 years: genetic proof from SP-C BRICHOS domain mutations. Am J Physiol Lung Cell Mol Physiol. 2013;305:L906–L911. doi: 10.1152/ajplung.00246.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2008;10:287–301. doi: 10.1089/ars.2007.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 36.Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182:774–783. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- 38.Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. 1989;170:727–737. doi: 10.1084/jem.170.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang-Hoover J, Sutton A, van Rooijen N, Stein-Streilein J. A critical role for alveolar macrophages in elicitation of pulmonary immune fibrosis. Immunology. 2000;101:501–511. doi: 10.1046/j.1365-2567.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–581. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 41.Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J Clin Invest. 1993;92:1812–1818. doi: 10.1172/JCI116771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217:492–502. doi: 10.1016/j.imbio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 43.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parks BW, Black LL, Zimmerman KA, Metz AE, Steele C, Murphy-Ullrich JE, Kabarowski JH. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J Lipid Res. 2013;54:1114–1123. doi: 10.1194/jlr.M035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabuyama Y, Suzuki T, Nakazawa N, Yamaki J, Homma MK, Homma Y. Dysregulation of very long chain acyl-CoA dehydrogenase coupled with lipid peroxidation. Am J Physiol Cell Physiol. 2010;298:C107–C113. doi: 10.1152/ajpcell.00231.2009. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimi N, Ikura Y, Sugama Y, Kayo S, Ohsawa M, Yamamoto S, Inoue Y, Hirata K, Itabe H, Yoshikawa J, et al. Oxidized phosphatidylcholine in alveolar macrophages in idiopathic interstitial pneumonias. Lung. 2005;183:109–121. doi: 10.1007/s00408-004-2525-0. [DOI] [PubMed] [Google Scholar]
- 47.Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care. 2011;56:1016–1028. doi: 10.4187/respcare.01125. [DOI] [PubMed] [Google Scholar]
- 48.Huizar I, Kavuru MS. Alveolar proteinosis syndrome: pathogenesis, diagnosis, and management. Curr Opin Pulm Med. 2009;15:491–498. doi: 10.1097/MCP.0b013e32832ea51c. [DOI] [PubMed] [Google Scholar]
- 49.Moore BB, Coffey MJ, Christensen P, Sitterding S, Ngan R, Wilke CA, McDonald R, Phare SM, Peters-Golden M, Paine R, III, et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J Immunol. 2000;165:4032–4039. doi: 10.4049/jimmunol.165.7.4032. [DOI] [PubMed] [Google Scholar]
- 50.Christensen PJ, Bailie MB, Goodman RE, O’Brien AD, Toews GB, Paine R., III Role of diminished epithelial GM-CSF in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2000;279:L487–L495. doi: 10.1152/ajplung.2000.279.3.L487. [DOI] [PubMed] [Google Scholar]