

Abstract
Gas exchange after birth is entirely dependent on the remarkable architecture of the alveolus, its formation and function being mediated by the interactions of numerous cell types whose precise positions and activities are controlled by a diversity of signaling and transcriptional networks. In the later stages of gestation, alveolar epithelial cells lining the peripheral lung saccules produce increasing amounts of surfactant lipids and proteins that are secreted into the airspaces at birth. The lack of lung maturation and the associated lack of pulmonary surfactant in preterm infants causes respiratory distress syndrome, a common cause of morbidity and mortality associated with premature birth. At the time of birth, surfactant homeostasis begins to be established by balanced processes involved in surfactant production, storage, secretion, recycling, and catabolism. Insights from physiology and engineering made in the 20th century enabled survival of newborn infants requiring mechanical ventilation for the first time. Thereafter, advances in biochemistry, biophysics, and molecular biology led to an understanding of the pulmonary surfactant system that made possible exogenous surfactant replacement for the treatment of preterm infants. Identification of surfactant proteins, cloning of the genes encoding them, and elucidation of their roles in the regulation of surfactant synthesis, structure, and function have provided increasing understanding of alveolar homeostasis in health and disease. This Perspective seeks to consider developmental aspects of the pulmonary surfactant system and its importance in the pathogenesis of acute and chronic lung diseases related to alveolar homeostasis.
Keywords: pulmonary, maturation, surfactant, lung development
Introduction to the Discoveries Series from Past ATS President
The American Thoracic Society (ATS) has a long history, originating as the American Sanatorium Association in 1905, which was established to promote the treatment and prevention of tuberculosis. Since then, the scope of our mission has widened, and the Society has become the premier professional society in respiratory medicine, with more than 15,000 members worldwide who are dedicated to advancing our clinical and scientific understanding of pulmonary diseases, critical illnesses, and sleep-related breathing disorders. Our members provide care for millions of people who suffer daily from asthma, chronic obstructive pulmonary disease, cystic fibrosis, sleep apnea, and lung diseases related to prematurity, to name a few.
In celebration of our 110th anniversary, the ATS journals and 2015 ATS International Conference will highlight many of the advances in patient care and research in adult and pediatric pulmonary, critical care, and sleep medicine. The ATS Discoveries Series is a new collection of articles and talks that features major scientific and clinical breakthroughs, which have changed the lives of the patients we treat, as told by leading scientists and clinicians. With input from our membership, the topics range from the ongoing global threat of tuberculosis to the origins of sleep apnea, from insights into asthma pathogenesis to the potential of lung regeneration.
The following article titled “Alveolar Development and Disease” by Jeffrey Whitsett, M.D., chief of the Section of Neonatology, Perinatal and Pulmonary Biology at Cincinnati Children's Hospital Medical Center, is the first of the series published in the American Journal of Respiratory Cell and Molecular Biology. I hope you enjoy learning about the seminal discoveries in respiratory medicine and their impact on patient care, now and in the future. Please be sure to read all of the articles in the Discoveries Series, which will appear not only in the “Red” journal, but also the American Journal of Respiratory and Critical Care Medicine and Annals of the American Thoracic Society during the coming months.
Thomas Ferkol, M.D.
Past President, American Thoracic Society
Introduction
Survival after birth is entirely dependent upon the delivery of oxygen and removal of carbon dioxide that is mediated by the efficient diffusion of gases across the alveolar epithelium and endothelial cells to blood in the pulmonary circulation. The embryonic lung is formed by the process of branching morphogenesis generating the conducting airways that lead to peripheral saccules from which alveoli are produced during the perinatal and postnatal periods of lung development. In late gestation and after birth, septation and continued lung growth creates the extensive alveolar surfaces mediating efficient gas exchange (Figure 1). Although incomplete, lung morphogenesis has proceeded normally in most preterm infants, but their survival is limited by the lack of pulmonary surfactant needed to reduce surface tension in the peripheral lung. Lack of surfactant activity in lung fluids obtained from infants dying from respiratory distress syndrome was first recognized by Avery and Mead in 1959 (1). Advances in biochemistry and biophysics demonstrated that “surfactant” activity, the ability of lipids to spread rapidly and remain stable during dynamic compression and decompression, resulted from the biophysical activities of phosphatidylcholine, which is synthesized and secreted by alveolar type II (ATII) epithelial cells. After secretion, lipid-rich membrane multilayers form at the interface between alveolar gases and the hydrated surface of the alveoli. The clinical utility of enhancing lung function by surfactant replacement and by inducing lung maturation with maternal antenatal glucocorticoid treatment demonstrated that lung structure is sufficient to support ventilation even in extremely premature infants. Nevertheless, in spite of intensive care, lung injury and remodeling cause a chronic lung disease, termed “bronchopulmonary dysplasia,” a common clinical complication experienced by extremely preterm infants. Because lung architecture is critical for its function, knowledge regarding the mechanisms mediating its formation and repair is relevant to the development of strategies to prevent and treat disorders that lead to loss of alveolar structure and function. The antibodies and complementary DNAs derived from surfactant proteins provided initial reagents used to identify the developmental processes involved in formation and differentiation of respiratory epithelial cells. Promoter-enhancer regions of genes encoding the surfactant proteins and their regulators have been useful in the study of lung biology, enabling deletion, addition, and mutation of genes in subsets of respiratory epithelial cells that have been used to uncover mechanisms involved in lung formation, maturation, and disease (2).
Figure 1.

Maturation of the alveolar epithelium. Subsets of cells from the anterior foregut endoderm expressing NKX2–1 (TTF-1) (green) form tubules that evaginate into the splanchnic mesenchyme to begin the process of branching morphogenesis (shown at embryonic day [E]10–E12). Sacculation and alveolarization create the alveolar structures seen at Postnatal Day (PND) 7. SOX2 and SOX9 are transcription factors defining the conducting versus peripheral progenitor cells of the respiratory epithelium, respectively. During mouse and human lung development, alveolarization, mediated by the process of septation, begins near the time of birth and continues during the postnatal period. Maturation of the peripheral respiratory epithelium is associated with differentiation of the airway epithelial cells (alveolar type II cells) and increased expression of genes associated with surfactant synthesis and homeostasis (e.g., Abca3, Sftpa, Sftpb, Sftpc, Sftpd, Napsa, and Gpr116). Continued septation subdivides the peripheral saccules into alveolar sacs that are highly vascularized (shown at PND7). Septa (PND7) are stained with homeodomain-only protein (HOPX) (green, alveolar type I), endomucin (red, endothelium), and α-smooth muscle actin (α-SMA; white, myofibroblasts). Lamellar bodies are secreted into the airways, unraveling to form tubular myelin as shown by electron microscopy (bottom right panel). Images provided by John Shannon, Susan Wert, and Joseph Kitzmiller.
Preparing for Lung Maturation
Survival at birth depends on (1) the architecture of the peripheral pulmonary acini and the maturation of the gas exchange region, (2) differentiation of squamous ATI cells and their close interface with endothelial cells of the pulmonary microvasculature system, (3) synthesis and secretion of surfactant lipids and proteins by ATII cells, and (4) clearance of lung fluids to establish ventilation (Figures 2 and 3). Whereas septation begins before birth, septation and alveolarization occur primarily in the postnatal period in most mammalian species. Significant progress has been made in identifying the cellular and molecular events mediating early lung morphogenesis. Respiratory epithelial cells are derived from progenitor cells in the anterior foregut, first identified by the expression of thyroid transcription factor 1 (TTF-1) (NKX2–1), a transcription factor expressed in endodermally derived epithelial cells that is critical for both lung formation and airway epithelial cell differentiation. Specification of proximal versus peripheral airway epithelial cells occurs early during foregut development, even before the appearance of the early lung buds (3). TTF-1 is expressed in the early lung buds and in the proximal stalk of the trachea and is required for lung formation (4, 5) (Figure 1). The important transcriptional and signaling programs underlying the remarkable stereotypic branching processes that generate the respiratory tract are increasingly understood (6–8). Completion of the airway branching heralds the increasing differentiation of the respiratory epithelium during the canalicular-saccular period of development, occurring from Embryonic Day (E) 16.5 to birth in the mouse (Figure 1). The maturation of respiratory epithelial cells includes the increasing differentiation of ATII and ATI cells, expression of surfactant lipids and proteins, that begins the “life cycle” of pulmonary surfactant, wherein synthesis, secretion, surface-active functions, recycling, and catabolism are established (Figure 4) (9).
Figure 2.

Immunofluorescence staining of HOPX (red; A and B), surfactant protein C (SFTPC) (green; A and B), smooth muscle action (ACTA2) (white; A–D), endomucin (EMCN) (red; C and D), and CSPG4 (green; C and D). CSPG4 is a pericyte marker. Lung images from E16.5 (A and C) and Postnatal Day 7 (B and D) mice are shown. Dramatic changes in peripheral lung architecture occur in the perinatal period.
Figure 3.
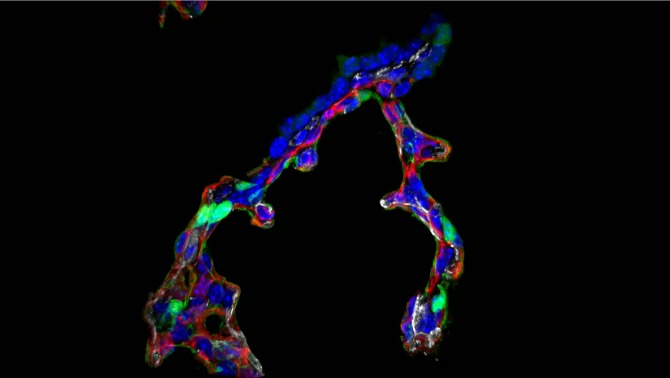
Immunofluorescence staining of HOPX (green, alveolar type I cell), endomucin (red, endothelium), and α-SMA (white, myofibroblasts) is shown in alveolar septa from Postnatal Day 7 mouse lung. Note the double capillary network and α-SMA staining at the septal tip.
Figure 4.

Surfactant metabolism. NKX2–1 (TTF-1) is a transcription factor critical for differentiation of type II epithelial cells and regulation of expression of Abca3, Slc34a2 (a phosphate transporter), and the surfactant proteins. Newly translated surfactant proteins (SPs) (proSP-B and proSP-C) and lamellar body proteins (ABCA3) traffic from the endoplasmic reticulum (ER) to the Golgi and subsequently to the multivesicular body (MVB); in contrast, SP-A and SP-D are constitutively secreted by the type II epithelial cells (dotted green arrows). Fusion of the MVB with the lamellar body (LB) is accompanied by proteolytic processing of SP-B and SP-C proproteins to their mature peptides. Newly synthesized surfactant phospholipids (DPPC, PG) are likely transported directly from the ER to the LB by lipid transfer proteins. The contents of the LB are secreted into the alveolar space, where they interact with SP-A to form tubular myelin and, ultimately, a phospholipid-rich film (surfactant) at the air–liquid interface. Cyclical expansion and compression of the bioactive film result in incorporation (green arrow) and loss (red arrows) of lipids/proteins from the multilayered surface film. Alveolar surfactant lipids and proteins are cleared through a granulocyte/macrophage colony-stimulating factor–dependent pathway that regulates alveolar macrophage differentiation and function. Surfactant remnants are also taken up by the type II epithelial cell and recycled to the LB via the MVB for resecretion, and a portion is degraded in lysosomes. SP-D plays an important role in regulating alveolar surfactant pool size, likely by enhancing its reuptake by type II epithelial cells; Gpr116 also regulates alveolar surfactant pool size by an unknown mechanism. The MVB integrates surfactant synthesis, secretion, recycling, and degradation pathways in the type II cell. Green arrows indicate biosynthetic pathways; red arrows indicate degradation and recycling pathways. Dotted arrows represent trafficking routes of proteins and lipids.
Transcriptional Regulation of Surfactant Lipids and Proteins before Birth
Pulmonary surfactant lipids and proteins are synthesized by ATII epithelial cells, which undergo dramatic changes in structure and function before birth. Increased synthesis of surfactant lipids is mediated in part by gene networks controlled by the concerted actions of a number of transcription factors that regulate target genes involved in the synthesis, routing, and secretion of surfactant lipids and proteins. SREBP, FOXA2, STAT3, and CEBPα interact in a gene network controlling surfactant lipid synthesis (10–12), increasing phosphatidylcholine synthesis and its remodeling to enrich for disaturated palmitoylphosphatidylcholine. A number of enzymes involved in surfactant lipid synthesis are induced before birth, including lysophosphatidylcholine acyl transferase, choline kinase, phosphocholine, cytidylyltransferase, acetylCoA synthase, fatty acid synthase, and others. This transcriptional network also regulates the transcription, processing, routing, and packaging of surfactant proteins (Figure 4). The role of TTF-1, encoded by the NKX2–1 gene, in perinatal ATII cell maturation and surfactant protein gene expression was initially recognized by its requirement for the transcriptional activation of the surfactant proteins (SP-A, SP-B, SP-C, and SP-D) and the surfactant lipid transporter (ABCA3) (13, 14), the latter being required for transport of phosphatidylcholine into lamellar bodies. Mutation of phosphorylation sites regulating TTF-1 activity inhibited perinatal lung maturation and synthesis of surfactant protein (15). TTF-1 interacts in a gene network with other transcription factors, including FOXA2, CEBPα, and nuclear factor of activated T-cells (NFAT)/calcineurin, that together regulate common gene targets involved in surfactant biosynthesis, fluid and electrolyte transport, and innate host defense. Bioinformatic analysis of lung RNA expression array data obtained after gene targeting or mutation of these critical transcription factors demonstrated their importance in perinatal lung maturation and surfactant synthesis (10–12, 15, 16). As a critical regulator of perinatal lung maturation, NKX2–1 is predicted to function with other transcription factors (e.g., HOPX, ETV5, GATA6, GRHL, FOXP2, and HMGA2) that together regulate many aspects of lung maturation, including ATI/ATII cell differentiation and surfactant synthesis (12). The direct binding of TTF-1 to cis-acting regulatory sites on target genes was identified by genome-wide CHIP (CHromatin Immuno-Precipitation) assays (17, 18), demonstrating its binding to sequences in the ABCA3, SFTPA, SFTPB, and SFTPC genes. Recent studies demonstrated that NKX2–1 and FOXA3 are regulated by a long-coding RNA (NANCI and FANCI) that modulates their expression in the thyroid and lung (19).
Establishing the Life Cycle of Pulmonary Surfactant
The increased synthesis of surfactant lipids and proteins in late gestation results in large intracellular surfactant lipid pools that are stored in lamellar bodies in ATII epithelial cells. Induction of surfactant synthesis in the perinatal period requires interactions between fibroblasts and type II epithelial cells that are mediated by the glucocorticoid receptor encoded by the Nr3c1 gene (20). Glucocorticoid receptor signaling mediates gene expression in fibroblasts derived from the splanchnic mesenchyme that signal to the respiratory epithelium to enhance both structural and biochemical aspects of perinatal lung maturation. At birth, intracellular surfactant in lamellar bodies is secreted into the peripheral lung saccules (Figure 4). Lamellar bodies containing SP-B and SP-C and surfactant lipid “unravel” and interact with SP-A and SP-D to create distinct lipid forms after ventilation, including (1) lamellar bodies, which are rich in phosphatidylcholine, SP-B, and SP-C; (2) tubular myelin, created by their interaction with SP-A in the presence of calcium; (3) multilayers of lipid films enriched in SP-B and SP-C, which spread over the alveolar surface to reduce surface tension; and (4) smaller lipid particles that are likely recycled by epithelial cells or catabolized by alveolar macrophages. SP-A, -B, -C, and -D each plays a distinct role in surfactant homeostasis, including surface tension reduction, innate host defense, and the regulation of surfactant forms and pool sizes (21). It is now well established that alveolar macrophages play an important role in the uptake and catabolism of both surfactant lipids and proteins in a process dependent on GM-CSF signaling. Mutations in GM-CSF receptors or auto-antibodies against GM-CSF impair alveolar macrophage differentiation, resulting in decreased clearance of surfactant lipids and proteins. Pulmonary alveolar proteinosis (PAP) is caused by mutations in the GM-CSF receptors or by neutralizing auto-antibodies against GM-CSF (for review, see Refs. 22–24). In both of these disorders, surfactant lipids and proteins accumulate to high levels, requiring the removal by lung lavage. The utility of inhaled GM-CSF for the treatment of PAP is under experimental study at present (25). Thus, the major components of the life cycle of surfactant are readily established after birth. Remarkably, surfactant pool sizes are much higher in the newborn infant than in the mature lung, with pool sizes decreasing to adult levels during the first weeks of life in a process dependent on SP-D and a G protein–coupled receptor, GPR116, both expressed by ATII cells (26, 27). Recent studies demonstrate that deletion of GPR116 dramatically enhances surfactant pool sizes, thus identifying an important component of the complex regulatory process involved in surfactant homeostasis (27). Because reduction of surface tension is required throughout life, lung injury, with resultant alveolar–capillary leak that inactivates surfactant activity, and the lack of surfactant production caused by ATII cell injury contribute to the pathogenesis of acute respiratory distress syndrome.
Hereditary Disorders of Surfactant Homeostasis
Elucidation of the proteins and genes regulating surfactant homeostasis provided genetic tools to identify mutations causing severe neonatal lung disease, which was previously considered “idiopathic” (for review, see Ref. 9) (Figure 5). Mutations in ABCA3 (OMIM 601615) (an ATP-dependent phospholipid transport protein required for the formation of lamellar bodies), SFTPB (OMIM 178640), and SFTPC (OMIM 178620) (both required for the normal spreading and stability of alveolar lipids and for intracellular processes in type II cells) were identified as the cause of acute respiratory failure in term newborn infants or interstitial lung disease (ILD) in children and older individuals (28–30). These disorders are classified as congenital pulmonary alveolar proteinosis, chronic pneumonitis of infancy, desquamative interstitial pneumonitis, nonspecific interstitial pneumonitis, and usual interstitial pneumonias (9), the variability and histopathology likely related to the type of mutation, age, modifier genes, and therapies used to support these critically ill patients. ABCA3, SP-B, and SP-C are critical for normal intracellular processes in ATII cells; therefore, surfactant replacement therapy is not effective in these disorders. Observations from gene-targeted mice and from in vitro experiments support the important role of chronic type II cell injury and the abnormal physical forces associated with the lack of surfactant activity in the pathogenesis of the tissue remodeling associated with the ILDs. Mutations or haploinsufficiency in NKX2–1 (TTF-1) cause a brain-lung-thyroid syndrome (OMIM 600635), with the majority of these patients developing severe interstitial lung disease. In older individuals, mutations in SFTPA2 (OMIM 178642), SFTPC, TERT (OMIM 61473), and TERC (OMIM 602322) are associated with type II cell injury, lung remodeling, emphysema, and ILDs. Taken together, the knowledge gained from the study of hereditary disorders of surfactant metabolism indicates the important role of the ATII cell injury and abnormal repair in the pathogenesis of ILD.
Figure 5.

Interstitial lung disorders caused by mutations in genes regulating surfactant homeostasis. Histology (hematoxylin and eosin) of lung tissue from patients with genetic disorders of the surfactant system. Neonatal lung tissue from patients with (A) SFTPB, (B) SFTPC, (C) ABCA3, (D) CSF2RA, and (E and F) NKX2–1 gene mutations. Scale bars: 200 μm. Reprinted by permission from Ref. 9.
Mutations in GM-CSF Receptor Signaling Causes Pulmonary Alveolar Proteinosis
Transgenic mice in which the genes encoding GM-CSF (Csf2) or its receptor (Csf2r) were deleted developed severe pulmonary pathology phenocopying idiopathic pulmonary alveolar proteinosis (iPAP), in which surfactant lipids and proteins accumulate, causing respiratory failure (for review, see Ref. 23). Defects in surfactant homeostasis in these mice were related to decreased clearance of surfactant lipids and proteins by alveolar macrophages, whose catabolic and innate immune functions were dependent upon GM-CSF signaling. GM-CSF activates the GM-CSF receptors via the transcription factors STAT5, PU.1, and PPARγ to control alveolar macrophage differentiation and function (22). Whereas most patients with iPAP have circulating antibodies against CSF2 that neutralize GM-CSF activity (OMIM 13906), pediatric forms of PAP are associated with mutations in CSF2RA (OMIM 306250) or CSF2RB (OMIM 138981). At present, PAP is treated by whole lung lavage. Correction of CSF2R signaling in alveolar macrophages is being considered for therapy of congenital PAP in the future (31, 32) in addition to GM-CSF for treatment of adult iPAP.
Conclusions
Pulmonary maturation depends on precisely choreographed interactions among multiple cell types, the timing of lung formation, and surfactant synthesis varying among diverse mammalian species. “Maturation” is a relative term because the growth and differentiation of the lung are not completed at birth, with survival being closely associated with the synthesis and secretion of adequate levels of pulmonary surfactant. Although historically “lung maturity” was strongly linked to pulmonary surfactant function in the newborn period, advances in critical care, ventilation, and surfactant replacement therapies have enabled the survival of extremely preterm infants whose lung structure and function are not yet mature, and their clinical course is often associated with tissue injury and structural remodeling, which cause bronchopulmonary dysplasia. Understanding the processes involved in surfactant homeostasis and the maturation of lung structure and function has helped inform the pathogenesis of lung remodeling associated with other interstitial lung diseases in children and adults, findings that provide the basis for prevention, diagnosis, and treatments to prevent lung injury and disruption of lung architecture and surfactant function associated with chronic lung diseases. The complexity of the multiple processes by which individual cell types interact in the formation and maintenance of the lung presents a daunting task for present-day and future cellular and molecular biology and physiology.
Footnotes
This work is supported by National Institutes of Health/National Heart, Lung and Blood Institute grants HL108907, HL110964, HL095580, HL122642, and HL119686 (J.A.W.) and R37-HL5625, HL61646, and HL086492 (T.E.W.).
Author Contributions: J.A.W. and T.E.W. conceived of and wrote the manuscript.
Originally Published in Press as DOI: 10.1165/rcmb.2015-0128PS on May 1, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Avery ME, Mead J. Surface properties in relation to atelectasis and hyaline membrane disease. AMA J Dis Child. 1959;97:517–523. doi: 10.1001/archpedi.1959.02070010519001. [DOI] [PubMed] [Google Scholar]
- 2.Rawlins EL, Perl AK. The a“MAZE”ing world of lung-specific transgenic mice. Am J Respir Cell Mol Biol. 2012;46:269–282. doi: 10.1165/rcmb.2011-0372PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci USA. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- 5.Lazzaro D, Price M, de Felice M, Di Lauro R. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development. 1991;113:1093–1104. doi: 10.1242/dev.113.4.1093. [DOI] [PubMed] [Google Scholar]
- 6.Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, Niklason L, Calle E, Le A, Randell SH, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzger RJ, Klein OD, Martin GR, Krasnow MA. The branching programme of mouse lung development. Nature. 2008;453:745–750. doi: 10.1038/nature07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol. 2015;10:371–393. doi: 10.1146/annurev-pathol-012513-104644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besnard V, Wert SE, Ikegami M, Xu Y, Heffner C, Murray SA, Donahue LR, Whitsett JA. Maternal synchronization of gestational length and lung maturation. PLoS One. 2011;6:e26682. doi: 10.1371/journal.pone.0026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridges JP, Schehr A, Wang Y, Huo L, Besnard V, Ikegami M, Whitsett JA, Xu Y. Epithelial SCAP/INSIG/SREBP signaling regulates multiple biological processes during perinatal lung maturation. PLoS One. 2014;9:e91376. doi: 10.1371/journal.pone.0091376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Wang Y, Besnard V, Ikegami M, Wert SE, Heffner C, Murray SA, Donahue LR, Whitsett JA. Transcriptional programs controlling perinatal lung maturation. PLoS One. 2012;7:e37046. doi: 10.1371/journal.pone.0037046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Besnard V, Xu Y, Whitsett JA. Sterol response element binding protein and thyroid transcription factor-1 (Nkx2.1) regulate Abca3 gene expression. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1395–L1405. doi: 10.1152/ajplung.00275.2007. [DOI] [PubMed] [Google Scholar]
- 14.Bohinski RJ, Di Lauro R, Whitsett JA. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol Cell Biol. 1994;14:5671–5681. doi: 10.1128/mcb.14.9.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeFelice M, Silberschmidt D, DiLauro R, Xu Y, Wert SE, Weaver TE, Bachurski CJ, Clark JC, Whitsett JA. TTF-1 phosphorylation is required for peripheral lung morphogenesis, perinatal survival, and tissue-specific gene expression. J Biol Chem. 2003;278:35574–35583. doi: 10.1074/jbc.M304885200. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Ikegami M, Wang Y, Matsuzaki Y, Whitsett JA. Gene expression and biological processes influenced by deletion of Stat3 in pulmonary type II epithelial cells. BMC Genomics. 2007;8:455. doi: 10.1186/1471-2164-8-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maeda Y, Tsuchiya T, Hao H, Tompkins DH, Xu Y, Mucenski ML, Du L, Keiser AR, Fukazawa T, Naomoto Y, et al. Kras(G12D) and Nkx2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest. 2012;122:4388–4400. doi: 10.1172/JCI64048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, Crowley D, Whittaker CA, Meyerson M, Kimura S, et al. Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell. 2013;50:185–199. doi: 10.1016/j.molcel.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herriges MJ, Swarr DT, Morley MP, Rathi KS, Peng T, Stewart KM, Morrisey EE. Long noncoding RNAs are spatially correlated with transcription factors and regulate lung development. Genes Dev. 2014;28:1363–1379. doi: 10.1101/gad.238782.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habermehl D, Parkitna JR, Kaden S, Brügger B, Wieland F, Gröne HJ, Schütz G. Glucocorticoid activity during lung maturation is essential in mesenchymal and less in alveolar epithelial cells. Mol Endocrinol. 2011;25:1280–1288. doi: 10.1210/me.2009-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16:27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135:223–235. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 24.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med. 2010;61:105–119. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tazawa R, Inoue Y, Arai T, Takada T, Kasahara Y, Hojo M, Ohkouchi S, Tsuchihashi Y, Yokoba M, Eda R, et al. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte-macrophage colony-stimulating factor therapy. Chest. 2014;145:729–737. doi: 10.1378/chest.13-0603. [DOI] [PubMed] [Google Scholar]
- 26.Bridges JP, Ludwig MG, Mueller M, Kinzel B, Sato A, Xu Y, Whitsett JA, Ikegami M. Orphan G protein-coupled receptor GPR116 regulates pulmonary surfactant pool size. Am J Respir Cell Mol Biol. 2013;49:348–357. doi: 10.1165/rcmb.2012-0439OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikegami M, Grant S, Korfhagen T, Scheule RK, Whitsett JA. Surfactant protein-D regulates the postnatal maturation of pulmonary surfactant lipid pool sizes. J Appl Physiol (1985) 2009;106:1545–1552. doi: 10.1152/japplphysiol.91567.2008. [DOI] [PubMed] [Google Scholar]
- 28.Dishop MK. Paediatric interstitial lung disease: classification and definitions. Paediatr Respir Rev. 2011;12:230–237. doi: 10.1016/j.prrv.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, Dell S, Fan LL, Hamvas A, Hilman BC, et al. American Thoracic Society Committee on Childhood Interstitial Lung Disease (the chiLDRN) An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188:376–394. doi: 10.1164/rccm.201305-0923ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol. 2009;12:253–274. doi: 10.2350/09-01-0586.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, Abe S, Trapnell C, Carey B, Moritz T, et al. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki T, Mayhew C, Sallese A, Chalk C, Carey BC, Malik P, Wood RE, Trapnell BC. Use of induced pluripotent stem cells to recapitulate pulmonary alveolar proteinosis pathogenesis. Am J Respir Crit Care Med. 2014;189:183–193. doi: 10.1164/rccm.201306-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]