

1. Introduction
The stereocontrolled construction of C–C bonds remains one of the foremost challenges in organic synthesis. At the heart of any chemical synthesis of a natural product or designed small molecule is the need to orchestrate a series of chemical reactions to prepare and functionalize a carbon framework. The advent of transition-metal catalysis has provided chemists with a broad range of new tools to forge C–C bonds and has resulted in a paradigm shift in synthetic strategy planning. The impact of these methods was recognized with the awarding of the 2010 Nobel Prize in Chemistry to Richard Heck, Ei-ichi Negishi, and Akira Suzuki for their seminal contributions to the development of Pd-catalyzed cross-coupling.
The potential of using transition-metal-catalyzed C–C bond formation to prepare enantioenriched molecules was immediately recognized by the synthetic chemistry community. Indeed, the first forays into enantioselective cross-coupling reactions occurred contemporaneously with the development of the transition-metal-catalyzed reactions themselves. Here we define transition-metal-catalyzed cross-coupling reactions as C–C bond forming reactions between an organic electrophile (typically an organic halide or pseudohalide, which in this review includes alcohols, amines, and their derivatives) and an organometallic reagent, mediated by a transition-metal catalyst.
This review will focus on transition-metal-catalyzed cross-coupling reactions to form C–C bonds, in which the stereogenic unit is defined by the C–C bond forming event. These reactions can be organized into two general categories.
The first group comprises enantioselective transition-metal-catalyzed cross-coupling reactions, which we define as reactions in which there is selective formation of one enantiomer over the other as defined by a nonracemic chiral metal catalyst. There are several different types of enantioselective cross-coupling reactions: those in which (a) racemic, C(sp3) organometallic reagents are stereoconvergently coupled to organic electrophiles, (b) racemic, C(sp3) organic electrophiles are stereoconvergently coupled to organometallic reagents, (c) achiral organic electrophiles are coupled to achiral organometallic reagents to produce chiral, nonracemic products, and (d) a prochiral starting material (either the organic electrophile or organometallic reagent) is desymmetrized. These reactions are schematically represented in Figure 1.
Figure 1.
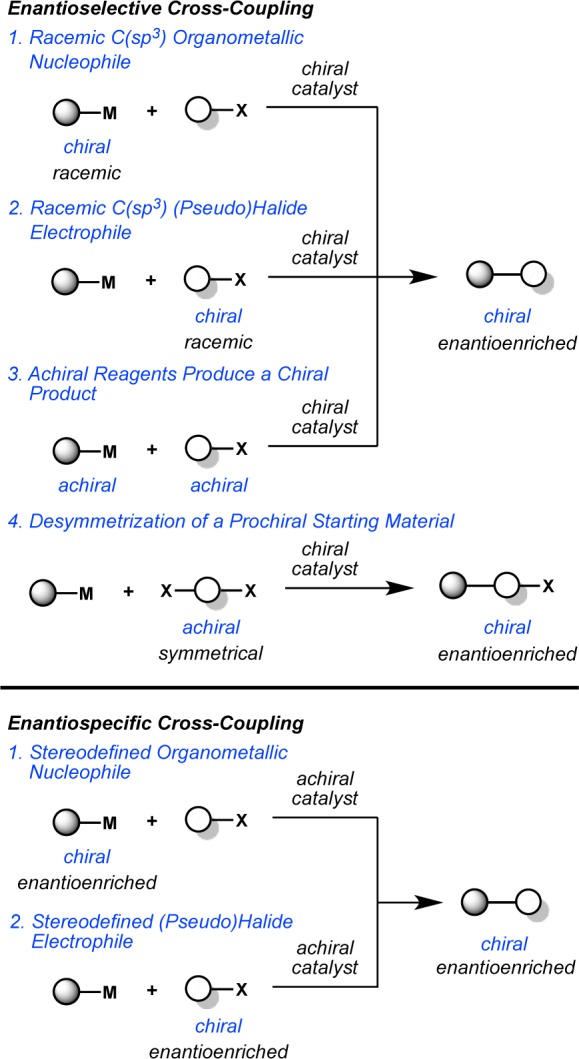
Strategies for enantiocontrolled cross-coupling.
The second group comprises enantiospecific transition-metal-catalyzed alkyl cross-coupling reactions, which we define as chirality exchange reactions in which the stereochemistry of a chiral, enantioenriched substrate defines the stereochemistry of the product. These reactions can be further categorized into those which involve the cross-coupling of (a) a stereodefined organometallic reagent with an electrophile or (b) a stereodefined electrophile with an organometallic reagent. These types of enantioselective and enantiospecific reactions have been used to prepare molecules exhibiting centro, axial, and planar chirality.
This review will encompass enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organic electrophiles and organometallic reagents, covering the literature published through the end of the year 2014. Carbon–carbon bond forming reactions that provide products with centro, axial, and planar chirality will be discussed. Diastereoselective reactions, in which an existing stereocenter in the substrate controls the formation of a second chiral center in the product, will not be evaluated. Correspondingly, the discussion of stereospecific reactions will be limited to substrates bearing only a single stereogenic center. Processes involving coupling reactions of isolated alkenes, α,β-unsaturated carbonyl compounds, or isolated alkynes will not be discussed here, including asymmetric Heck1 and oxidative Heck2 reactions, conjugate addition reactions, and asymmetric ring-opening reactions of oxabicyclic compounds.3 Stoichiometric transition-metal-mediated reactions will also not be covered. Although allylic substitution reactions using enolates and related soft nucleophiles are beyond the scope of this review, transition-metal-catalyzed allylic substitution reactions using organometallic reagents will be discussed. Cu-catalyzed asymmetric allylic substitution reactions prior to 2008 have been well-reviewed elsewhere and will not be included here.4−6
2. Enantioselective Transition-Metal-Catalyzed Alkyl Cross-Coupling Reactions
Despite promising initial reports, highly enantioselective transition-metal-catalyzed alkyl cross-coupling reactions were slow to develop, in part because of the general challenges encountered in Pd-catalyzed alkyl cross-coupling reactions. For Pd and other metals that react by polar, two-electron mechanisms, sec-alkylmetallic reagents are typically slower than their n-alkyl or C(sp2)-hybridized counterparts to undergo transmetalation.7 Similarly, sec-alkyl electrophiles are frequently slow to undergo oxidative addition to Pd.8 Moreover, in either case, the resulting sec-alkyl transition-metal complexes can suffer from rapid, nonproductive β-hydride elimination. Thus, the successful realization of enantioselective transition-metal-catalyzed alkyl cross-coupling reactions has resulted from fundamental studies of the factors, particularly ligands, that control and influence the efficiency of these elementary steps. In addition, a renewed interest in Ni catalysts, which can engage with sec-alkyl halides through single-electron oxidative addition mechanisms, has resulted in a rapidly increasing number of enantioselective alkyl cross-coupling reactions.
2.1. Reactions of Secondary Alkylmetallic Reagents
Early efforts to develop enantioselective transition-metal-catalyzed alkyl cross-coupling reactions focused primarily on the use of configurationally labile sec-alkylmetallic species such as organomagnesium and organozinc reagents. In general, the configurational stability of an organometallic reagent correlates to the electronegativity of the metal, with less electronegative metals resulting in more configurationally labile sec-alkyl reagents.9−11 For example, sec-alkylmagnesium reagents have been shown to racemize above −10 °C, whereas the corresponding sec-alkylboron reagents are configurationally stable indefinitely at room temperature.12 In principle, fast equilibration between the two enantiomers of a sec-alkylmetallic reagent or between two diastereomers of a chiral transition-metal complex could enable enantioselective cross-coupling through a dynamic kinetic asymmetric transformation (DYKAT), in which the newly formed stereogenic center is controlled by the chirality of the metal catalyst (Figure 2).
Figure 2.
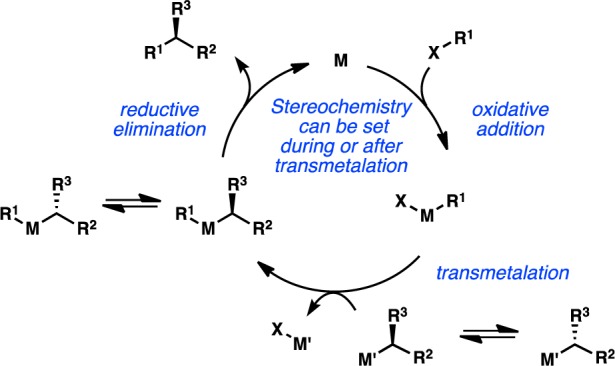
Stereochemical outcome of cross-coupling with secondary nucleophiles.
Enantioselective reactions of configurationally stable sec-alkylmetallic reagents can arise from catalyst-controlled kinetic resolution processes, wherein the relative rates of transmetalation for the two enantiomers of the chiral organometallic reagent are substantially different. In this case, an excess of the organometallic reagent must be used to obtain the cross-coupled product in good yield. A third possibility involves a stereoablative mechanism, in which the initial configuration of the starting material is destroyed and then reset by the chiral catalyst during the reaction.
2.1.1. Organomagnesium Reagents
In 1972 Corriu and Kumada independently reported the Ni-catalyzed cross-coupling between alkylmagnesium halides and aryl or vinyl halides;13−15 shortly thereafter the first studies aimed at utilizing chiral transition-metal complexes to catalyze these reactions enantioselectively were reported.16 In 1973 and 1974, respectively, Consiglio and Kumada independently reported that the complex generated from nickel halide salts and the chiral bidentate phosphine ligand DIOP (L1) catalyzes the reaction between sec-butylmagnesium bromide or chloride and bromo- or chlorobenzene to give product 1 with promising enantioinduction (Figure 3).17,18 These results were an important proof of concept for the area of enantioselective cross-coupling; however, because low yields of product were obtained, it remains ambiguous whether these reactions proceed by kinetic resolution of the sec-alkylmagnesium reagent or through a DYKAT. It was subsequently reported that Prophos (L2) provides improved enantioinduction and higher yields of 1.19 The identity of the halogen on both the organic halide and the organometallic reagent significantly influenced the absolute configuration and the enantiomeric excess (ee) of 1. Further improvements were observed when Norphos (L4) was employed as the chiral ligand, providing 1 in 50% ee.20,21 A carbohydrate-derived chiral ligand (L3) was also reported to deliver 1 in good ee, although with poor yields.22
Figure 3.
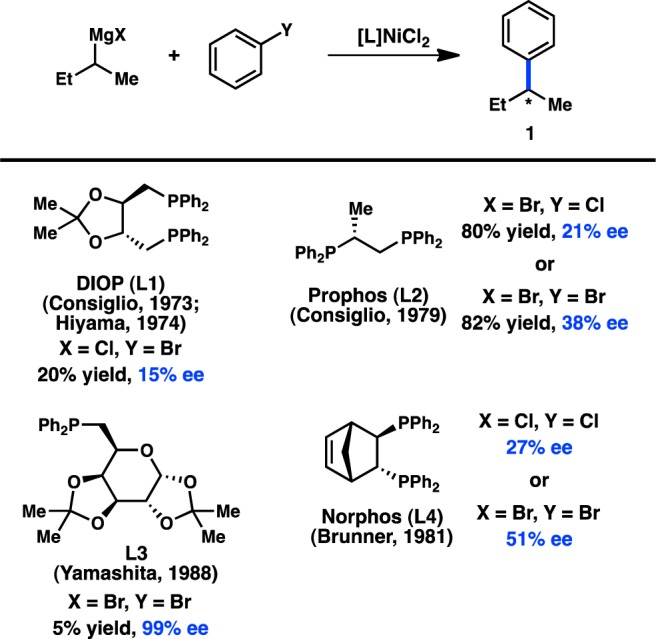
Stereoconvergent arylation of sBu Grignard reagents.
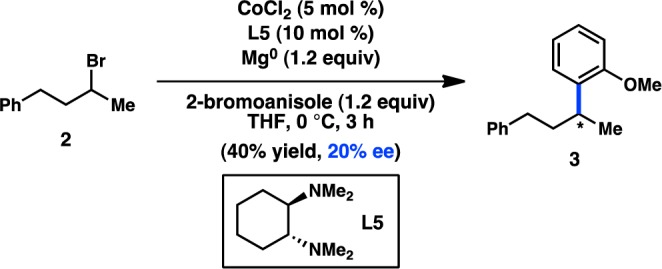
In 2009, Jacobi von Wangelin and co-workers reported the in situ generation of a secondary Grignard reagent that can subsequently undergo a Co-catalyzed asymmetric cross-coupling with promising enantioinduction (Scheme 1).23 Additional ligand development and expansion of the substrate scope are imperative, but this initial result represents a solid advance for in situ Grignard formation in stereoconvergent cross-couplings.
Scheme 1. Stereoselective Coupling of a Grignard Reagent Prepared in Situ.
Concurrent with their efforts to develop enantioselective cross-coupling reactions of sec-butyl Grignard reagents, Kumada and co-workers investigated the Ni-catalyzed enantioselective coupling between α-methylbenzyl Grignard reagents and vinyl halides (Figure 4). DIOP (L1) and the axially chiral Naphos (L6) ligand systems provided the product with low enantioinduction.18,24 Following up on Kumada’s studies, Brunner and co-workers reported that the use of Norphos (L4) in this reaction furnished 4 in 95% yield and 67% ee.25
Figure 4.
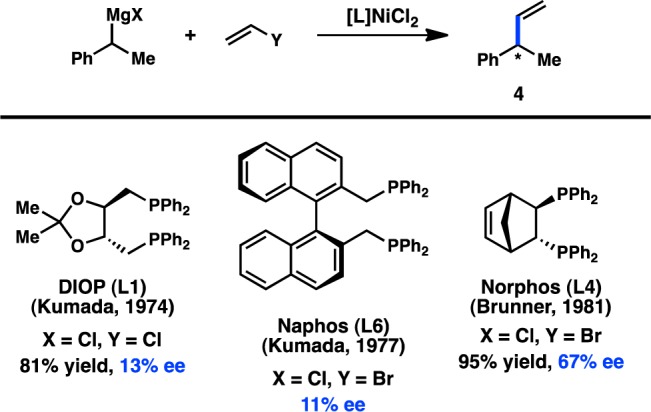
Stereoconvergent vinylation of benzylic Grignard reagents.
Since Kumada’s initial report, the majority of studies have focused on identifying new ligands to improve the selectivity in the coupling between α-methylbenzyl Grignard reagents (5) and vinyl bromide. Whereas the early studies focused on the use of bidentate bisphosphine ligands, which delivered modest levels of enantioinduction, later efforts turned to chiral P,N ligands. Kumada, Hayashi, and co-workers reported that chiral (β-aminoalkyl)phosphines—easily prepared from enantiopure amino acids—delivered exceptionally high yields for the cross-coupling between 5 and vinyl bromide (Figure 5).26−28 Interestingly, whereas the alkyl substitution on the ligand backbone exhibited little influence on the yield of the reaction, it dramatically impacted the enantioselectivity: increasing the steric profile of the ligand raised the ee from 38% when the chiral tertiary substituent was Me (L7) to 94% when this group was tBu (L10). To probe the origin of asymmetric induction, the isomeric P,N ligand L11 was designed. Under the same reaction conditions, L11 delivered 4 in only 25% ee. Moreover, the analogous bisphosphine L12 provided no enantioinduction, suggesting a critical role for the amino group. A proposed catalytic cycle for this reaction is shown in Figure 6, which involves precoordination between Grignard reagent 5 and the amino group of the ligand to give complex 7. The authors hypothesize that this coordination could selectively direct the transmetalation of a single enantiomer of the organometallic reagent, although the importance of this interaction has been debated.29
Figure 5.

Chiral ligands developed for the enantioselective cross-coupling of α-methylbenzyl Grignard reagents. The above ee values are culled from several different studies in which the reactions were conducted under varying conditions; comparisons should be drawn with caution.
Figure 6.
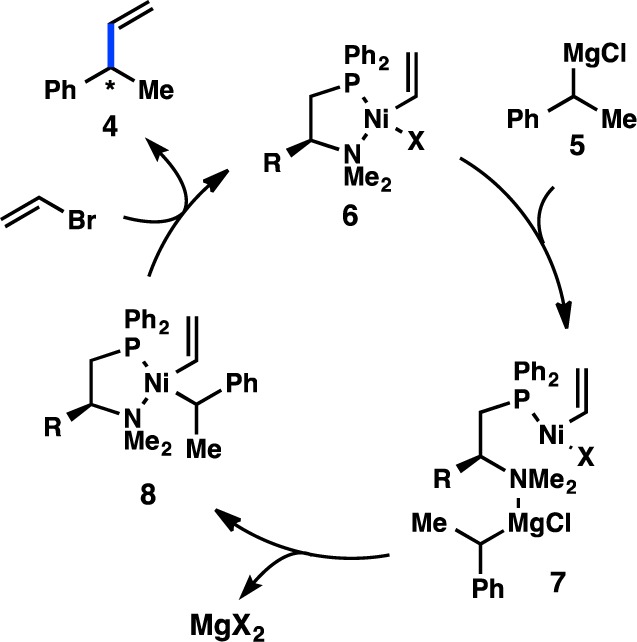
Proposed catalytic cycle for the enantioselective coupling of α-methylbenzyl Grignard reagents.
Elaborating on this concept, Kellogg and co-workers investigated the use of (β-aminoalkyl)phosphine ligands bearing pendant heteroatoms, such as those derived from lysine or methionine.30,31 The authors reported a reversal of the stereochemical outcome in the presence of exogenous zinc halide salts (Figure 7). Control experiments using pregenerated (α-methylbenzyl)zinc bromide did not support the intermediacy of an organozinc species; instead it is possible that coordination between the Lewis acidic zinc halide and the side chain heteroatom could alter or disrupt the ability of the amino group to direct the transmetalation event.
Figure 7.
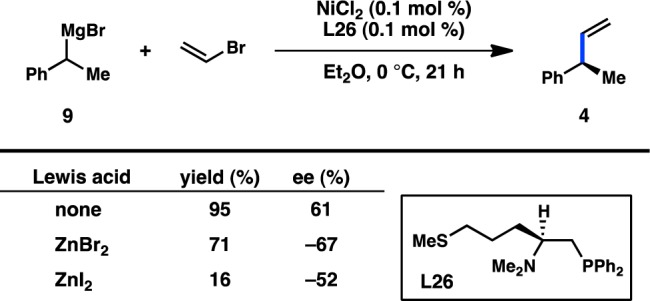
Addition of exogenous zinc halide salts reverses the sense of enantioinduction when sulfur-containing ligand L26 is used.
The importance of an amino directing group on the chiral ligand was also reported by Kumada, Hayashi, and co-workers, during their investigations of ferrocenylphosphines in the Ni-catalyzed coupling between α-methylbenzyl Grignard reagent 5 and vinyl bromide (Figure 5). These bidentate P,N ligands possess both centrochirality at carbon and planar chirality. The ligand PPFA (L13) furnished 4 in an excellent 99% yield and 63% ee.32−35 The ee of the product was determined to remain roughly constant over the course of the reaction.36 A structure–activity relationship study revealed that FcPN (L14), lacking centrochirality but maintaining planar chirality, gave 4 in 60% ee, demonstrating the dominant role of planar chirality in this system. EPPF (L15), which possesses neither centrochirality nor the dimethylamino group, delivered 4 in only 4% ee, validating the importance of the amino group and supporting a role for precoordination as proposed in Figure 6. Further evidence for the significance of a coordinating group comes from L16, which possesses a methoxy moiety instead of a dimethylamino group and provides 4 in 57% ee. Diphosphine BPPFA (L17), which could potentially coordinate through phosphorus in a bidentate fashion, also provides 4 in 65% ee. The similarity of the ee data obtained with L13 and L17 suggests that they both coordinate the metal in the same fashion, likely through a P–N mode. Consistent with this observation, changing the steric bulk on the amine of L13 gives a range of ee values for 4 (see L20), whereas changing the steric environment of the phosphine does not significantly perturb the selectivity (see L19). Homologated ligand L18 delivers 4 in poor ee.37 Pd catalysts were also investigated and were shown to give results comparable to those of Ni (Figure 8).34
Figure 8.
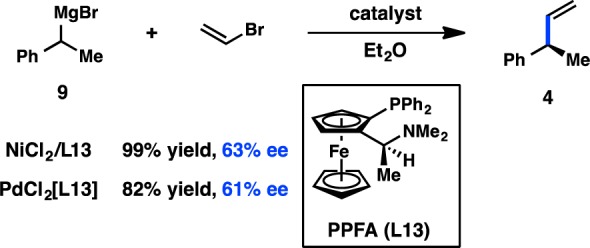
Use of the P–N ligand PPFA provides similar results in both Ni- and Pd-catalyzed transformations.
Several other ligand families have been developed for the enantioselective preparation of 4 (Figure 5). Catalysts generated from macrocyclic sulfides (L21) and nickel salts have been shown to impart moderate enantioselectivity, possibly through a simple kinetic resolution.38,39 The use of pyrrole-containing P,N ligand L22 or phosphine L23 delivers 4 in 32% and 68% ee, respectively, under Ni catalysis.40,41 Using Pd catalysis, the P,N ligand L24, containing both planar and centrochirality, gives improved results with respect to PPFA (L13).42 High ee can also be achieved with phosphine–quincoridine L25.43
Despite the advances made through ligand tuning when vinyl bromide is used as an electrophile, the scope of the asymmetric alkyl cross-coupling is poor. Disubstituted alkenes were typically found to be less enantioselective; for example, the reaction of (E)-bromostyrene using PPFA (L13) as the ligand delivered 10 in only 52% ee and moderate yield (Figure 9).34,44−46 Although the yield could be improved using the simpler aminophosphine L27, the ee of 10 decreased.47,48L28, designed to induce axial chirality upon coordination to a transition metal, was able to induce 76% ee for 10.49,50 Moderate enantioselectivity could also be attained with phosphine–oxazoline ligand L29.51,52 Knochel and co-workers have reported C2-symmetric ferrocenylphosphine L30 as capable of delivering excellent ee for the coupling of bromostyrene, although the reaction scope is still limited.53
Figure 9.
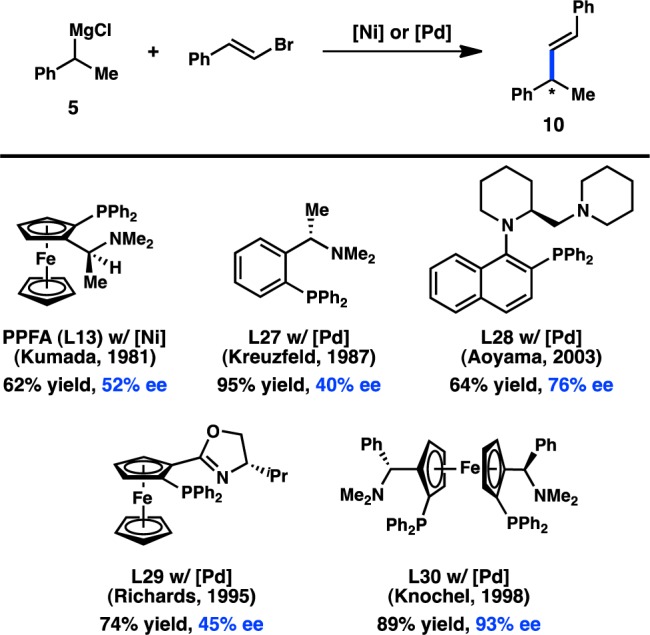
Asymmetric Kumada–Corriu cross-coupling of bromostyrene.
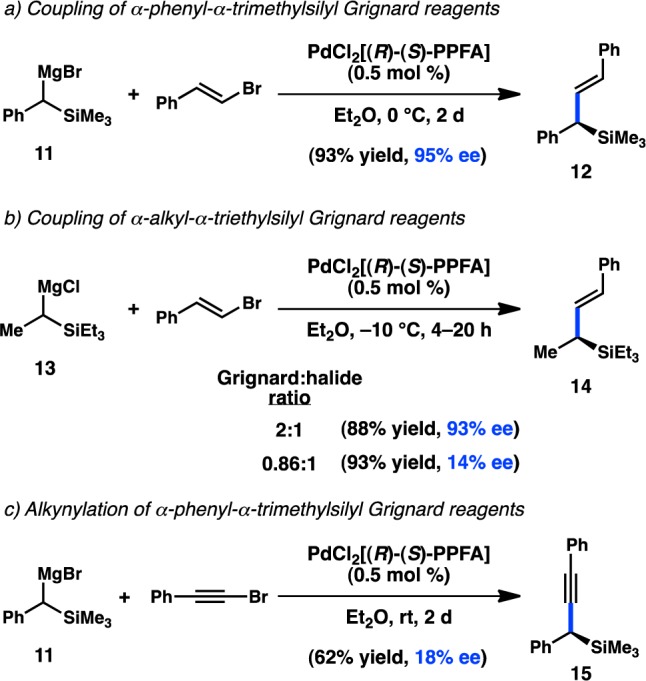
The asymmetric cross-coupling of organomagnesium reagents has been extended to α-trialkylsilyl Grignard reagents (11 and 13). When vinyl halides are used as the coupling partners, the products are allylsilanes, versatile reagents for the construction of C–C bonds. In initial studies, Kumada, Hayashi, and co-workers reported that Ni catalysts delivered poor yields of the desired allylsilane. However, the chiral PdCl2[PPFA] complex furnished 12 in 93% yield and 95% ee (Scheme 2a).54,55 (E)-Vinyl bromides were found to provide higher selectivities than the corresponding (Z)-substrates, and the enantioselectivity was independent of the ratio of Grignard reagent to vinyl bromide. In contrast, the coupling of alkyl-substituted Grignard 13 proceeded in 93% ee when excess organomagnesium reagent was employed, but the ee fell precipitously when Grignard 13 was used as the limiting reagent (Scheme 2b). These findings might suggest that, for Grignard 13, the rate of racemization is slow relative to the rate of C–C bond formation, resulting in a simple kinetic resolution instead of a DKR. A similar kinetic resolution had been observed previously in the diastereoselective coupling of nonbenzylic Grignard reagents.56 Lastly, an ee of 18% could be achieved in the alkynylation of 11 in the presence of PdCl2[PPFA] (Scheme 2c).55,57
Scheme 2. Diphosphine Ligands in the Coupling of α-Silyl Grignard Reagents.
2.1.2. Organozinc Reagents
The pioneering studies of enantioselective transition-metal-catalyzed alkyl cross-coupling reactions were initially performed using Ni catalysts and organomagnesium reagents—species expected to exhibit configurational lability (see section 2.1.1). Advances in the development of the Negishi cross-coupling subsequently enabled the use of organozinc reagents in asymmetric alkyl cross-coupling reactions, with Hayashi, Kumada, and co-workers reporting the first examples in 1983.58 Preliminary studies were carried out on the coupling of the organozinc chloride prepared from transmetalation of 5 with ZnCl2; however, Ni catalysts were determined to be poorly reactive. On the other hand, the combination of Pd and PPFA (L13) delivered 4 in 85% ee (Figure 10a). Significantly, when the organometallic was prepared by direct insertion of Zn0 into the organic chloride, the same enantioselectivity was achieved, albeit with a lower yield. Such an outcome implicates RZnCl as the transmetalating agent, rather than ZnCl2 behaving as a Lewis acid that can otherwise affect the transformation.30 Lower selectivities are obtained with the corresponding Grignard reagent under similar conditions. Additional improvements in ligand design revealed that 4 is formed in 93% ee when L31 is used.59 Despite a growing interest in the enantioselective cross-coupling reactions of organozinc reagents over the past three decades, successful efforts to expand further upon the enantioselective alkyl Negishi cross-coupling have been limited. Recently, Reisman and co-workers reported the Pd-catalyzed coupling between thioester 17 and organozinc 16 to form ketone 18 using chiral phosphoramidite L32 (Figure 10b).60 Although the enantioselectivity of the transformation is still low, the study represents a proof of concept for the possibility of employing organozinc reagents in enantioselective acyl cross-coupling reactions.
Figure 10.
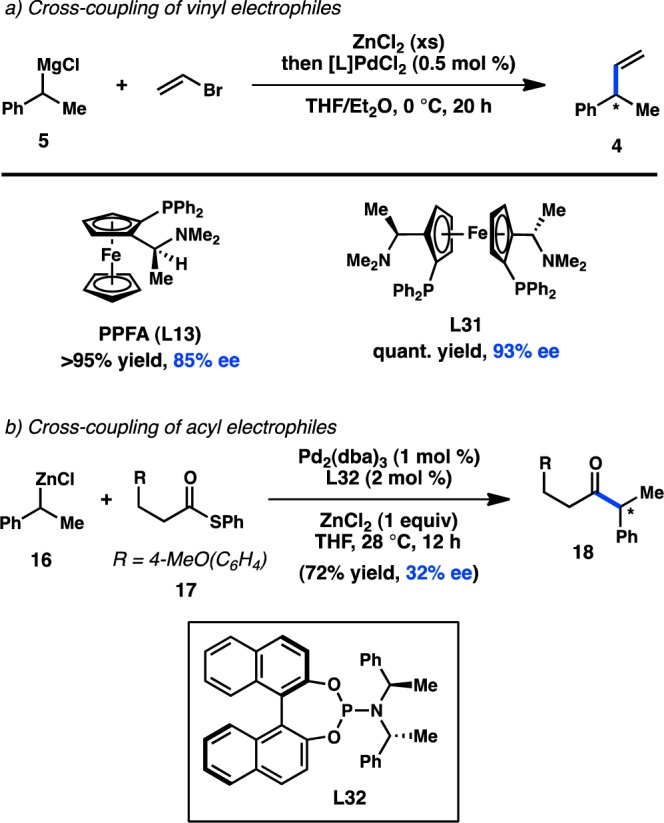
Enantioselective alkyl Negishi-type cross-couplings.
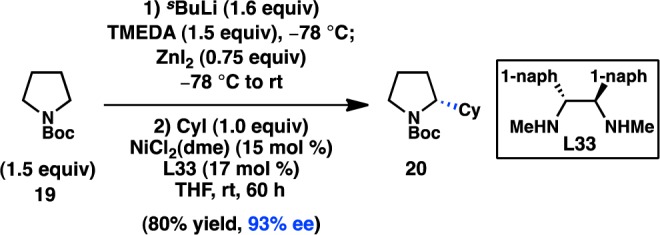
In a seminal 2013 report, Fu reinvestigated the Negishi cross-coupling of α-zincated N-Boc-pyrrolidine, which Campos and co-workers had previously shown can undergo stereospecific Pd-catalyzed cross-coupling to deliver enantioenriched α-arylpyrrolidine products (see section 3.1.5).61 Under Ni catalysis, in the absence of a chiral ligand, coupling of the stereodefined organozinc reagent with cyclohexyl iodide produced the coupled product in almost racemic form. Alternatively, when the chiral Ni/L33 complex was used as the catalyst, coupling of racemic 19 with cyclohexyl iodide furnished 20 with high ee in a stereoconvergent fashion, representing the first enantioconvergent alkyl–alkyl coupling of a racemic organometallic reagent (Scheme 3).62 Mechanistic studies have determined that this stereoconvergence does not arise from a series of β-hydride elimination/alkene insertion processes of the organometallic reagent.
Scheme 3. Enantioselective Functionalization of Pyrrolidine.
2.1.3. Organoboron Reagents
Trifluoroborate salts are often used in the Suzuki–Miyaura cross-coupling because of their improved stability with respect to boronic acids and esters. The two-electron mechanism of transmetalation typically believed to be operative in Suzuki–Miyaura reactions innately favors transmetalation in a stereospecific manner. However, Molander and co-workers hypothesized that transmetalation through a single-electron pathway could favor transfer of a C(sp3)-hybridized alkyl fragment via a stereoconvergent, radical process. To generate a radical from an organoboron reagent, the authors envisaged a dual catalysis mechanism in which Ni-catalyzed cross-coupling and Ir-catalyzed photoredox events occur synergistically (Figure 11).63 In an important proof of concept, chiral bioxazoline (BiOX) L34 was used to furnish 23 in 50% ee. Electron transfer to an excited-state *IrIII complex from an organoboron species would generate an alkyl radical. The alkyl radical can then combine with a chiral NiII complex to form a NiIII species that can reductively eliminate the desired product. The resulting NiI can be reduced by IrII to complete both catalytic cycles. Additional investigations toward asymmetric catalysis would be valuable.
Figure 11.
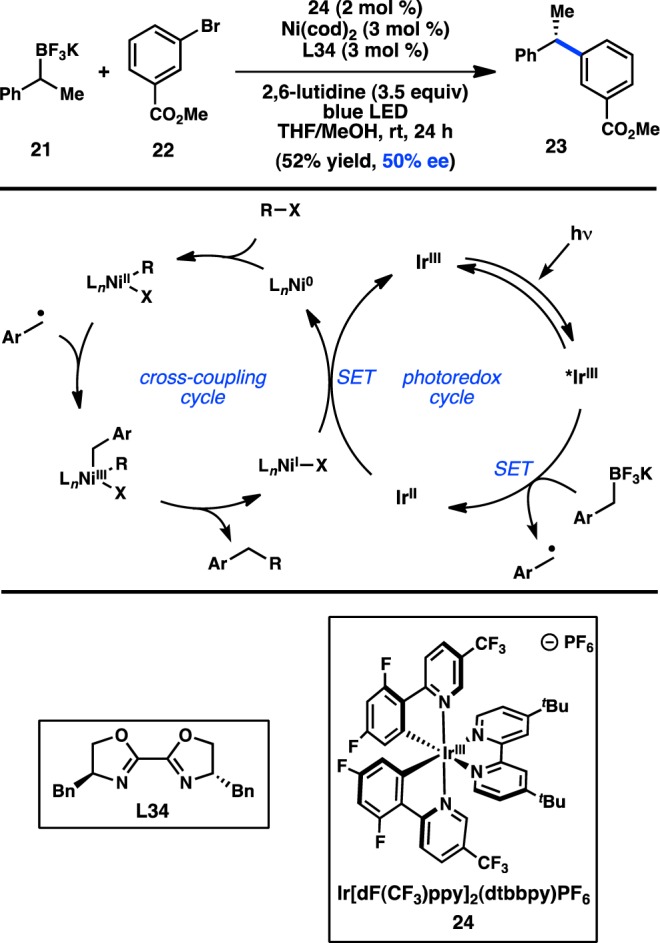
Dual catalysis approach to asymmetric cross-coupling.
2.2. Reactions of Secondary Alkyl Electrophiles
The challenges associated with oxidative addition of sec-alkyl electrophiles, as well as the propensity for alkyl transition-metal complexes to undergo rapid β-hydride elimination, conspired to make the cross-coupling of these electrophiles difficult to realize using Pd, which had emerged as the metal of choice for cross-coupling in the 1980s. In the early 2000s, researchers began reinvestigating first-row transition metals for the cross-coupling of sec-alkyl halides and organometallic reagents.8 Following the first reports of alkyl cross-coupling to form stereogenic C(sp3) centers, the systematic examination of asymmetric induction in these processes became a chief objective. In these systems, catalysts that favor a single-electron oxidative addition mechanism may undergo a stereoconvergent oxidative addition to set the ultimate stereochemistry of the product (Figure 12). Alternatively, rapidly equilibrating mixtures of diastereomeric transition-metal complexes can result in preferential transmetalation or reductive elimination of one diastereomer over the other.
Figure 12.
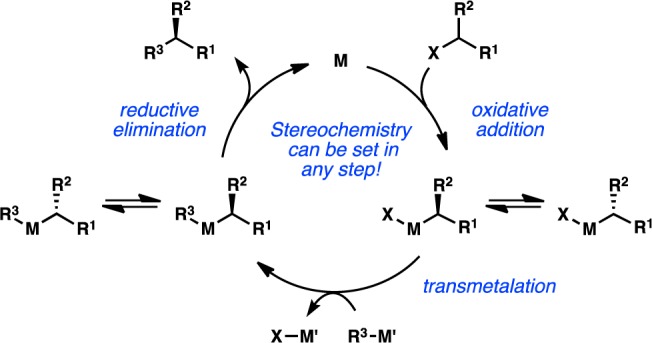
Stereochemical outcome of cross-coupling with secondary electrophiles.
2.2.1. With Organomagnesium Reagents
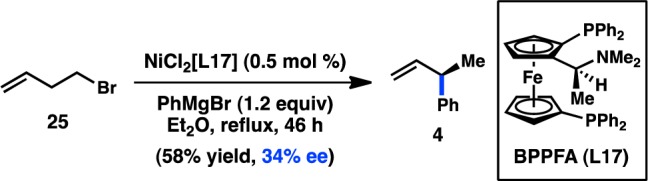
The earliest example of an enantioselective transition-metal-catalyzed cross-coupling reaction between an alkyl electrophile and an organomagnesium reagent was disclosed by Kumada and co-workers in 1977, the result of a surprising alkyl group isomerization observed during the coupling between homoallylic halide 25 and PhMgBr (Scheme 4).64 In the presence of the chiral catalyst NiCl2[BPPFA], 4 was formed in 34% ee. Whereas the isomerization of secondary organometallic reagents to primary species is a well-known side reaction in cross-coupling chemistry, the inverse isomerization is much more rarely observed.65 Although this preliminary result was not further developed by Kumada and co-workers, it presaged the explosion of asymmetric cross-couplings of sec-alkyl electrophiles that would emerge in the literature nearly two decades later.
Scheme 4. Primary-to-Secondary Isomerization in Asymmetric Cross-Coupling.
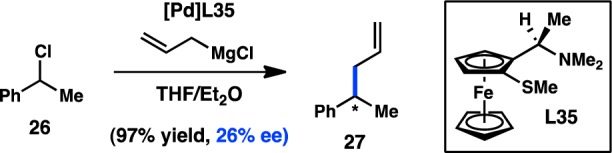
In 1988, the Pd-catalyzed enantioselective cross-coupling between allylmagnesium chloride and racemic (1-chloroethyl)benzene (26) was reported by Brubaker and co-workers. A variety of bidentate S,N and Se,N ligands bearing planar and centrochirality were prepared and used to generate chiral Pd complexes.66−68 The complex derived from L35 catalyzed the formation of 27 in a high yield and modest, yet promising ee (Scheme 5). Mechanistic studies to elucidate whether the enantioenriched product arises from a stereoconvergent process were not disclosed. The same group later studied Ni complexes of these ligands in the same transformation, obtaining similar levels of enantioinduction.
Scheme 5. Asymmetric C(sp3)–C(sp3) Kumada–Corriu Cross-Coupling.
The first synthetically useful enantioselective, stereoconvergent cross-coupling between a sec-alkyl electrophile and a Grignard reagent was reported two decades later by Fu and co-workers. In this seminal report, the combination of NiCl2(dme) and bidentate bis(oxazoline) ligand L36 or L37 was found to promote the coupling of α-halo ketones 28 and arylmagnesium halides to give α-aryl ketones (Figure 13).69 Notably, the reaction can be run at some of the lowest temperatures reported for the cross-coupling of alkyl electrophiles (−60 °C); the low temperature prevents the racemization of ketone product 29 through enolization by the Brønsted basic Grignard reagent. Both alkyl and aryl ketones can be prepared by this method, and these products can be diastereoselectively derivatized to access chiral alcohols and amines.70
Figure 13.
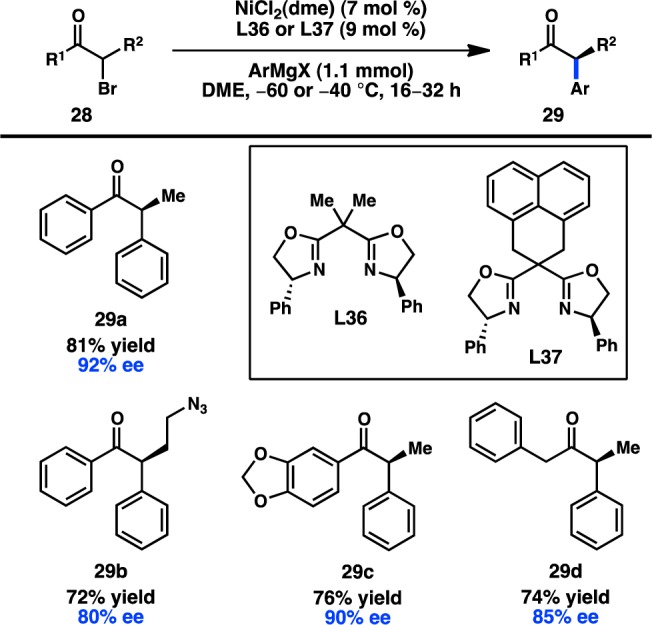
Stereoconvergent Kumada–Corriu coupling of α-halo ketones.
2.2.2. With Organozinc Reagents
In 2005, two reports from the Fu laboratory demonstrated the first utilization of secondary alkyl electrophiles in highly enantioselective cross-coupling reactions. In one example, treatment of α-bromo amide 30 with an alkylzinc reagent and a Ni/L38 catalyst delivered 31 in good yield and high ee (Figure 14a).71 The identity of the amide substituents played a key role in achieving high enantioselectivity. When the organozinc reagent is used as a limiting reagent, the α-bromo amide is recovered as a racemate, suggesting that the reaction does not proceed by a kinetic resolution. In a second example by Fu and co-workers, the Ni/L38-catalyzed coupling of 1-bromoindanes and alkyl halides produced chiral indane 33 in good yield and high ee (Figure 14b).72 The use of acyclic 1-(1-bromoethyl)-4-methylbenzene furnished 33c with more modest enantioselectivity. In both cases, only primary organozinc reagents were compatible with the reaction conditions. A computational investigation by Lin and co-workers proposed that a NiI/NiIII mechanism consisting of transmetalation/oxidative addition/reductive elimination is more energetically favorable than a Ni0/NiII mechanism.73 The enantioselectivity of the reaction was also correlated to the difference in free energy between the two transition states for reductive elimination.
Figure 14.
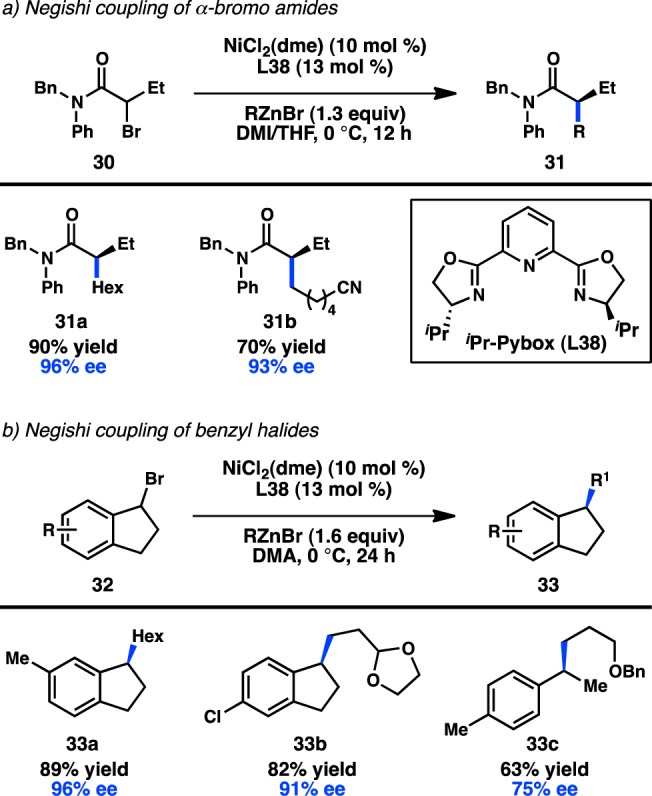
Seminal reports of stereoconvergent cross-couplings of secondary alkyl halides.
Despite Fu’s promising results for the asymmetric, stereoconvergent Negishi cross-coupling of alkylzinc reagents, the extension to arylzinc species proved challenging. After a lengthy investigation, it was discovered that Ni/L39 complexes catalyze the cross-coupling between propargyl halide 34 and Ph2Zn to furnish 35 in a high yield and ee (Figure 15a).74 Because relatively few diarylzinc reagents are commercially available, the group sought to identify other arylzinc reagents that were effective for this transformation. Unfortunately, the use of arylzinc halides or in situ-prepared diarylzincs, generated from transmetalation of the corresponding organolithium or -magnesium reagent, was unsuccessful. However, the group determined that ArZnEt, prepared from ArB(OH)2 and Et2Zn, could react to provide comparable results. A recent mechanistic investigation supports a radical chain pathway in which a bimetallic oxidative addition to propargyl bromide 34 takes place.75 In contrast to the stereospecific Pd-catalyzed coupling of propargyl halides, no allene formation, arising from SN2′ oxidative addition, was observed.76
Figure 15.
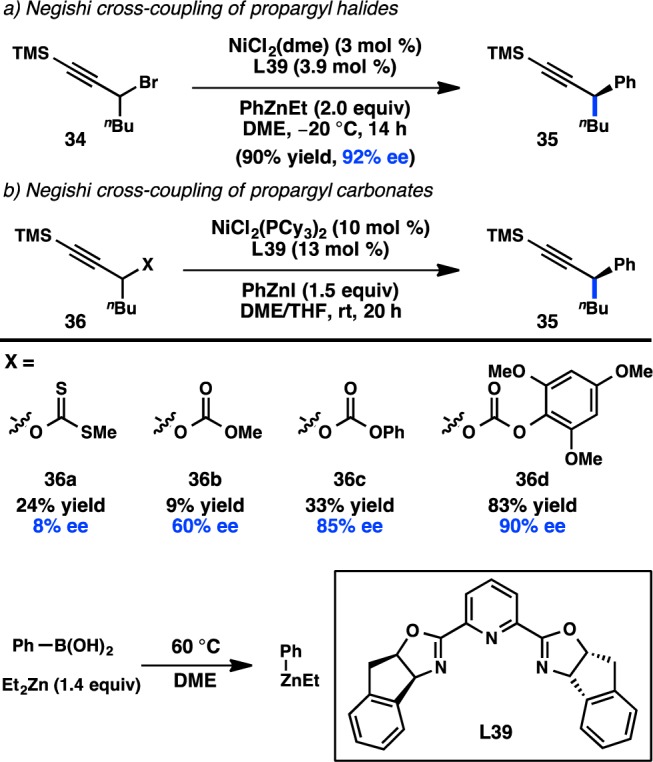
Stereoconvergent Negishi cross-coupling of propargylic electrophiles.
Organic halides are frequently prepared from the corresponding alcohols, and for certain substrates this functional group interconversion can be low yielding. Recognizing the synthetic advantage of using oxygen-based electrophiles directly in cross-coupling reactions, Fu and colleagues turned their attention to the asymmetric cross-coupling of propargylic alcohol derivatives. Hypothesizing that the reaction would proceed through a radical-based oxidative addition to Ni, a xanthate was chosen as a potential leaving group because of its propensity toward radical cleavage in Barton–McCombie-type transformations. However, these substrates performed poorly, producing 35 in low yield and ee (Figure 15b).77 On the other hand, simple carbonate 36b underwent cross-coupling with improved enantioselectivity. Further investigation revealed that both the yield and ee could be improved by use of aryl-substituted carbonates, with 36d delivering 35 in 83% yield and 90% ee. The optimized reaction conditions proved to be general not just for propargyl carbonates, but also for the coupling of propargyl chlorides and bromides.
In 2013, Fu and co-workers published a stereoconvergent Negishi coupling of benzylic mesylates that could be prepared from the corresponding alcohols immediately prior to the coupling and used without purification (Figure 16).78 Bioxazoline L40 was identified as the optimal ligand, with more traditional Pybox and Box ligands delivering poor enantioselectivity. LiI was employed to allow in situ displacement of the mesylate to form a reactive benzylic iodide. A wide substrate scope was demonstrated for the cross-coupling; a slight erosion of ee is observed when R = Me. Although several stereospecific routes to diarylalkanes have been developed to date,79−85 this reaction provides a complementary approach.
Figure 16.
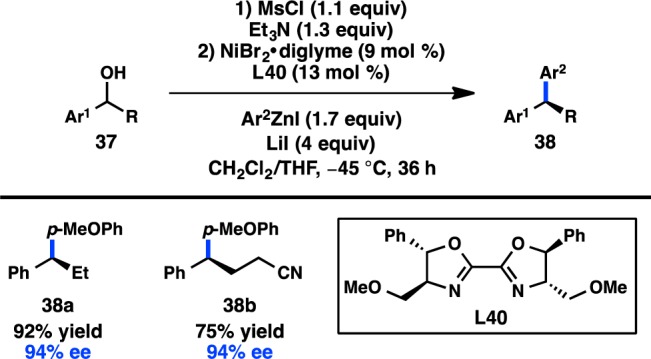
Stereoconvergent Negishi cross-coupling of benzyl alcohol derivatives.
A long-term objective in the area of enantioselective alkyl cross-coupling is to couple sec-alkyl electrophiles with sec-alkylmetallic reagents. The Fu laboratory made a significant advance toward this objective in 2012 when they reported the asymmetric Negishi cross-coupling between benzylic bromide 39 and cyclic organozinc halides (Figure 17).65 Isoquinoline–oxazoline ligand L41 delivered the products in high yields and ee’s, in contrast to the more commonly employed Pybox and Box ligands. Acyclic secondary organozinc halides resulted in a mixture of branched and linear products; surprisingly, primary organozinc halides also resulted in a mixture of branched and linear products.
Figure 17.
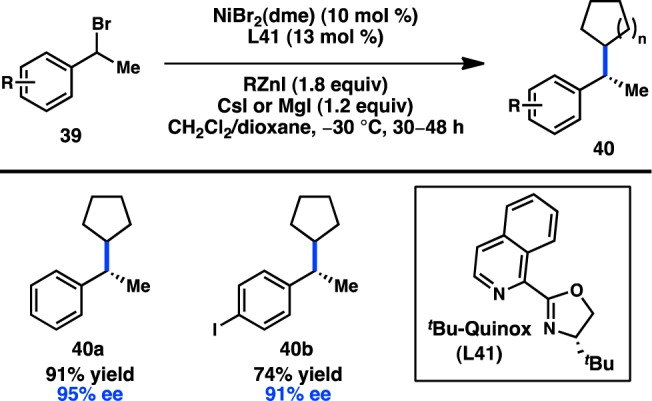
Enantioconvergent Negishi cross-coupling of secondary organozinc reagents.
Prior to their disclosure of the enantioselective cross-coupling between α-bromo ketones and aryl Grignard reagents (see Figure 13), the Fu laboratory developed a Ni/L42-catalyzed asymmetric cross-coupling of α-bromo ketones and arylzinc reagents (Figure 18a).70 The low basicity of the organozinc reagent, as well as a reduced reaction temperature, accounts for the configurational stability of the potentially sensitive tertiary stereocenter in 42. The synthesis of dialkyl ketones proceeded with lower enantioinduction; however, this substrate limitation is addressed by their subsequently developed Kumada–Corriu conditions.69 A recent modification of the reaction conditions has permitted the use of α-halo-α-fluoro ketones, enabling the asymmetric formation of tertiary fluorides (Figure 18b).86
Figure 18.
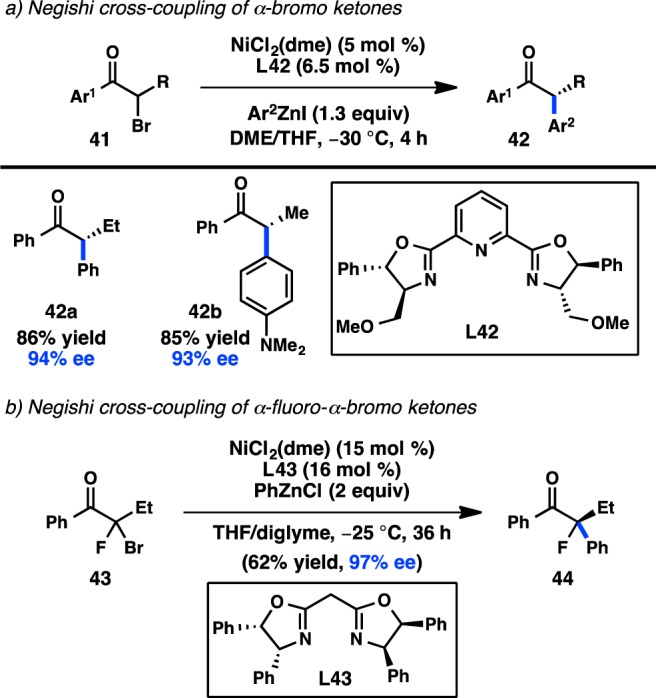
Asymmetric Negishi cross-coupling of α-halo ketones.
The Fu group has further expanded the scope of alkyl electrophiles amenable to Ni-catalyzed stereoconvergent Negishi cross-coupling to include α-bromo nitriles.87 Coupling of α-bromo nitrile 45 and R2Zn in the presence of NiCl2(dme) and L44 at −78 °C furnishes 46 in high yield and ee (Figure 19a).88 For the first time, alkenylzinc reagents were suitable coupling partners, delivering 46b in 94% yield and 91% ee. Somewhat unexpectedly, a variant of 46 containing a pendant alkene failed to cyclize under the reaction conditions, in contrast to what was observed in the related coupling of simple unactivated halide electrophiles.89,90 A more comprehensive mechanistic analysis is thus required to elucidate the mechanism of oxidative addition for the given transformation.
Figure 19.
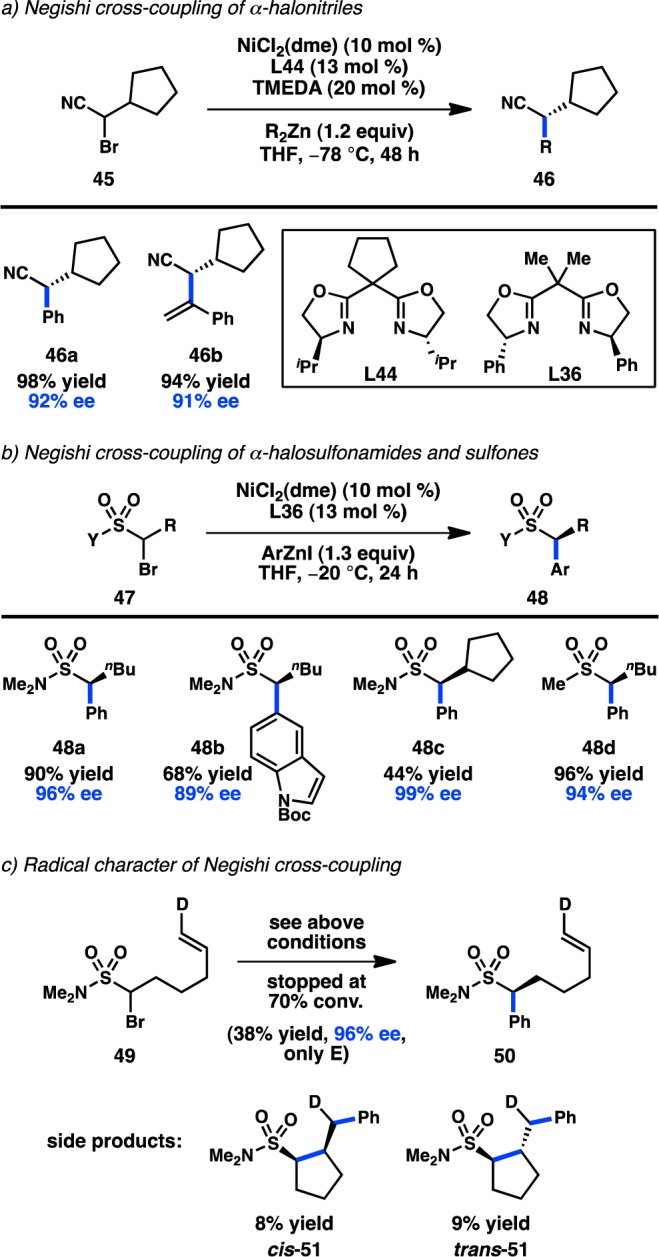
Other directing groups in asymmetric Ni-catalyzed Negishi cross-coupling.
The previous examples of Ni-catalyzed stereoconvergent Negishi cross-coupling reactions from the Fu laboratory have focused on the use of activated secondary electrophiles; in 2014, they reported the coupling between α-halo sulfonamides (47) and arylzinc reagents (Figures 19b).91 Because sulfonyl groups do not significantly stabilize α-radicals, 47 can be considered as an unactivated electrophile. Investigations of the substrate scope revealed that sulfones are also suitable substrates without any change in the reaction conditions, furnishing 48d in high yield and ee. Subjection of radical clock substrate 49 to the reaction conditions provided a mixture of 50, cis-51, and trans-51; the ratio of uncyclized product to cyclized product was found to linearly increase with increased Ni loading. These data could suggest that the reaction proceeds through a noncaged radical species, and also illustrates the dichotomy between the coupling of electrophiles 45 and 47.
2.2.3. With Organoboron Reagents
Seminal contributions to the transition-metal-catalyzed enantioselective cross-coupling of sec-alkyl electrophiles with organoboron reagents have been made by the Fu laboratory. Shortly after disclosing the Ni-catalyzed cross-coupling of sec-alkyl electrophiles with alkylboranes to prepare racemic products,92 Fu and co-workers reported that use of catalytic Ni(cod)2 in conjunction with chiral 1,2-diamine ligand L45 enabled the enantioselective coupling of homobenzylic bromides (52) with organoboranes (Figure 20a).93 The Ni catalyst was proposed to engage in a secondary interaction with the benzylic substituent on 52, allowing differentiation between the two alkyl groups of the starting material. Although a variety of homobenzylic bromides were tolerated, poor enantioselectivity was attained in the formation of 53b. Fu hypothesized that the ether might also interact with the Ni catalyst, leading to poor asymmetric induction. On the basis of this hypothesis, the group subsequently reported that carbamate-protected halohydrins (54) can also be coupled with alkylboranes in high enantioselectivity using a chiral 1,2-diamine L46 (Figure 20b).94 Modified conditions permitted the enantioselective coupling of a homologated halohydrin. Further expansion of the substrate scope determined that halides (56) bearing proximal arylamines as directing groups can be coupled with alkylboranes in high enantioselectivity as well (Figure 20c).95 The reaction was found to be directed by the nitrogen atom of the arylamine group.
Figure 20.
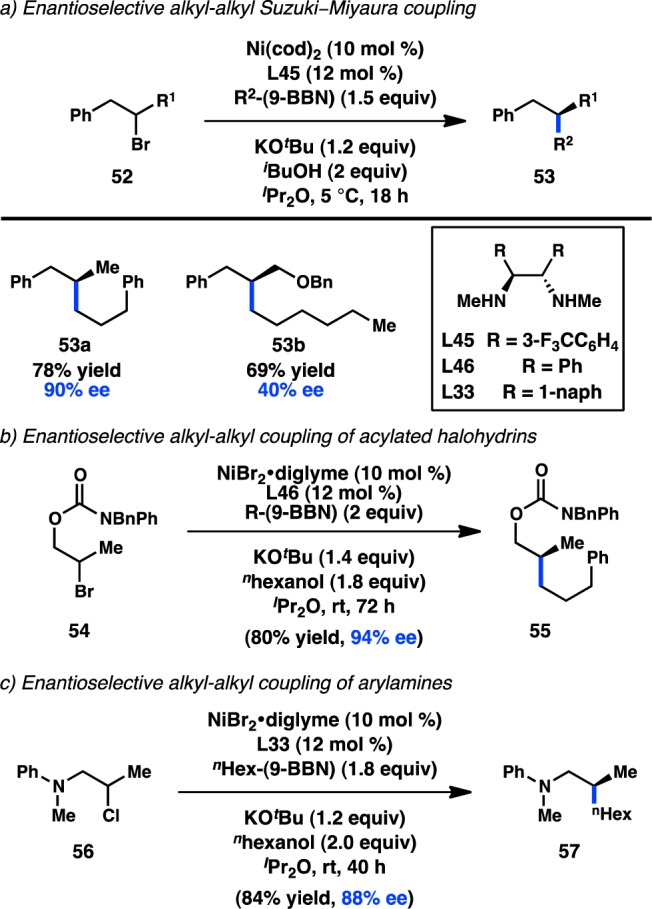
Enantioconvergent Ni-catalyzed alkyl–alkyl Suzuki–Miyaura coupling.
The early examples of enantioconvergent alkyl–alkyl Suzuki–Miyaura couplings all involved alkyl halide substrates with a directing group capable of coordinating the Ni center. Subsequent efforts turned to identifying new directing groups and to exploring how far removed the directing group could be from the reacting C–halide bond. Illustrating that distal functional groups are still capable of directing highly enantioselective reactions, both γ- and δ-chloro amides were shown to undergo Suzuki–Miyaura cross-coupling with good asymmetric induction to form 58 and 59, respectively (Figure 21).96 Various halides proximal to protected amines, such as carbamates or sulfonamides, were also optimized toward enantioconvergent cross-coupling.97,98 After confirming that the oxygen of the sulfonamide was the key directing atom, Fu and co-workers examined sulfone-containing electrophiles and reported that good enantioselectivity can still be maintained for these substrates.97
Figure 21.
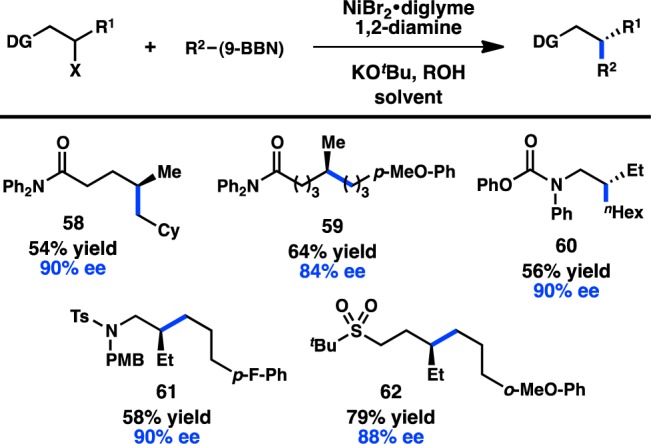
Examples of directing groups for the enantioconvergent Suzuki–Miyaura coupling.
In addition to the Ni-catalyzed cross-coupling of organomagnesium and organozinc reagents to α-halocarbonyl compounds, the Fu laboratory has identified conditions for the enanatioselective coupling between α-halo amides and arylboron reagents. After first investigating several different amides, it was found that the combination of NiBr2·diglyme and L45 catalyzed the coupling between α-chloro amides (63) and Ar(9-BBN) reagents to furnish 64 in good yields and high ee’s (Figure 22).99 The identity of the amide substituents was important for good enantioinduction: diphenyl amides and Weinreb amides delivered nearly racemic products. In contrast to previous stereoconvergent couplings of secondary electrophiles, a modest kinetic resolution of 63 was observed. Further studies confirmed an irreversible oxidative addition step. The Fu laboratory has also shown that γ-halo amides can be arylated with Ph(9-BBN) in good ee but only moderate yield.96
Figure 22.
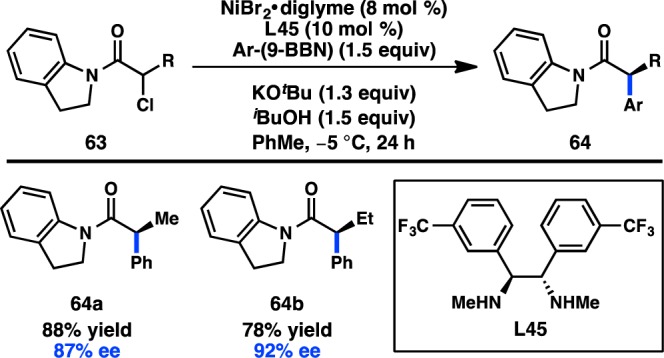
Asymmetric Suzuki–Miyaura coupling of α-halo amides.
Building off their growing mechanistic understanding of Ni-catalyzed stereoconvergent alkyl cross-coupling reactions, Fu and co-workers have developed a cascade cyclization/cross-coupling to forge two C–C bonds in one step with both excellent ee and high diastereomeric ratio (dr) (Figure 23).100 Key to this transformation was the insight that a “transmetalation first” mechanism could be operative, and that organonickel complex 67 might undergo migratory insertion faster than oxidative addition of the alkyl halide electrophile. This theory was validated in the Ni-catalyzed asymmetric cascade cyclization/cross-coupling reaction between arylborane 65 and several simple alkyl bromides, in which heterocyclic products 66 were obtained in excellent ee. Realizing the compatibility of their reaction conditions with those previously optimized for coupling of γ-halo amides (see Figure 21), a γ-halo amide was also used as an electrophile.96 Remarkably, a single Ni complex controls the stereochemical outcome of two distinct C–C bond forming processes, providing product 66c in good yield, good dr, and excellent ee.
Figure 23.
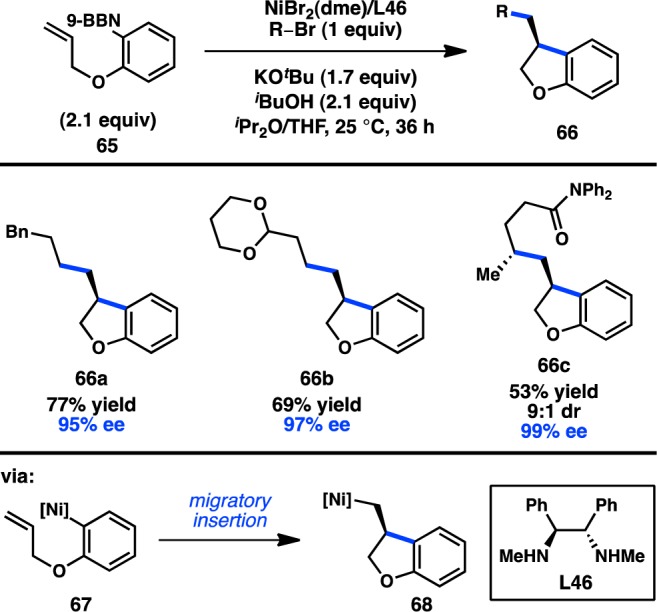
Asymmetric cascade cyclization/cross-coupling.
The Doyle laboratory has focused on expanding the scope of electrophiles suitable for transition-metal catalysis, investigating the cross-coupling reactions of acetals and N,O-acetals. These efforts led to the discovery that Ni(cod)2 catalyzes the addition of various arylboroxines to N,O-acetal 69, presumably via the intermediacy of quinolinium ion 71.101 When chiral phosphoramidite L47 is used as a supporting ligand, 70 is formed in 52% ee (Figure 24). A unique oxidative addition mechanism, in which the Lewis acidic boroxine promotes ionization of the leaving group and results in an SN1-type addition of Ni0, was discovered for this coupling.102 A wider survey of ligands showed that improved ee could be realized with TADDOL-based phosphonite L48.103 In an extension of this work, the addition of arylzinc reagents into pyridinium ions was subsequently reported.104
Figure 24.
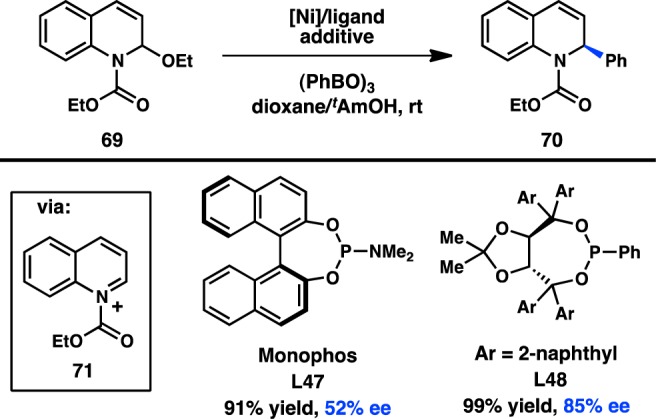
Asymmetric addition into quinolinium ions.
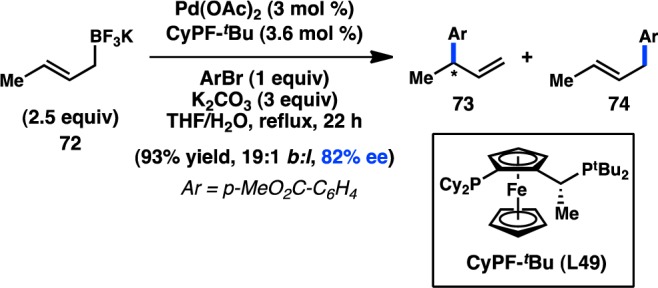
In 2006, Yamamoto, Miyaura, and co-workers reported a novel strategy for the preparation of enantioenriched products from achiral starting material. Bidentate phosphine ligands were used to allow high γ-selectivity in the Pd-catalyzed asymmetric cross-coupling between allylic trifluoroborate salts and aryl bromides.105 A subsequent evaluation of bulky, chiral ferrocenylphosphine ligands revealed that Josiphos-type ligand L49 could promote formation of 73 in 82% ee while still maintaining a high selectivity for γ-addition (Scheme 6).106 DFT studies support an SE2′ (open) transition state for transmetalation, whereas the corresponding closed transition state was of slightly higher energy;107 transmetalation was also proposed to be the stereochemistry-determining step, which is followed by a fast reductive elimination to forge the desired product. Similar γ-selectivity was observed in the stereospecific couplings of allylic boronates108 and silanes (see sections 3.1.1 and 3.1.3).109,110
Scheme 6. Enantioselective Cross-Coupling of Allylic Trifluoroborate Salts.
2.2.4. With Organosilicon Reagents
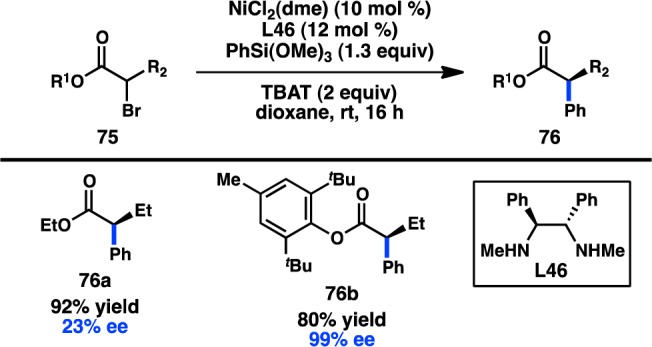
Only a single example of an asymmetric cross-coupling between sec-alkyl organic halides and organosilicon reagents has been reported to date. Fu and colleagues developed a Ni/L46-catalyzed stereoconvergent coupling of α-bromo esters (75) and arylsiloxanes to furnish α-aryl esters in good yields and with high enantioinduction (Figure 25).111 Although simple ethyl esters gave good yield but poor ee, the use of the BHT (butylated hydroxytoluene) ester resulted in formation of 76b in a remarkable 99% ee. The nature of the fluoride source and the steric profile of R2 also affected the level of enantioinduction. In the same paper, the optimized reaction conditions were extended to the coupling of alkenylsilanes as well.
Figure 25.
Stereoconvergent coupling of arylsilanes.
2.2.5. With Organozirconium Reagents
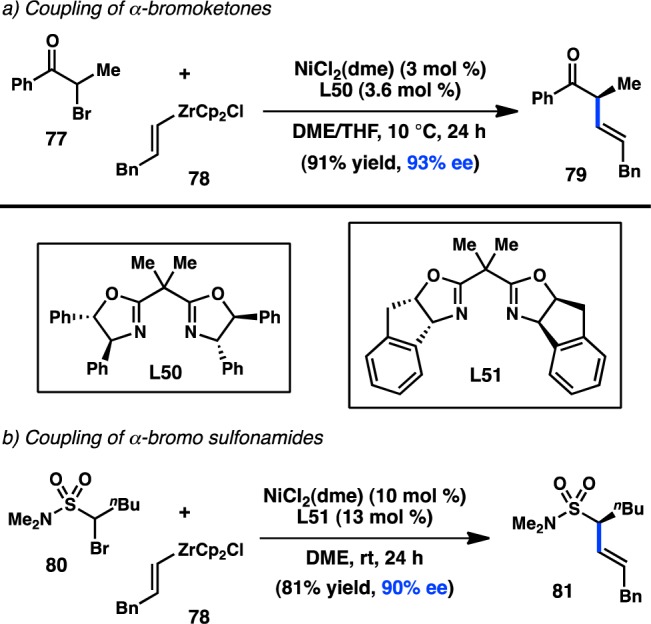
Alkenylzirconium complexes are attractive alkenylmetallic species for use in organic synthesis because they can be easily prepared from Schwartz’s reagent and an alkyne. Although Fu has disclosed a remarkable variety of stereoconvergent arylation reactions, most of the reaction conditions could not easily be extended to the cross-coupling of alkenylmetal species, with alkenylsilicon111 and -zinc88 reagents being the most promising. In 2010, Fu and co-workers published the Ni/L50-catalyzed asymmetric cross-coupling of alkenylzirconium reagents and α-bromo ketones, allowing access to 79 in 93% ee (Figure 26a).112 The versatility of this approach has been exemplified by the efficient coupling of both aryl alkyl ketones and dialkyl ketones under the same conditions. Alkenylzirconium complexes have also been shown to react with α-bromo sulfonamides in high yield and ee (Figure 26b).91
Figure 26.
Stereoconvergent coupling of alkenylzirconium reagents.
2.2.6. With Organoindium Reagents
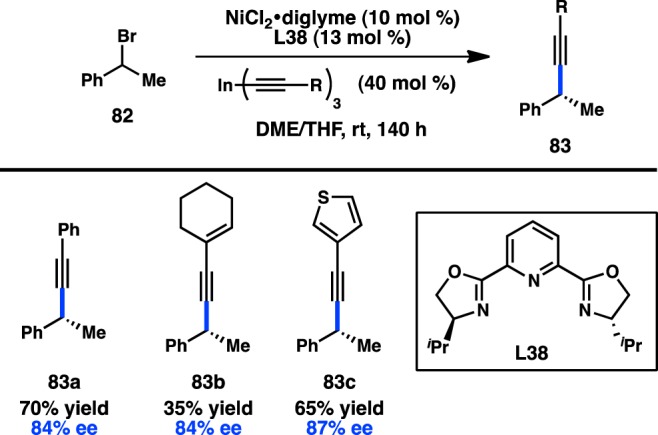
Shortly after the publication of Fu’s seminal examples of Ni-catalyzed stereoconvergent cross-coupling reactions between sec-alkyl electrophiles and either C(sp3)- or C(sp2)-hybridized organometallic reagents,71,72 Sestelo, Sarandeses, and co-workers investigated the asymmetric coupling between C(sp)-hybridized organometallic reagents and benzylic bromides. Alkynylindium reagents exhibited clean cross-coupling under Ni catalysis, and were selected for further study. Pybox ligand L38 was optimal, delivering cross-coupled product 83 in up to 87% ee for several different alkynes (Figure 27).113 Further work on the asymmetric coupling of C(sp) organometallic reagents has not been disclosed.
Figure 27.
Alkynylmetallic reagents in stereoconvergent cross-coupling.
2.3. Transition-Metal-Catalyzed Desymmetrization Reactions
One approach to generating enantioenriched products through transition-metal-catalyzed alkyl cross-coupling reactions is to perform desymmetrization reactions of meso-compounds. In this case, the C(sp3)-hybridized carbon at the site of C–C bond formation is not necessarily stereogenic; instead, the C–C bond formation is used to break symmetry through a catalyst-controlled process, giving rise to a molecule with centrochirality. Most of the work in this area has focused on the desymmetrization of meso-electrophiles; however, some researchers have investigated the desymmetrization of meso-bisorganometallic reagents or processes that involve desymmetrization by C–H functionalization.
2.3.1. Organozinc Reagents
The desymmetrization of meso-anhydrides has emerged as a robust method for the synthesis of enantiopure products.114−116 Rovis and co-workers117 have developed a monofunctionalization of cyclic anhydrides through a Ni-catalyzed Negishi coupling with Et2Zn.118 The transformation was sensitive to the bite angle of the ligand and required an electron-deficient styrene additive, which has been demonstrated by Knochel to accelerate reductive elimination over β-hydride elimination.119,120 On the basis of these initial findings, the authors sought to develop a desymmetrizing Negishi reaction of meso-cyclic anhydride 84, and determined that the catalyst prepared from of Ni(cod)2 and iPr-PHOX (L52) furnished 85 in 79% ee (Scheme 7).121 Surprisingly, omission of the p-(trifluoromethyl)styrene additive reduced the ee to 4%, prompting Rovis and co-workers to examine the mechanism of the reaction more closely. Kinetic analysis of the reaction revealed two competing mechanisms for the formation of 85 (Figure 28).122 One occurred in the absence of styrene and proceeded with low enantioselectivity (cycle B). The other involved coordination of styrene and provided 89 in high ee (cycle A). For both reactions, the rate-determining step was found to be oxidative addition. However, in contrast to the initial proposal that p-(trifluoromethyl)styrene would accelerate reductive elimination, it was instead shown to increase the rate of oxidative addition. Although the origin of this rate enhancement is unclear, it was hypothesized that p-(trifluoromethyl)styrene might coordinate to Ni and facilitate deligation of cod, providing a three-coordinate Ni complex capable of undergoing oxidative addition. The kinetic analysis determined that cycle A proceeds approximately 4 times faster than cycle B and is roughly consistent with the somewhat modest enantioselectivities obtained under these conditions.
Scheme 7. Alkylative Desymmetrization of meso-Anhydrides.
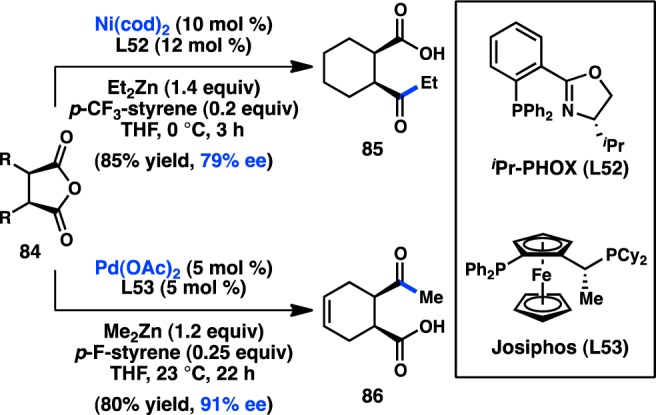
Figure 28.
Competing mechanisms in the Ni-catalyzed desymmetrization of meso-anhydrides.
The Pd-catalyzed desymmetrization of succinic anhydrides was also developed by Rovis and co-workers (Scheme 7). Treatment of 84 with Me2Zn in the presence of Pd(OAc)2 and the bidentate phosphine Josiphos (L53) furnished 86 in 91% ee; the use of p-fluorostyrene as an additive was crucial to achieving the high level of enantioselectivity.123
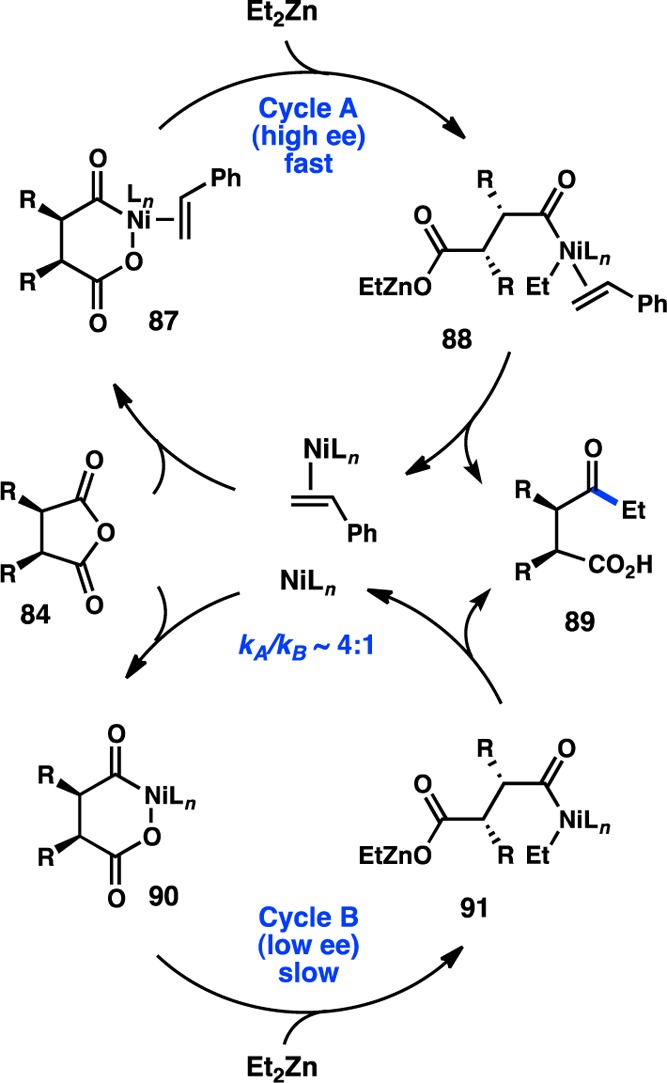
One hurdle in the Pd-catalyzed desymmetrization was the catalyst’s sensitivity to halide salts, meaning that the organozinc reagent could not be prepared in situ from the more readily available organomagnesium or -lithium reagents. Investigation of Rh catalysts in the desymmetrization of glutaric anhydride, a substrate that reacts poorly in the presence of Pd, revealed that high yields and good ee’s could be attained, allowing access to syn-deoxypolypropionate synthons (Figure 29).124−126 This reaction is proposed to proceed through a RhI/RhIII catalytic cycle, with transmetalation occurring prior to oxidative addition of the anhydride. Furthermore, the organozinc reagent could now be prepared in situ. The desymmetrization of glutaric anhydrides is an enantioselective alternative to Breit’s enantiospecific Kumada–Corriu coupling for the synthesis of deoxypolypropionates.127
Figure 29.
Rh-catalyzed desymmetrization of glutaric anhydrides.
Efforts by Rovis and co-workers to effect an alkylative desymmetrization of meso-anhydrides have been complemented by attempts to use arylzinc reagents in similar transformations. In the presence of a Pd/Josiphos catalyst system, 1,4-ketoacid 95 could be prepared from commercially available Ph2Zn in good yield and high ee (Figure 30).123 In contrast to Rovis’s alkylation method, excess fluorostyrene is not necessary to achieve a high enantioinduction. Although Ph2Zn was reactive in the Pd-catalyzed transformation, lower yields and ee’s were observed when the organozinc reagent was prepared in situ from the corresponding organolithium. In fact, simple exposure of 94 and Ph2Zn to LiX under the standard reaction conditions led to a sharp drop in enantioselectivity. As a result, Rovis next turned to Rh complexes, which are well-known to be less prone to interaction with Lewis bases such as halides. After optimization toward a chiral phosphoramidite ligand, 96 could be furnished in 85% yield and 87% ee.125,128 A variety of in situ-prepared arylzinc reagents can be coupled under the Rh-catalyzed conditions with uniformly good enantioselectivity, leading the authors to propose that the stereochemistry-determining step occurs independent of the organometallic reagent. These reaction conditions could not be extended to the coupling of alkylzinc reagents.
Figure 30.
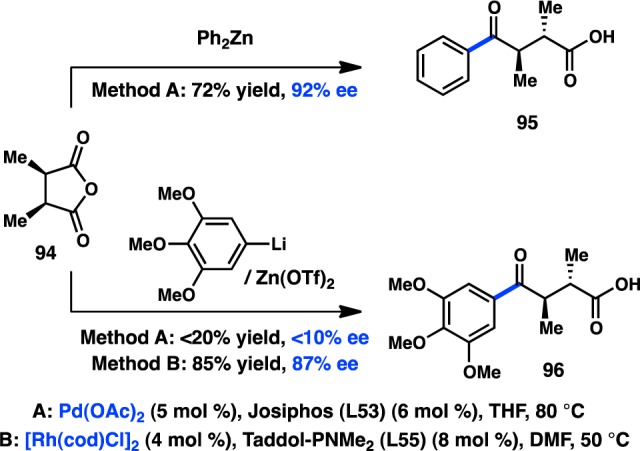
Pd- and Rh-catalyzed desymmetrization with arylzinc reagents.
2.3.2. Organoboron Reagents
In 1998, Shibasaki and co-workers reported a Pd-catalyzed intramolecular enantiotopic group-selective Suzuki–Miyaura coupling of alkylboranes (97) to prepare exo-methylenecyclopentanes, but the highest ee value obtained was 31% (Figure 31a).129 More recently, Morken demonstrated that prochiral diboronate 99 can be cross-coupled with an aryl halide in the presence of Pd(OAc)2 and phosphoramidite L56 to forge benzylic boronate 100 with good enantioinduction (Figure 31b).130 Enantioenriched boronate 100 can undergo a subsequent stereospecific cross-coupling to generate an enantioenriched diarylalkane.79 Prior studies by Shibata and co-workers had demonstrated that geminal diboronate 99 is activated toward transmetalation and proposed that the “ate” complex of one boronate can coordinate to Pd and assist in an SE2 transmetalation of the second boronate.131 The resulting monoboronate 100 lacks this mode of activation, avoiding the formation of diarylated products.
Figure 31.
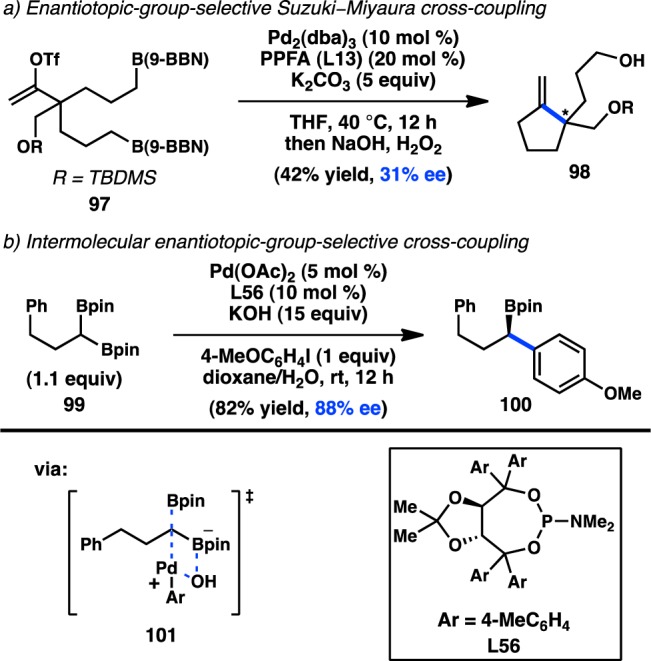
Enantiotopic-group-selective cross-coupling.
In a distinct desymmetrization approach, Willis and co-workers reported the asymmetric Suzuki–Miyaura cross-coupling of meso-ditriflate 102.132 The catalyst generated from Pd(OAc)2 and chiral biarylylphosphine L57 furnished monoarylated 103 bearing a stereodefined quaternary center (Figure 32). Even though the yield of the transformation was moderate, good enantioselectivity was still accomplished. The remaining triflate on 103 was shown to serve as a versatile handle for further diversification of the reaction products.
Figure 32.
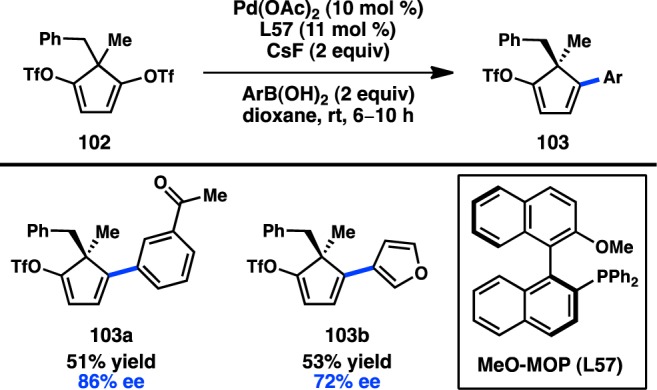
Enantioselective desymmetrization of a meso-ditriflate.
Yu and co-workers reported the first enantioselective C–H activation/cross-coupling via a desymmetrization process in 2008. Prochiral pyridyldiarylmethane 104 was selected for these studies because of the relatively low temperatures required for C–H activation.133 Following a thorough ligand study, the use of monoprotected amino acid L58 in conjunction with Pd(OAc)2 was found to impart a high degree of asymmetric induction for the C–H activation/cross-coupling between 104 and butylboronic acid to give 105 (Figure 33a). The N-protecting group on L58 was a critical element for generating high ee. The reaction is hypothesized to proceed through concerted metalation/deprotonation transition state 106, in which the unreactive aryl group is positioned anti to the carbamate protecting group of the ligand.134
Figure 33.
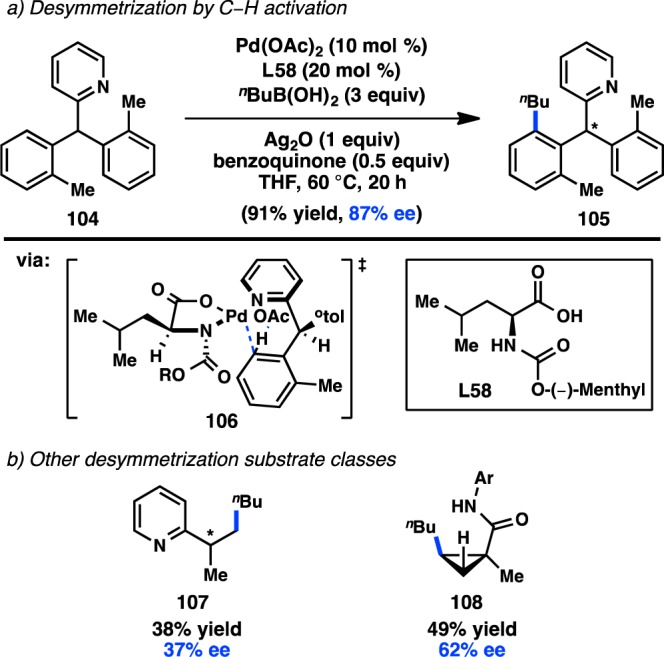
Desymmetrizing enantioselective C–H activation/cross-coupling with alkyl boronic acids.
Yu and co-workers were subsequently able to expand this chemistry to the desymmetrization of cyclopropanes and cyclobutanes. After extensive re-engineering of the ligand, the combination of Pd(OAc)2 and monoprotected amino acid L59 was found to catalyze C–H arylation of cyclopropanamide 109 with phenylboronate ester to produce 110 in good yield and high ee (Scheme 8a).134 Unfortunately, these conditions could not be extended toward the less acidic C–H bonds of a cyclobutane. The authors hypothesized that a more strongly coordinating ligand could increase stereoselection and reactivity in the C–H activation of cyclobutanamide 111. Using the more Lewis basic hydroxamic acid L60, 112 could be produced with a high level of enantioinduction (Scheme 8b).135 A related ligand also enabled the desymmetrization of prochiral methyl groups in acyclic amides to forge β-arylated products in modest ee.135
Scheme 8. C–H Activation of Strained Cycles.
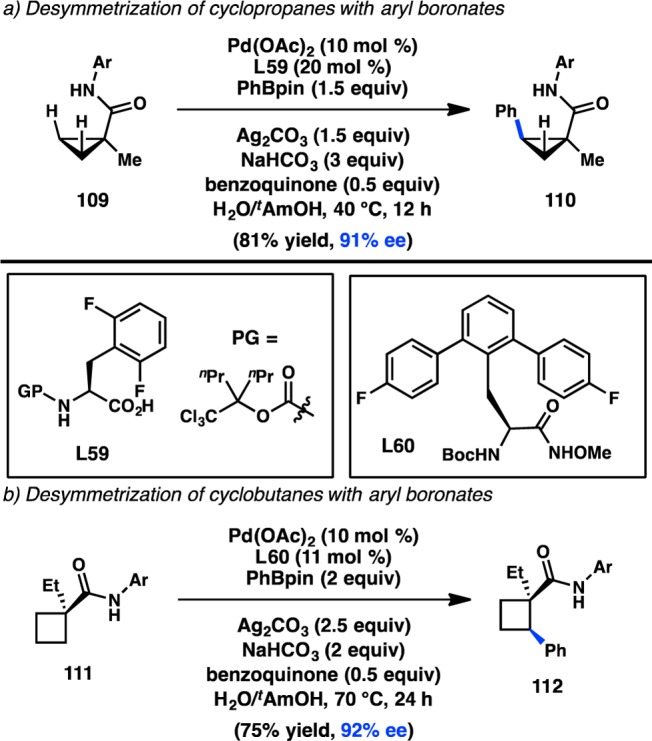
3. Enantiospecific Transition-Metal-Catalyzed Alkyl Cross-Coupling Reactions
The considerations taken into account to invoke enantiospecific cross-coupling reactions are similar to those discussed for enantioselective C–C bond formation. Configurationally stable sec-alkylsilicon, -boron, and -tin species have received more attention than harder to access organometallic reagents derived from magnesium and zinc. In the transmetalation step, competition between open and closed transition states can erode the ee of the final product. Following transmetalation, fast reductive elimination is needed to outcompete racemization pathways that would deliver products with reduced stereofidelity. With respect to secondary electrophiles, catalysts that undergo two-electron oxidative addition are more likely to proceed with high stereospecificity; retention or inversion can be influenced by the presence of coordinating groups proximal to the site of oxidative addition. Although Pd is the most studied transition metal for enantiospecific cross-coupling, Ni catalysis has recently gained popularity for its ability to cross-couple unconventional pseudohalide electrophiles.
3.1. Reactions of Secondary Alkylmetallic Reagents
3.1.1. Organosilicon Reagents
The first investigation of a transition-metal-catalyzed cross-coupling of an enantioenriched sec-alkylmetallic reagent was reported by Hiyama and co-workers in 1990 and focused on the coupling of C(sp3) organosilanes. The high configurational stability of organosilanes, coupled to the development of asymmetric hydrosilylation reactions in the 1980s, allowed ready access to the requisite enantioenriched organosilane starting materials.136 Pd-catalyzed arylation of benzylic trifluorosilane 113 at 50 °C in THF produced 115 with nearly complete retention of configuration (Figure 34).137 Alternatively, increasing the reaction temperature to 90 °C furnished ent-115, formed with inversion of configuration in moderate enantiospecificity (es). An analysis of solvent effects for the reaction showed that incorporating HMPA as a polar cosolvent also led to inversion of configuration but with reduced stereochemical fidelity. To explain the divergent stereochemical outcomes, the authors propose two competing modes of transmetalation. At lower temperatures, transmetalation is hypothesized to occur through a four-centered, SE2(cyclic) transition state (116), resulting in retention of configuration. The transition state is organized by a critical fluoride bridge between Pd and a pentacoordinate silane. Treatment with a large excess of TBAF leads to inhibition of product formation, presumably because of the presence of a hexacoordinate silane incapable of accommodating transition state 116. Raising the reaction temperature is thought to facilitate cleavage of the fluoride bridge and accelerate transmetalation through an SE2(open) transition state (117), leading to inversion of configuration via backside attack. Similarly, polar cosolvents such as HMPA can also disrupt the cyclic transition state and favor an SE2(open) mechanism.
Figure 34.
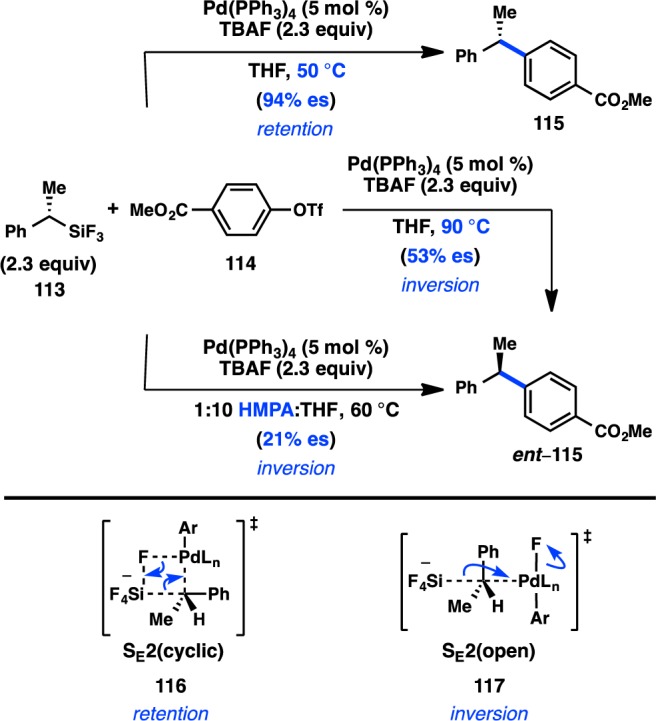
Stereochemical outcome of secondary silanes in the Hiyama cross-coupling.
Following their studies of Pd-catalyzed enantiospecific cross-coupling reactions of benzyltrifluorosilanes, in 1994, Hiyama and co-workers reported the stereospecific coupling of enantioenriched allyltrifluorosilanes with aryl triflates (Figure 35).109 Coupling of silane 118 and triflate 119 using Pd(PPh3)4 as the catalyst and CsF as a fluoride source furnished 120 with retention of configuration. However, the solvent polarity and the nature of the fluoride source determined the stereospecificity of the reaction. Replacing CsF with the nonmetallic fluoride source TASF delivered ent-120, the product of inversion. In addition, the use of DMF as a polar coordinating solvent provides ent-120 with inversion regardless of the fluoride source. As in the coupling of benzylic trifluorosilanes,137 two competing models for transmetalation were proposed by Hiyama. In the presence of CsF, a fluoride bridge between Pd, Cs, and the trifluorosilane promotes syn SE′ transmetalation to Pd, leading to retention of configuration. Disruption of the fluoride bridge in polar, coordinating solvents, or by use of nonmetallic fluoride sources, would result in anti SE′ transmetalation to Pd, resulting in inversion of stereochemisty.
Figure 35.
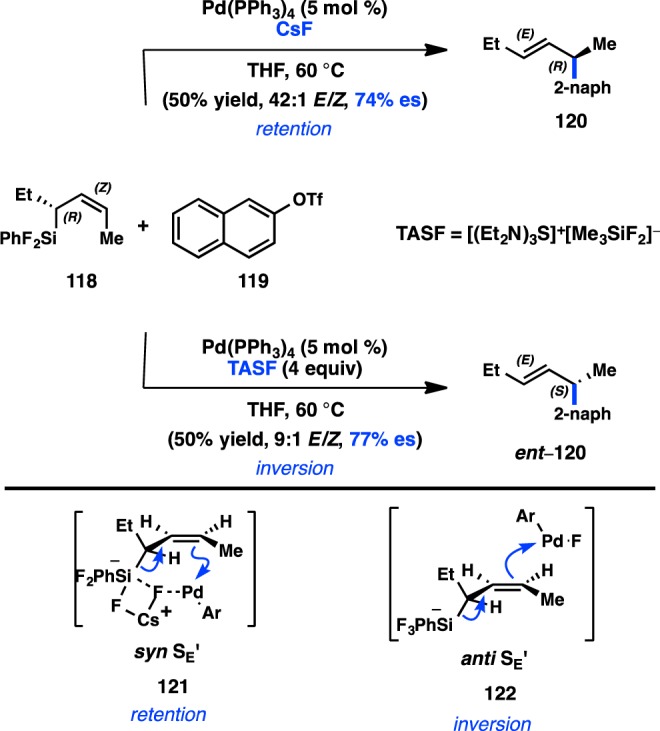
Stereochemical outcome of allylsilanes in the Hiyama cross-coupling.
More recently, Denmark and co-workers extended Hiyama’s work to more synthetically tractable allylic silanoate salts.110 High γ-selectivity in the Pd-catalyzed enantiospecific coupling of 123 with bromobenzene was achieved by using π-acidic diene L61 as the ligand, which is hypothesized to accelerate reductive elimination and preclude deleterious σ–π isomerization (Figure 36). The regioselectivity is consistent with syn SE′ transmetalation via pseudochair conformation 125, in which the Si–O–Pd linkage directs addition of Pd to the γ-position. Favored transition state 125 places the iBu group in a pseudoequatorial position, leading to retention of stereochemistry and formation of the (E)-alkene; transition state 126, which would furnish the (Z)-alkene, suffers from a destabilizing 1,3-diaxial interaction.
Figure 36.
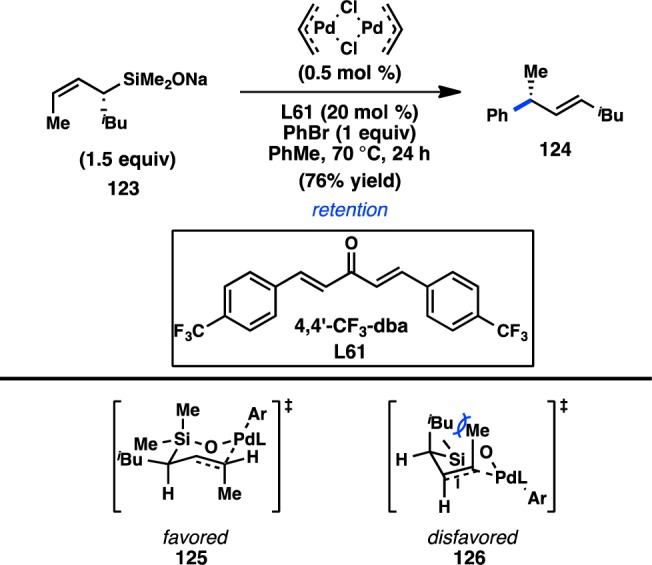
Stereochemical outcome of allylsilanoates in the Hiyama–Denmark cross-coupling.
3.1.2. Organotin Reagents
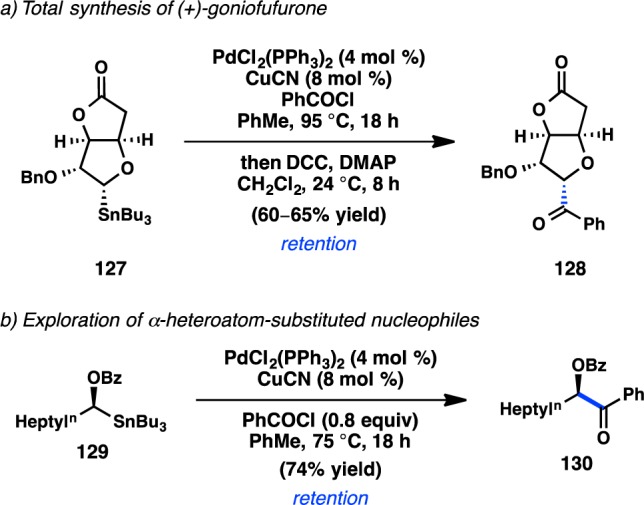
The first study of an enantiospecific alkyl Stille cross-coupling to generate a tertiary stereogenic center was reported in 1993 by Falck and co-workers as part of their synthetic efforts toward the anticancer natural product (+)-goniofufurone. In a key step of the synthesis, enantiopure α-stannylated tetrahydrofuran 127 was coupled with benzoyl chloride in the presence of catalytic PdCl2(PPh3)2 and CuCN to deliver 128 with retention of configuration (Scheme 9a). This result was in contrast to Stille’s initial studies of organostannane transmetalation using ((S)-(−)-(α)-deuteriobenzyl)tributyltin, which underwent transmetalation with inversion of configuration.138 A follow-up investigation revealed that enantioenriched α-stannylated benzoate 129 also underwent Pd-catalyzed stereoretentive cross-coupling with benzoyl chloride (Scheme 9b).139 An investigation of scope revealed that both aroyl chlorides and alkanoyl chlorides bearing coordinating groups couple smoothly under the reaction conditions. A Cu-free reaction afforded the product in lower yield but with no erosion of stereochemistry. Coordinating groups have been demonstrated to accelerate the rate of transmetalation of alkylstannane reagents;140 it is possible that the α-alkoxy substituent favors transmetalation through an SE2(cyclic) transition state.
Scheme 9. Seminal Stereospecific Stille Cross-Couplings.
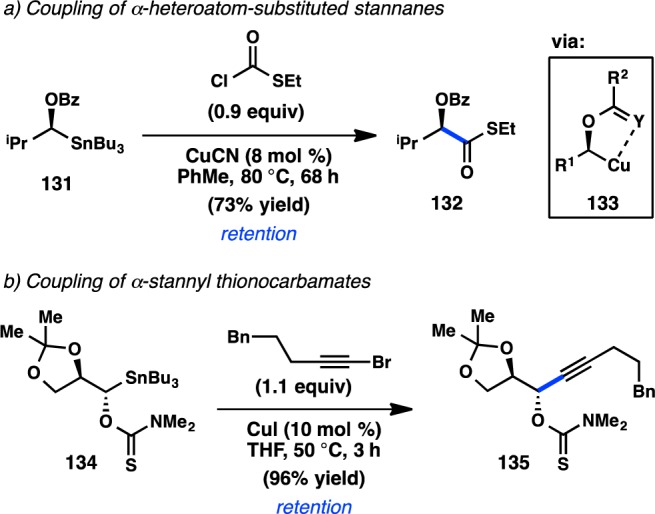
Subsequently, Falck disclosed that use of Cu as the sole catalyst also provided good yields for the coupling of α-stannylated benzoate 131 with S-ethyl chlorothioformates (Scheme 10a).141,142 The resulting products exhibited retention of configuration. The reaction scope was studied using racemic Sn reagents bearing an array of α-heteroatom directing groups; good yields were obtained for reactive electrophiles such as allyl bromides and chloroformates, but lower yields were attained with aryl and vinyl halides. Sulfur substituents on either coupling partner were found to increase yields and reaction rates and could suggest the intermediacy of chelated organocopper species such as 133 during the catalytic cycle. Alkynylation of 134, bearing a proximal thionocarbamate group, occurs in excellent yield and with retention of configuration (Scheme 10b).143,144 The reaction proceeds without any β-elimination of stannane 134.
Scheme 10. Cu-Catalyzed Stereospecific Stille Cross-Coupling.
In collaboration with Liebeskind and co-workers, Falck investigated the Cu-catalyzed cross-coupling of complex α-amino thioesters 136 with α-stannylated thionocarbamate 137 (Figure 37).145 The formation of α,α,α′α′-tetrasubstituted ketone 138 occurred in excellent yield and dr, with retention of configuration at the thionocarbamate-bearing stereocenter. Surprisingly, the reaction proceeded efficiently with catalytic CuTC, despite the very strong Cu–S bond that was expected to form following oxidative addition. The authors propose a mechanism in which Sn/Cu transmetalation takes place to provide stabilized organocopper intermediate 140. An oxidative addition/reductive elimination sequence furnishes coupled product 142 and a copper thiolate complex (143). Typically, copper thiolates of this type are catalytically unreactive; however, in this case a second transmetalation between 144 and SnBu3X forms a tin thiolate and regenerates the active copper catalyst. The second transmetalation is presumably driven by the strength of the Sn–S bond (464 kJ mol–1) relative to the Cu–S bond (274 kJ mol–1).
Figure 37.
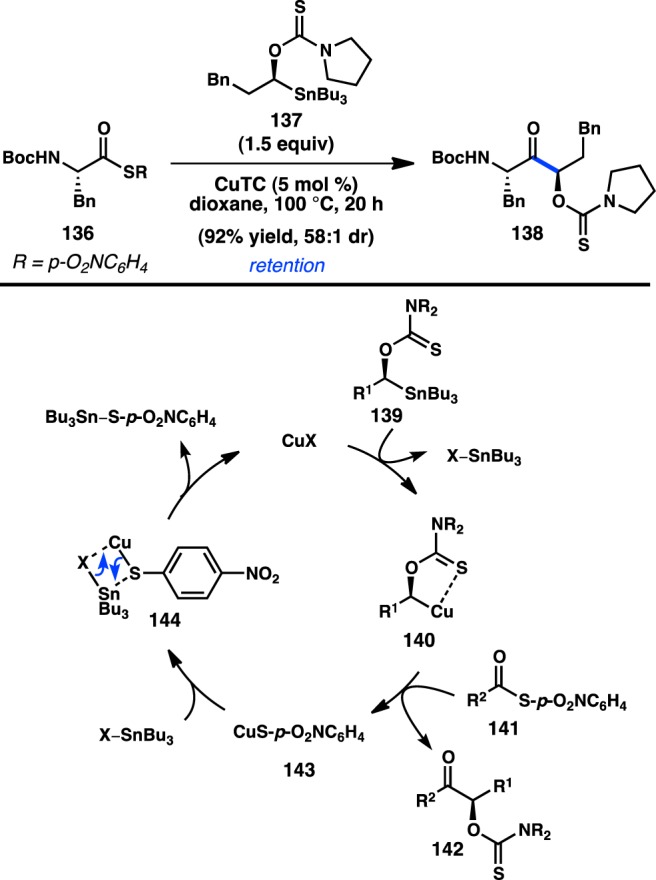
Cu-catalyzed cross-coupling of α-amino thioesters.
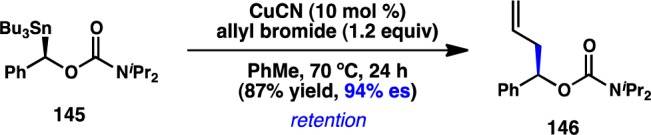
Hoppe and co-workers have developed an enantioselective preparation of α-carbamoyl benzylstannanes (e.g., 145) and explored the use of these nucleophiles in Cu-catalyzed stereospecific allylic alkylations. Using CuCN as the catalyst, coupling of 145 and allyl bromide proceeds in good yield and with retention of configuration (Scheme 11).142 Good levels of es were observed. The carbamate group was postulated to stabilize the organocopper intermediate through coordination, promoting configurational stability at the stereogenic C–Cu bond. Substituted allylic electrophiles reacted with low dr but maintained excellent stereospecificity at the benzylic center.
Scheme 11. Stereospecific Substitution of α-Carbamoyl Benzylstannanes.
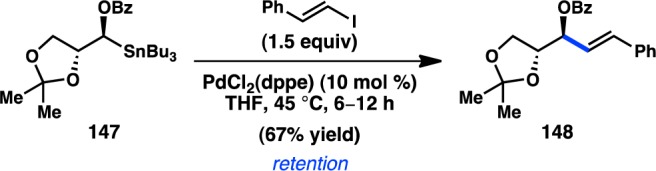
Despite the advances made in Cu catalysis, these conditions still performed relatively poorly when utilized to couple aryl or vinyl electrophiles. Falck and co-workers then turned to other transition-metal catalyst systems and discovered that α-stannylated benzoate 147 and β-iodostyrene readily undergo stereospecific Pd-catalyzed cross-coupling (Scheme 12).146 A control experiment revealed that the adjacent stereocenter in 147 had no effect on the stereochemical outcome of the reaction. Alkenyl bromides and triflates, as well as aryl iodides and bromides, cross-couple in moderate yield and with retention of configuration.
Scheme 12. Pd-Catalyzed and Cu-Free Stereospecific Stille Cross-Coupling.
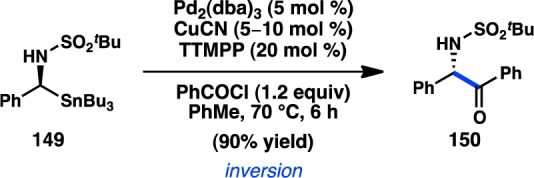
The stereospecific Stille cross-coupling reactions developed by Falck and co-workers focused primarily on the use of α-oxygenated organostannanes. In contrast, the coupling of α-amino organostannanes has received less attention. Chong and co-workers have disclosed a diastereoselective addition of Bu3SnLi to imines bearing a chiral tert-butanesulfinamide auxiliary.147 The sulfinamide products of the reaction were not active toward Stille cross-coupling conditions. Fortunately, oxidation to sulfonamide 149 allowed cross-coupling with benzoyl chloride in the presence of catalytic Pd2(dba)3 and ligand tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP) (Scheme 13). The high donicity of the ligand was thought to prevent deleterious β-hydride elimination. Significantly, analysis of the reaction products revealed that 150 was formed with inversion of configuration, consistent with an SE2(open) transition state and Stille’s original mechanistic studies.
Scheme 13. Stereoinvertive Stille Cross-Coupling.
Whereas the previous examples of cross-coupling reactions of sec-alkylstannanes benefit from α-heteroatoms that enforce the stereospecificity of transmetalation, Hoppe and co-workers have reported a stereospecific Stille coupling of allystannane 151, which benefits from a γ-directing group (Figure 38). Treatment of 151 with iodobenzene and catalytic Pd(PPh3)4 provides 152, the result of inversion of configuration, as the major product.148 Unfortunately, the yields are modest because of the competing formation of 153. The minor product, 153, is presumably formed via π-allyl complex 154; isomerization of 154 to γ-substituted Pd complex 155 can then result in [3,3] rearrangement to 156. Reductive elimination from vinylpalladium complex 156 furnishes minor product 153, which is notably formed with complete enantiospecificity. Thus, a direct SE2′ mechanism is not proposed for the formation of 153.
Figure 38.
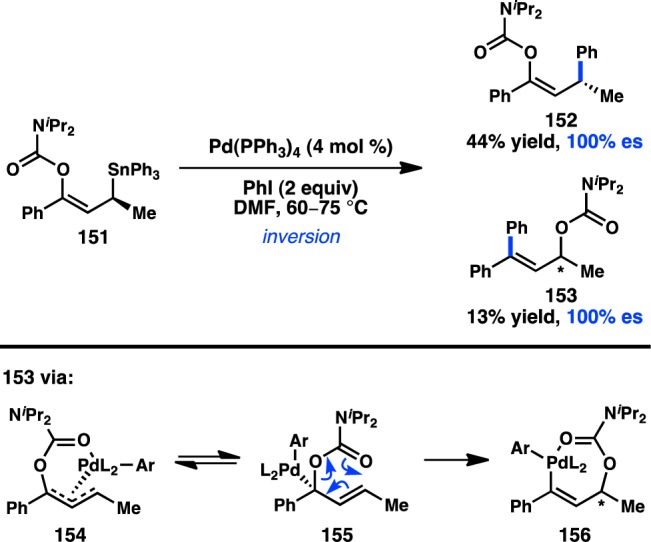
Stille cross-coupling of allyltin reagents.
Recently, Biscoe and co-workers reported the first enantiospecific Stille cross-coupling of simple alkylstannanes. Integral to the success of the reaction was the use of alkylazastannatranes as the organotin coupling partner. Most Stille couplings utilize trialkylstannyl groups, wherein the alkyl groups act as dummy ligands because of their slow rate of transfer to a transition-metal catalyst. However, alkylazastannatranes have been shown to transfer alkyl groups selectively; intramolecular coordination of the nitrogen to tin activates the apical alkyl group toward transmetalation.149 Reaction optimization for the coupling between 157 and aryl halides identified JackiePhos (L62) as providing good levels of conversion and minimal alkane isomerization (Figure 39).150 The coupling proceeds with retention of configuration and is notably tolerant of a range of heteroaryl halide partners. New methods for the enantioselective construction of simple alkylazastannatranes are necessary to realize the full potential of this method.
Figure 39.
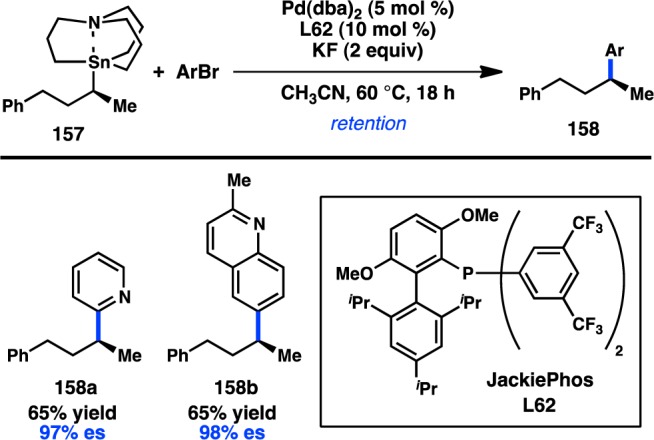
Stereospecific Stille cross-coupling of unactivated secondary alkyltin reagents.
3.1.3. Organoboron Reagents
The alkyl cross-coupling of stereodefined C(sp3) organoborane reagents is a synthetically appealing transformation that, for several reasons, has been somewhat challenging to develop. In general, alkylboron species undergo transmetalation at a slower rate than their vinyl, aryl, and alkynyl counterparts. The increased steric hindrance of the sec-alkylboron species required for stereospecific cross-coupling reactions further exacerbates the challenge of the transmetalation step. Moreover, until the past decade, there were relatively few methods to prepare these reagents in enantioenriched form.
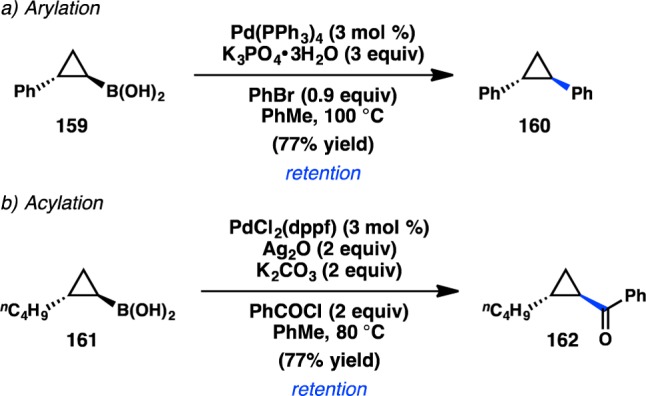
Early efforts to develop cross-coupling reactions of sec-alkylboron reagents focused on cyclopropylboronic acids because the Walsh orbitals of a cyclopropyl ring impart extra p character to the C–C bonds, resulting in a greater C(sp2) character of the C–B bond. Cyclopropylmetallic species also benefit from a high-energy barrier for β-hydride elimination. In 1996, Deng and co-workers reported a Suzuki–Miyaura cross-coupling of cyclopropylboronic acids and aryl bromides, observing that trans-cyclopropyl starting materials provided trans-products.151 Later, both Deng and Charette demonstrated that racemic cis-cyclopropyl trifluoroborates also react with retention of configuration.152,153 In 1998, Deng disclosed an asymmetric route to 159 via a diastereoselective cyclopropanation of an alkenylboronate ester followed by hydrolysis to form the free boronic acid.154 Treatment of 159 with bromobenzene under Deng’s previously optimized conditions delivered 160 in good yield and with retention of stereochemistry, indicating that transmetalation from boron to Pd occurs with retention of configuration at the carbon atom (Scheme 14a).155−158 This example represented the first report of a stereoretentive Suzuki–Miyaura cross-coupling reaction of a secondary organoboron species. Later studies illustrated that both aryl and heteroaryl triflates were competent coupling partners;159,160 acylation of 161 was also achieved using Ag2O as a base (Scheme 14b).161
Scheme 14. Stereospecific Suzuki–Miyaura Coupling of Cyclopropylboronic Acid.
Despite the promising reactivity of cyclopropylboronic acids, it was not until 2009 that Crudden and co-workers were able to develop the first stereospecific Suzuki–Miyaura cross-coupling of acyclic, stereodefined sec-alkylboronates. Prior studies in the Crudden laboratory had established an enantioselective Rh-catalyzed hydroboration of styrenes to furnish secondary benzylic boronates in high ee.162 Exposure of hydroboration product 163 to Pd-catalyzed cross-coupling conditions delivered 164 with retention of configuration (Scheme 15a).79 Ag2O was critical to enhance the rate of transmetalation of the sec-alkylboronate; it was later found that the combination of Ag2O and K2CO3 enhances enantiospecificity.163 An 8:1 ligand:metal ratio was required to maintain good yields, although further increasing the phosphine loading led to reduced enantiospecificity. Whereas primary benzylboronate esters react in yields comparable to that of 163, primary alkylboronates were shown to be unreactive under the optimal conditions;164 these results highlight the importance of the adjacent aryl group of 163 to promote transmetalation. Employing similar reaction conditions, triarylmethane 166 could be prepared from boronate ester 165 (Scheme 15b).165 The use of 2,2-dimethylpropanediol-derived boronic esters (Bneop) instead of pinacol boronates was required to synthesize starting material 165 efficiently via Aggarwal’s asymmetric 1,2-metalate rearrangement.166
Scheme 15. Suzuki–Miyaura Cross-Coupling of Benzylic Boronates.
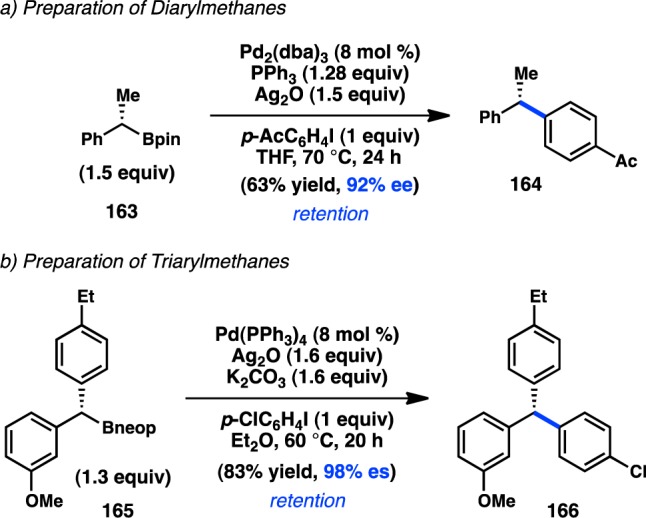
Realizing the important role of adjacent π-unsaturation for efficient cross-coupling of secondary organoboronate esters, Crudden and co-workers next investigated the cross-coupling of secondary allylic boron species. After developing a cross-coupling of racemic allylic boronate esters,164 Crudden collaborated with the Aggarwal laboratory to prepare enantioenriched 167 from the corresponding carbamate by a lithiation–borylation route.108 Pd-catalyzed reaction of 167 with iodobenzene provided 168 with complete retention of stereochemistry and high γ-selectivity (Figure 40). The authors propose that the cross-coupling proceeds through a syn SE′ mechanism wherein the Pd–O–B linkage in 170 directs γ-addition to form 171. Reductive elimination from 171 forms γ-product 175; alternatively, isomerization through π-allylpalladium complex 172 would lead to α-product 174. Deuterium labeling studies confirmed that the rate of reductive elimination is faster than the rate of isomerization. The aforementioned mechanism is consistent with proposals for the coupling of stereogenic allyltrifluorosilanes and allylsilanoates (see section 3.1.1).109,110 Suginome and co-workers have disclosed a cross-coupling of homoallylic boronates that also proceeds with retention of configuration.167
Figure 40.
Regioselective cross-coupling of allylic boronates.
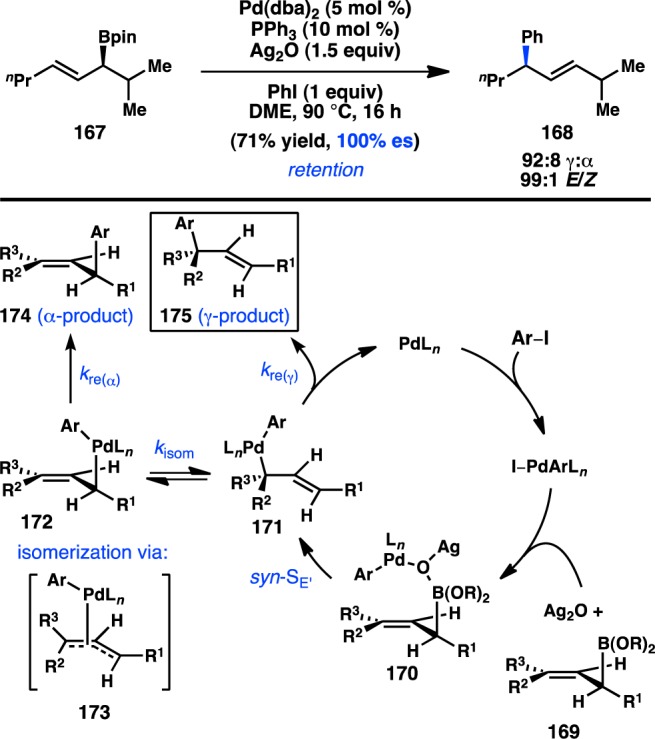
Aggarwal and co-workers concurrently investigated the asymmetric preparation of tertiary propargylic boronate esters through their lithiation–borylation method. After synthesis of 176 in high ee, the tertiary boronate was subjected to modified Crudden conditions to deliver γ-addition product 177 exclusively with minimal formation of the protodeboronated product (Figure 41).76 This reaction represented the first example of a tertiary organoboron species being employed in a stereospecific Suzuki–Miyaura coupling. In analogy to the coupling of allylboronates, the reaction is expected to proceed through a syn SE′ mechanism in which the “ate” complex of 178 can direct Pd toward γ-addition.
Figure 41.
Tetrasubstituted allene formation from Suzuki–Miyaura cross-coupling.
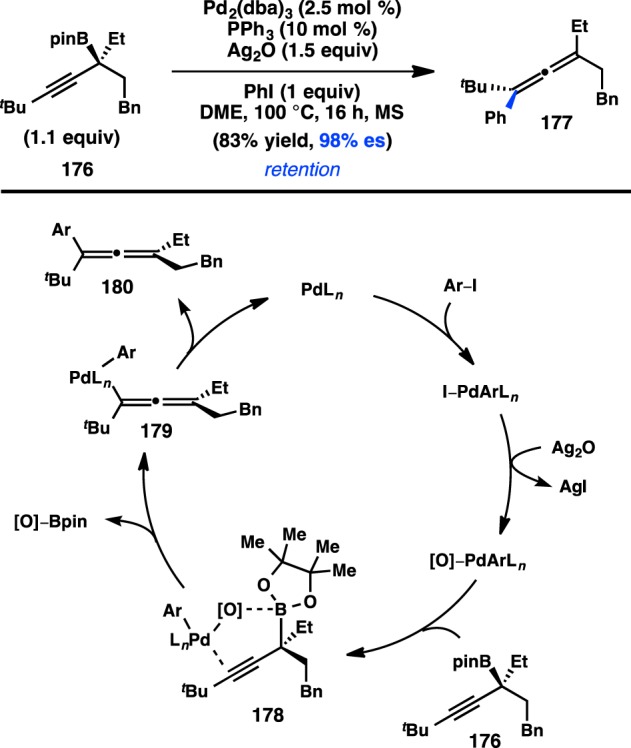
Following the early reports by Crudden, Suginome and co-workers disclosed the Pd-catalyzed cross-coupling of enantioenriched [α-(acylamino)benzyl]boronic ester 181. Optimization studies of the Suzuki–Miyaura cross-coupling of 181a and aryl bromides established that use of Pd/L63 and K2CO3 delivered the product with moderate stereofidelity (Figure 42).168 Incorporation of a bulkier protecting group on nitrogen, as in 181b, increased the enantiospecificity to 97%. Aryl and heteroaryl bromides, as well as aryl chlorides, could thus be coupled with 181b in high yields and excellent es. Stereochemical assignment of the amide products revealed that the reaction occurred with inversion of configuration. Suginome proposed that strong intramolecular coordination between the carbonyl and the boron atom of 181 would enforce transmetalation to Pd through an SE2(open) mechanism, as shown in 183. In contrast, a stereoretentive cross-coupling would proceed by transmetalation via four-centered SE2(cyclic) transition state 184, which would require decoordination of the carbonyl group from the boron atom of 181.
Figure 42.
Stereoinvertive cross-coupling of [α-(acylamino)benzyl]boronic esters.
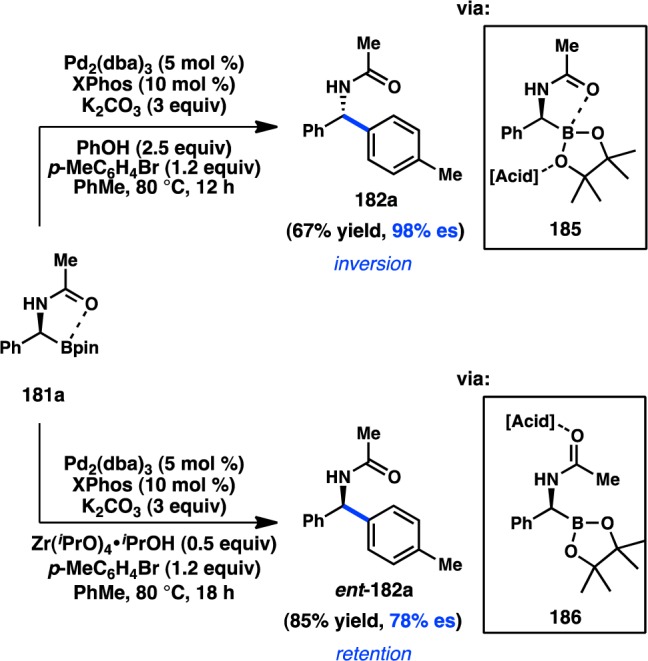
In a follow-up study, Suginome investigated how additives might affect the stereochemical course of cross-coupling reactions of 181a. Addition of protic acids was found to increase the es of the stereoinvertive coupling of 181a, with phenol providing 182a in 98% es (Scheme 16).169 The authors propose that phenol hydrogen bonds to the oxygen atom of the pinacol ligand (185), making the boron more electropositive and strengthening the dative interaction between the carbonyl and boron, thereby enforcing an SE2(open) transition state. On the other hand, the Lewis acid Zr(iPrO)4·iPrOH was found to reverse the stereochemical outcome of the reaction, delivering ent-182a in 78% es and with retention of configuration. In this case, coordination of the amide of 181a to the exogenous Lewis acid is proposed to disrupt the intramolecular coordination of the carbonyl to boron (186), favoring instead stereoretention through an SE2(cyclic) transition state.
Scheme 16. Additive Effects in Coupling of [α-(Acylamino)benzyl]boronic Esters.
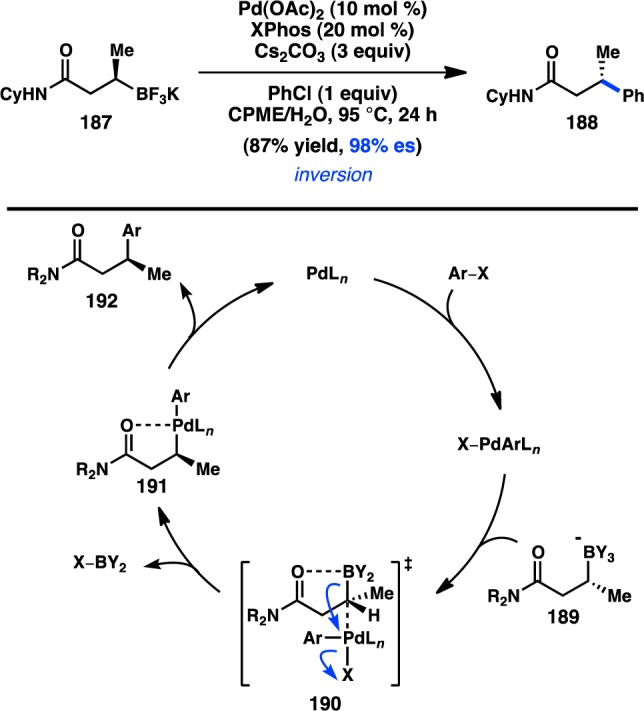
A second stereoinvertive Suzuki–Miyaura cross-coupling was concurrently developed by Molander and co-workers in 2010. Enantioenriched trifluoroborates such as 187 are readily prepared by asymmetric β-borylation of the corresponding α,β-unsaturated amides.170 Trifluoroborate 187 was found to undergo Pd-catalyzed cross-coupling with chlorobenzene to give 188 in high yield and es, and with inversion of stereochemistry (Figure 43). The use of the bulky Buchwald ligand XPhos (L63) was found to prevent the formation of products resulting from β-hydride elimination or isomerization.171 Amide 187 can be coupled with a variety of aryl and heteroaryl chlorides; a reduced yield is obtained using bromobenzene, and no product is observed when using iodobenzene.172 However, substrates containing a ketone or ester in place of the amide in 187 did not deliver any of the desired cross-coupling products. To explain the key role of the amide, Molander proposes a stereochemical model similar to that of Suginome, in which intramolecular dative coordination between boron and the carbonyl of 187 favors an SE2(cyclic)-type mechanism for stereoinvertive transmetalation. In addition, formation of five-membered chelate 191 might stabilize the resulting organometallic complex, preventing β-hydride elimination by precluding formation of the required syn-coplanar conformation. Janesko and co-workers used DFT calculations to find that the bulky XPhos protects the coordinatively unsaturated PdII intermediate from β-hydride elimination while also accelerating reductive elimination from 191 by interrupting the five-membered chelate.173
Figure 43.
Stereoinvertive Suzuki–Miyaura coupling of β-trifluoroborato amides.
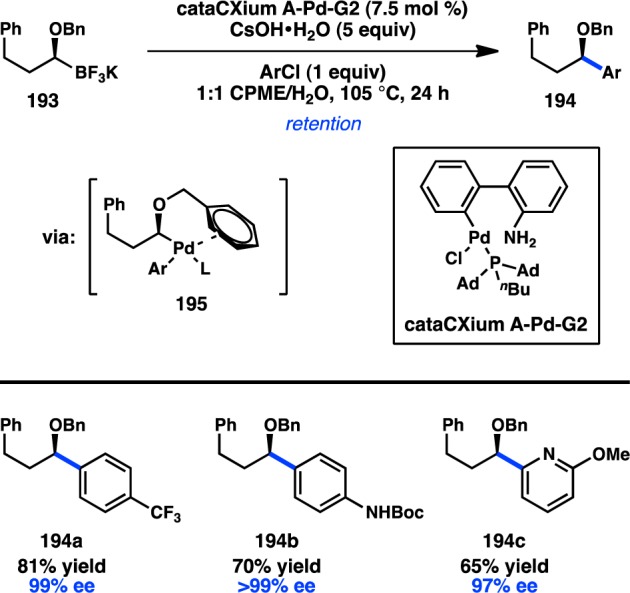
Molander also investigated the cross-coupling of [1-(benzyloxy)alkyl]boron species 193 with aryl chlorides using the Pd precatalyst system cataCXium A-Pd-G2 (Figure 44). Prior research by Falck and others demonstrated that Stille cross-couplings of α-heteroatom-substituted organotin reagents can proceed with high stereospecificity (see section 3.1.2). In the present case, a benzyl ether was utilized to facilitate coordination to Pd in an η2 binding mode following oxidative addition (195).174 It was hypothesized that this interaction could stabilize the organometallic intermediate, disfavoring β-hydride elimination. Indeed, a variety of aryl and heteroaryl chlorides could be coupled in high yield and with excellent stereospecificity. These cross-coupling reactions proceed with retention of stereochemistry, in analogy to the cross-coupling of benzylic, allylic, and propargylic boronates studied by Crudden and Aggarwal.76,79,108,163−165
Figure 44.
Stereoretentive coupling of a [1-(benzyloxy)alkyl]boron compound.
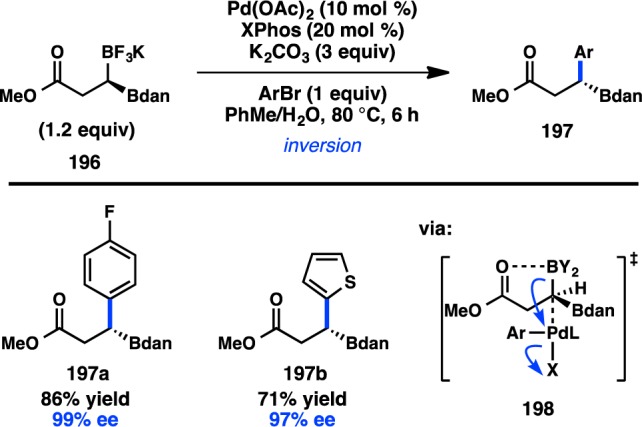
Recent advances in catalytic asymmetric borylation reactions have provided access to an array of new enantioenriched C(sp3) organoboron compounds. In 2011, Hall and co-workers disclosed the enantioselective preparation of geminally substituted 1,1-diboron compound 196, which contains differentially protected boron groups (Figure 45).175 Subsequent exposure of 196 to Suzuki–Miyaura cross-coupling conditions resulted in a chemoselective arylation of the BF3K group to form 197 with inversion of configuration. Notably, a substrate bearing a Bpin group in place of the BF3K group results in a racemic product. As previously discussed by Molander in the coupling of β-boryl amides, the cross-coupling is proposed to proceed through an SE2(open) transition state (198). Although Molander observed poor reactivity with ester directing groups, it is hypothesized that the α-Bdan group exerts a cooperative effect with the ester to stabilize transition state 198.131 However, when a simple homobenzylic 1,1-diboron compound was subjected to the reaction conditions, the cross-coupled product was obtained in only 15% yield and reduced enantiospecificity, illustrating the importance of the ester directing group.176
Figure 45.
Stereoinvertive coupling of geminal diborons.
All of the above examples employ enantioenriched organoboron reagents activated by a proximal π-system or Lewis basic directing group. The Biscoe laboratory has focused on the development of enantiospecific cross-coupling reactions of simple, secondary organometallic reagents.150 Building from pioneering work by Molander177 and van der Hoogenband,178 Biscoe and co-workers discovered that Pd/P(tBu)3 precatalyst 201 catalyzed the cross-coupling of sec-alkyl trifluoroborates (199) with aryl chlorides to provide products with branched to linear ratios as high as >200:1 (Figure 46).179 Both boronic acids and trifluoroborate salts were competent reaction partners, and products bearing interesting heterocyclic scaffolds could be prepared with good efficiency. Examination of the stereochemical outcome revealed that the transformation proceeds with inversion of stereochemistry. Previous stereospecific Suzuki–Miyaura cross-couplings have been shown to proceed with either retention or inversion of stereochemistry; it is possible that the stereochemical outcome of this transformation reflects the innate mechanism of transmetalation for unactivated secondary alkylboron reagents.
Figure 46.
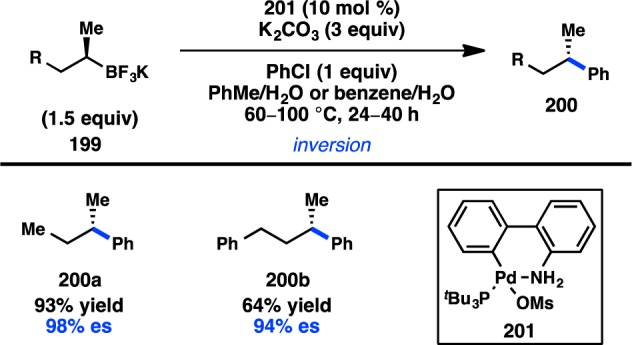
Stereoinvertive coupling of unactivated acyclic organoboron reagents.
3.1.4. Organomagnesium Reagents
The configurational lability of the C–Mg bond has hindered the development of stereospecific cross-coupling reactions of stereodefined sec-alkylmagnesium reagents. Although the racemization of hard organometallic compounds can be attenuated at low temperatures and in certain solvents,9,180 most syntheses of Grignard reagents proceed via alkyl radicals and preclude formation of enantioenriched material.181 Hoffmann and co-workers developed a route to stereodefined Grignard reagent 204 by employing a sulfoxide/Mg exchange followed by a carbenoid homologation reaction (Scheme 17).12 The half-life of 204 was measured to be ∼5 h at −10 °C. Hoffmann utilized the stereogenicity of 204 as a mechanistic probe for transition-metal-catalyzed Kumada–Corriu cross-couplings.182 Under Ni or Pd catalysis, stereochemical information is maintained throughout the reaction, supporting a polar mechanism for transmetalation. On the other hand, Fe- and Co-catalyzed reactions provided 205 in reduced ee, indicating the presence of single-electron transfer pathways.
Scheme 17. Stereospecific Coupling of Secondary Organomagnesium Compounds.
3.1.5. Organozinc Reagents
As with organomagnesium reagents, stereospecific cross-coupling of enantioenriched sec-alkylzinc reagents has been limited by the asymmetric preparation of the organozinc coupling partner. Oxidative addition of Zn into C–X bonds often occurs via a radical process and does not proceed with stereochemical fidelity. In contrast, transmetalation of ZnII salts with organolithium or organoboron compounds occurs with retention of configuration. Knochel and co-workers have pioneered a boron/zinc exchange for diastereoselective cross-couplings, but progress toward using these reagents in enantiospecific processes has been slow.183−186 Lithium/zinc transmetalation has enjoyed more success in enantiospecific cross-coupling. Studies by Beak and co-workers demonstrated that treatment of N-Boc-pyrrolidine 19 with sBuLi and (−)-sparteine (L64) results in stereodefined organolithium complexes that are stable at low temperatures (Scheme 18).187 These complexes can react with an array of electrophiles to form C–C bonds; they also readily undergo transmetalation with ZnII to form stereodefined organozinc compounds.
Scheme 18. Asymmetric Deprotonation To Form Chiral Lithium Reagents.
In 2006, Campos and co-workers reported the synthesis and coupling of organozinc reagent 207, which could be warmed to room temperature without loss of configurational integrity (Figure 47).61,188 In the presence of Pd(OAc)2 and P(tBu)3, a variety of aryl and heteroaryl halides could be cross-coupled in good yields and with excellent enantiospecificity. Use of the bulky, electron-rich, monodentate ligand P(tBu)3 enables oxidative addition of the aryl halide at low temperature, while also mitigating the β-hydride elimination pathway. The reaction was applied to the synthesis of a glucokinase activator and performed on the kilogram scale.189 Efforts to increase the scope of the reaction demonstrated that 207 could be coupled with alkenyl bromides in good yield and retention of stereochemistry to form 208c.190
Figure 47.
Pyrrolidine functionalization via stereoretentive Negishi cross-couplings.
Although asymmetric deprotonation allows access to enantioenriched pyrrolidine 206, challenges have been encountered when attempting to apply this procedure to similar heterocycles, such as piperidines.191 An alternative approach involves a dynamic thermodynamic resolution (DTR) of 2-lithiopiperidines.192,193 In the DTR of 209, racemic organolithium 210 is prepared at low temperature by treatment with sBuLi (Figure 48). Chiral ligand L* is added, and the reaction temperature is subsequently raised. Diastereomers 211 and epi-211 may reach a thermodynamic equilibrium by carbanion inversion, permitting a single stereoisomer (in this case, epi-211) to be preferentially populated. The reaction temperature is then lowered to a point of configurational stability (k2, k3 ≫ k1, k–1), effectively freezing the thermodynamic equilibrium. Treatment with an electrophile leads to product formation with an ee similar to the ratio of 211 and epi-211 present from the thermodynamic equilibrium.
Figure 48.

Dynamic thermodynamic resolution (DTR).
Gawley and co-workers have applied a DTR technique to prepare enantioenriched piperidylzinc species. Dilithiated diaminoalkoxide ligand L65 was employed catalytically in the presence of superstoichiometric achiral TMEDA, enabling a catalytic dynamic resolution (CDR)194 of 2-lithiopiperidine. Transmetalation to Zn followed by a Cu-mediated Negishi cross-coupling afforded benzylation product 214 in moderate yield and excellent ee (Figure 49a).195,196 The Cu-catalyzed protocol was useful for benzylation and allylation but was not amenable to cross-coupling with aryl or vinyl halides. However, Pd-catalyzed cross-coupling under the conditions developed by Campos for the arylation of pyrrolidines furnished the 2-aryl- or 2-vinylpiperidines in good ee (Figure 49b).197,198 Although heteroaryl bromides were reactive under the coupling conditions, conversion was sluggish and heating to 60 °C was required. Enantioselectivity studies reveal a moderate loss of stereochemical integrity as the reaction temperature is increased from 22 to 60 °C, implicating a slow racemization process for the intermediate organozinc reagent. In contrast, Campos’s coupling of pyrrolidines occurs at 60 °C without any erosion of ee.61
Figure 49.
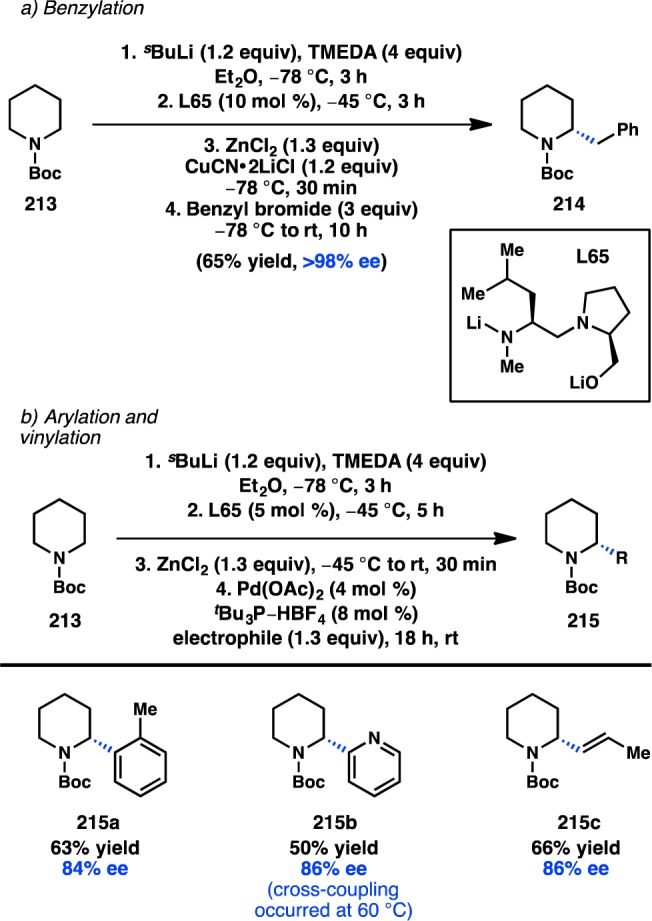
Piperidine functionalization via stereoretentive Negishi cross-couplings.
3.2. Reactions of Secondary Alkyl Electrophiles
3.2.1. With Organomagnesium Reagents
Although the Kumada–Corriu coupling was first reported over 40 years ago, the development of stereospecific reactions between enantioenriched sec-alkyl electrophiles and organometallic reagents has only recently gained traction. In 1990, Larchevêque and co-workers demonstrated that α-hydroxy esters react with stoichiometric cuprate reagents with inversion of configuration.199 Building off this work, Breit and co-workers reported a Zn-catalyzed cross-coupling of enantioenriched α-hydroxy ester triflates and alkylmagnesium chlorides in 2008 (Figure 50).200 The enantioenriched triflate electrophiles were easily accessed from enantiopure lactic acid or α-amino acids. The cross-coupling proceeded with excellent transfer of chirality and inversion of configuration. In an extension of this method, an iterative cross-coupling protocol was developed for the preparation of (oligo)deoxypropionates.127 The reaction is proposed to proceed through a triorganozincate species that is generated in situ from the ZnCl2 catalyst and the Grignard reagent. The triorganozincate is thought to be more nucleophilic than a simple dialkylzinc because of its enhanced polarization of the C–Zn bond. The reaction likely also benefits from activation of the triflate by coordination to Mg. SN2 displacement of the triflate provides product 219 with inversion of configuration.
Figure 50.
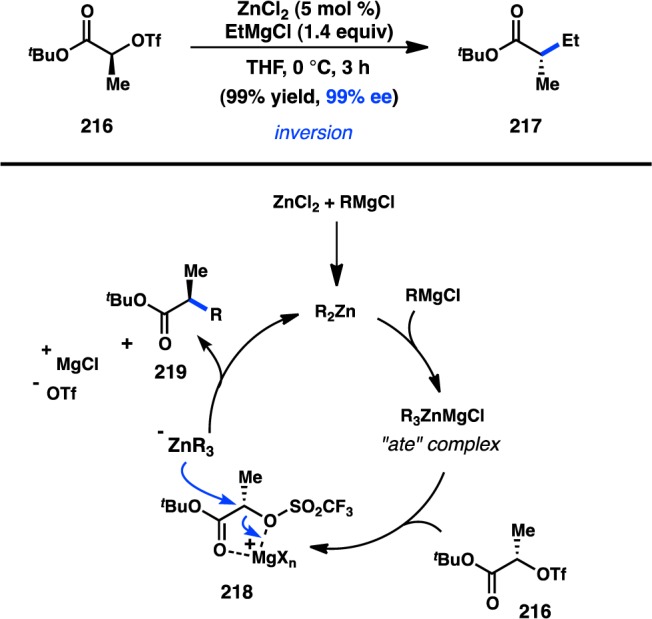
Zn-catalyzed enantiospecific C(sp3)–C(sp3) cross-coupling.
In 2009, Carretero and co-workers reported a Pd-catalyzed cross-coupling between enantioenriched secondary benzylic bromides and aryl or vinyl Grignard reagents (Scheme 19).201 Xantphos (L66), possessing a very wide bite angle of 111°, was crucial to preventing β-hydride elimination. The cross-coupling of enantioenriched bromide 82 proceeded with good enantiospecificity and full inversion of configuration. Although this was an important demonstration of an enantiospecific cross-coupling of a sec-alkyl electrophile, the required enantioenriched benzylic bromides can be challenging to prepare in high enantiomeric excess.
Scheme 19. Stereospecific Kumada–Corriu Coupling of Secondary Benzyl Bromides.
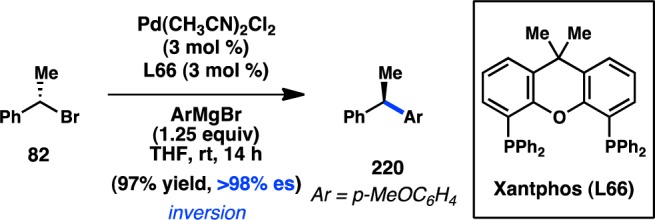
In 2011, Jarvo and co-workers reported the first stereospecific Ni-catalyzed Kumada–Corriu cross-coupling of a sec-alkyl ether.80 In the presence of Ni/phosphine complexes, enantioenriched benzylic ethers (221) could be coupled to MeMgI in good yields and high ee and with inversion of configuration (Figure 51a). The initially disclosed reaction scope was limited to electrophiles bearing extended aromatic systems that promote efficient oxidative addition through π-benzyl formation. The method was also limited to Grignard reagents that lacked β-hydrogens because of competing β-hydride elimination. A second-generation protocol was developed for the more general coupling of alkylmagnesium halides and benzylic ethers (Figure 51b).82 The ability to use easy-to-prepare enantioenriched sec-alkyl ethers as electrophiles provides practical advantages over the previously discussed coupling by Carretero and co-workers.
Figure 51.
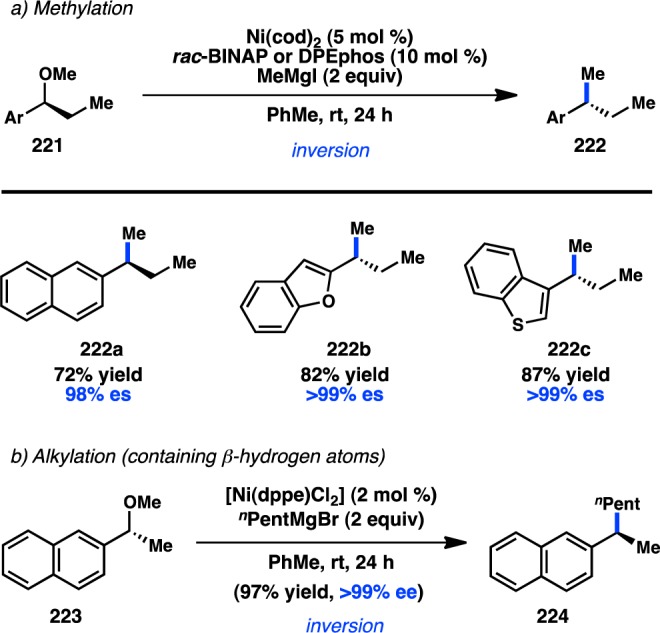
Stereospecific Kumada–Corriu cross-coupling of benzylic ethers.
The aforementioned Kumada–Corriu coupling suffered from a limited substrate scope because of the inability of the Ni catalyst to add oxidatively into benzylic ethers that lacked a π-extended system. To promote cross-coupling of simple benzylic ethers, Jarvo and co-workers hypothesized that incorporation of a more coordinating leaving group could facilitate oxidative addition by a traceless activation strategy. Following electrophile optimization, the use of 2-methoxyethoxy ethers (225) was found to enable Kumada–Corriu coupling of simple benzylic substrates; bidentate chelation of Mg is proposed to activate the C–O bond toward oxidative addition (Figure 52).81 Under these conditions, both diphenyl- and phenylcarbinol derivatives could be coupled enantiospecifically with inversion of configuration.
Figure 52.
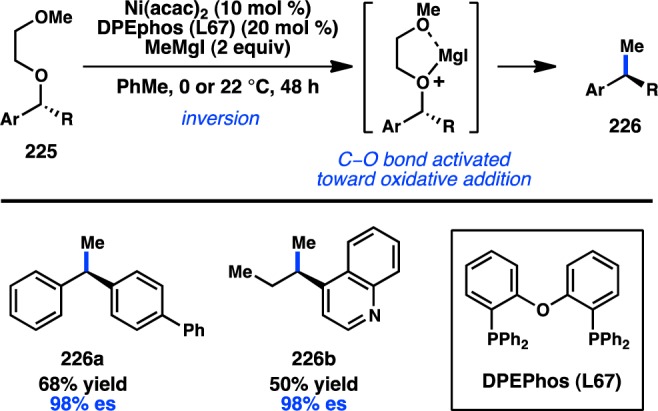
Traceless directing group in the Kumada–Corriu cross-coupling.
Jarvo and co-workers have recently succeeded in further extending their Ni-catalyzed enantiospecific cross-coupling method80,81 to the arylation of diarylmethanol derivatives. The yield and enantiospecificity of the transformation were heavily dependent on both the identity of the phosphine ligand and the electrophile (Figure 53).202 When DPEPhos was employed in the cross-coupling of methyl ether 227a, product was obtained in modest yield and poor es. The main side product was the homodimer of 227a. Modification of the electrophile to a traceless 2-methoxyethoxy directing group (227b) improved the yield of 228, but with only a slight increase in es. However, the combination of substrate 227b and 1,8-bis(diphenylphosphino)octane (dppo) resulted in clean coupling to deliver 228 in good yield and perfect es. The reaction is limited to substrates containing π-extended systems and cannot be used in the synthesis of diarylalkanes. Jarvo’s strategy to prepare triarylmethanes represents a conceptually similar but reverse-polarity approach compared to that of Crudden.165
Figure 53.
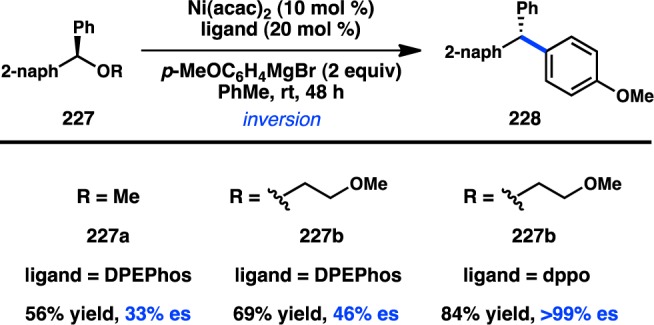
Preparation of enantioenriched triarylmethanes by Kumada–Corriu coupling.
In a rare example of a cross-coupling to give products with vicinal tertiary centers, Liu and co-workers reported the stereospecific Cu-catalyzed reaction of secondary tosylates with primary or secondary alkylmagnesium halides; the reactions proceed with complete inversion of configuration (Figure 54).203 These findings are consistent with mechanistic studies by Kambe and co-workers, which had previously demonstrated that Cu-catalyzed cross-couplings of primary alkyl halides with Grignard reagents occur in an SN2 fashion.204 Excellent chemoselectivity was realized in the Cu-catalyzed reaction: competition studies indicated that secondary alkyl bromides or tosylates selectively underwent cross-coupling in the presence of an aryl bromide.
Figure 54.
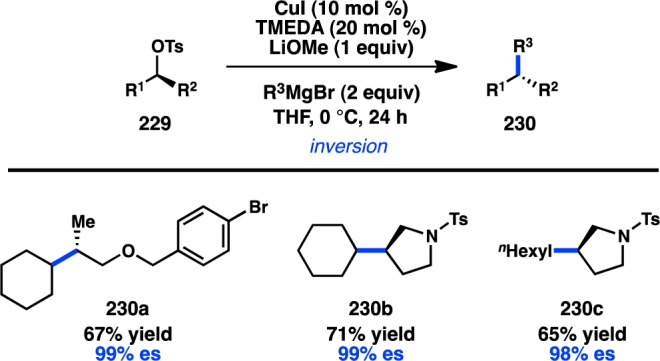
Cu-catalyzed stereospecific Kumada–Corriu cross-coupling.
3.2.2. With Organozinc Reagents
The first stereocontrolled Cu-catalyzed cross-coupling of an alkylzinc reagent was reported by Ready and co-workers in 2004.205 Treatment of α-chloro ketone 231 with an alkylzinc species in the presence of catalytic Cu(acac)2 furnished 232 with inversion of configuration (Scheme 20). It was imperative for the zinc reagent to be prepared via transmetalation of the corresponding Grignard compound for the reaction to proceed efficiently. The observed inversion of configuration supports an SN2 mechanism for the alkylation and disfavors intermediate enolate formation.
Scheme 20. Stereospecific Cu-Catalyzed Negishi Cross-Coupling.
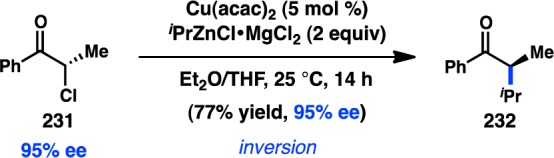
In addition to their development of the cross-coupling between sec-alkyl ethers and organomagnesium reagents (see Figure 51), Jarvo and co-workers have investigated Ni-catalyzed cross-coupling reactions with ZnMe2.206 Whereas their previous Kumada–Corriu coupling proceeded with simple methyl ether electrophiles, it was reasoned that the decreased nucleophilicity of organozinc species would require a more reactive electrophile as a coupling partner. A traceless directing group strategy was adopted, and a screen of leaving groups bearing different chelating moieties identified 2-methylthio ester 233 as optimal (Scheme 21a). The complex generated from NiCl2(dme) and DPEPhos catalyzed the cross-coupling between electrophiles such as 233 and ZnMe2 in high yields and with complete inversion of configuration. To extend the transformation to ZnEt2, ester 234 was employed to mitigate β-hydride elimination and hydrogenolysis products that were observed when using 233 (Scheme 21b). Unfortunately, the decreased reactivity of organozinc reagents currently limits the method to activated electrophiles bearing π-extended systems.
Scheme 21. Traceless Directing Group for Alkylation of Benzylic Esters.
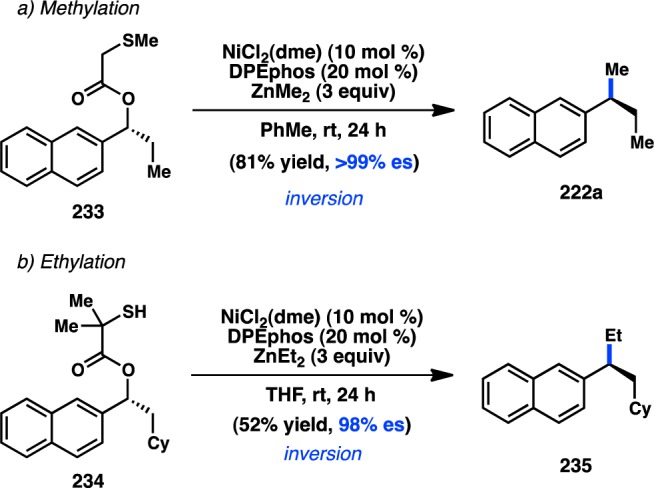
Having identified that esters behave as feasible electrophiles for stereospecific Negishi couplings, Jarvo and co-workers turned their attention to the cross-coupling of aryl-substituted lactones. The authors demonstrated that treatment of lactone 236 with their modified Negishi methylation conditions delivered ring-opened product 237 in good yield and excellent enantiospecificity (Figure 55).207 Several heteroaryl-substituted lactones were also shown to react well under the optimized reaction conditions.
Figure 55.
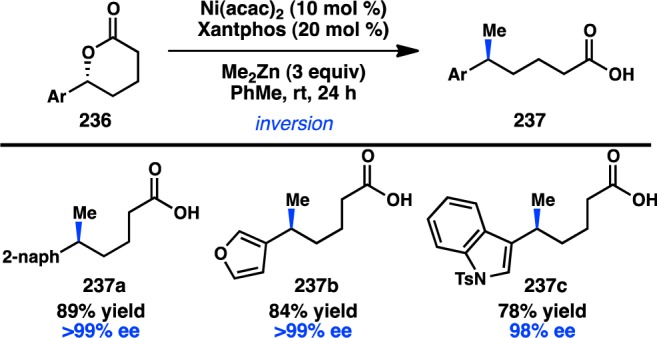
Stereospecific ring-opening of aryl-substituted lactones.
3.2.3. With Organoboron Reagents
Despite the general popularity of Suzuki–Miyaura cross-coupling reactions, examples of stereospecific couplings of sec-alkyl electrophiles have only recently been studied. In 2007, Asensio and co-workers reported the Pd-catalyzed Suzuki–Miyaura coupling of enantioenriched α-bromo sulfoxide 238 with PhB(OH)2 (Scheme 22).208 The α-arylated sulfoxides 239 were produced with inversion of configuration. Surprisingly, the diastereomer of 238 does not deliver any desired product. The authors hypothesize that coordination of Pd to the sulfinyl group is important for oxidative addition; an unfavorable gauche interaction could potentially destabilize the transition state for oxidative addition of the unreactive diastereomer. A computational study supports this hypothesis, finding that coordination of the sulfinyl group to Pd lowers the energy barrier for oxidative addition to the C–Br bond through an SN2 mechanism.209
Scheme 22. Stereospecific Suzuki–Miyaura Cross-Coupling of a Secondary Electrophile.
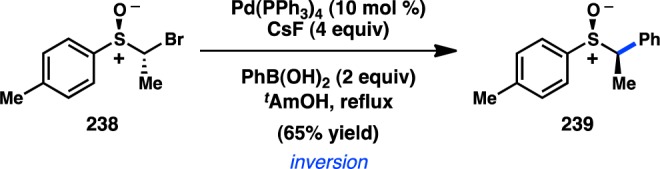
Employing a similar strategy, Falck and co-workers developed the first enantiospecific Suzuki–Miyaura cross-coupling of a sec-alkyl halide in 2010. Key to their success was the use of an α-cyanohydrin triflate, where the proximal nitrile can accelerate oxidative addition by coordinating to Pd in a fashion analogous to that of the sulfinyl group in Asensio’s system. Exposure of 240 and PhB(OH)2 to Pd complex 242 delivered 241 in excellent yield and with inversion of configuration, while minimizing formation of acrylonitrile side products (Scheme 23).87 Subsequent computational studies by Janesko and Pudasaini support the critical role of the Lewis basic nitrile in both accelerating the rate of oxidative addition to Pd and slowing the rate of β-hydride elimination from alkylpalladium intermediates.210 Other organoborons such as boronate esters, BF3K salts, and MIDA boronates were unreactive under the reported conditions.
Scheme 23. Enantiospecific Suzuki–Miyaura Cross-Coupling of α-Cyanohydrin Triflate.
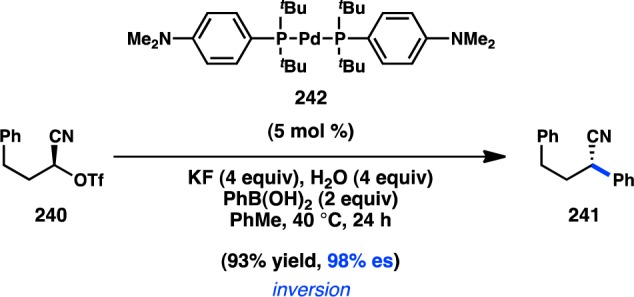
Given the abundance of commercially available organoboron reagents, Jarvo and co-workers sought to extend the scope of their Ni-catalyzed stereospecific cross-coupling reactions to the use of organoboron coupling partners.202 However, Suzuki–Miyaura cross-coupling reactions of sec-alkyl ethers were expected to require harsher conditions because of the slower rates of transmetalation with organoboron reagents. Nonetheless, when diaryl carbamates (e.g., 243) and strong base are employed, Ni-catalyzed enantiospecific cross-coupling to boronate esters is possible (Scheme 24).211 Systems lacking an extended π-system could be coupled when pivalates were used as the leaving group. Surprisingly, the identity of the ligand completely overturned the stereochemical outcome of the reaction. When PCy3 was used, the cross-coupling proceeded with retention of configuration, contrary to Jarvo’s previous findings in the Kumada–Corriu coupling. The use of nBuOH as an additive played a key role in enhancing the enantiospecificity. When SIMes, an NHC ligand, was employed, ent-245 was produced with inversion of configuration. Although additional studies are necessary, a preliminary proposal by the authors suggests that PCy3-mediated reactions of 243 occur through initial coordination of Ni to the carbamate followed by a syn, or retentive, oxidative addition; alternatively, the SIMes-mediated reaction could proceed through a stereoinvertive SN2′-type oxidative addition.
Scheme 24. Stereodivergent Synthesis of Triarylmethane.
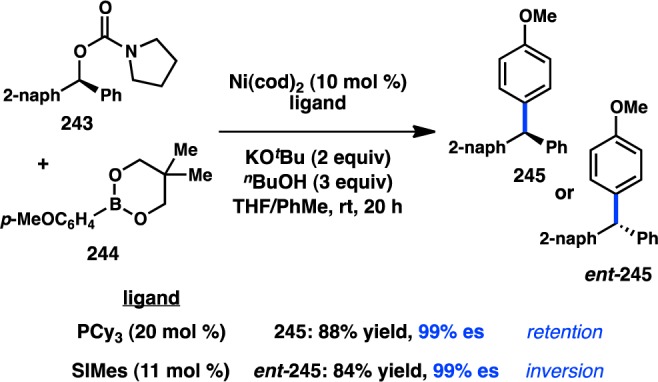
Concurrent with the studies by Jarvo described above, Watson and co-workers reported similarly intriguing stereodivergent behavior in the cross-coupling of benzhydryl pivalates and arylboronic acids. Treatment of pivalate 246 and PhB(OH)2 with Ni/PCy2Ph furnished 247 with retention of configuration and moderate es (Scheme 25).83 In contrast, nearly exclusive inversion of configuration was observed under ligand-free coupling conditions. These results are consistent with Jarvo’s hypothesis that Ni/phosphine complexes can coordinate to carbamate or ester groups and provide a syn oxidative addition. Under Watson’s ligand-free conditions, both diaryl- and triarylalkanes can be accessed in high yield and with inversion of configuration.
Scheme 25. Stereodivergent Coupling of Benzyl Pivalates.
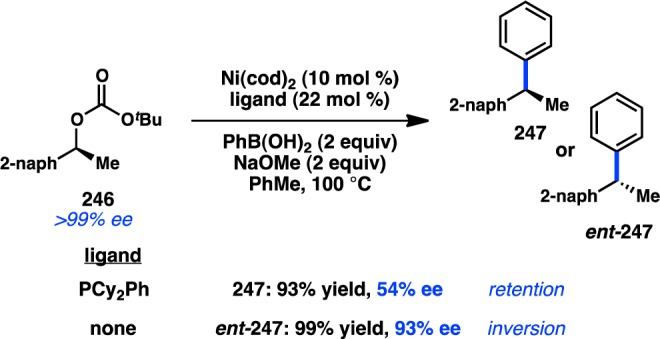
Watson and co-workers have also investigated the stereospecific Suzuki–Miyaura cross-coupling reactions of benzylammonium salts in an interesting example of oxidative addition to a C(sp3) C–N bond. Employing Ni/P(o-tol)3 as the catalyst, a variety of diarylalkanes could be prepared in good yield and high es, with inversion of configuration (Figure 56a).84 The stereochemical outcome is opposite that observed with benzylic pivalates when Ni/phosphine complexes are utilized. Subsequent efforts to extend the scope of the reaction to heterocycles and substrates lacking π-extended systems revealed that Ni(cod)2, in the absence of an exogenous ligand, catalyzes the desired cross-coupling reaction with high enantiospecificity (Figure 56b).85 Under these modified conditions, heterocyclic and alkenylboronic acids were coupled in good yields and simple α-phenylammonium triflates could also be coupled.
Figure 56.
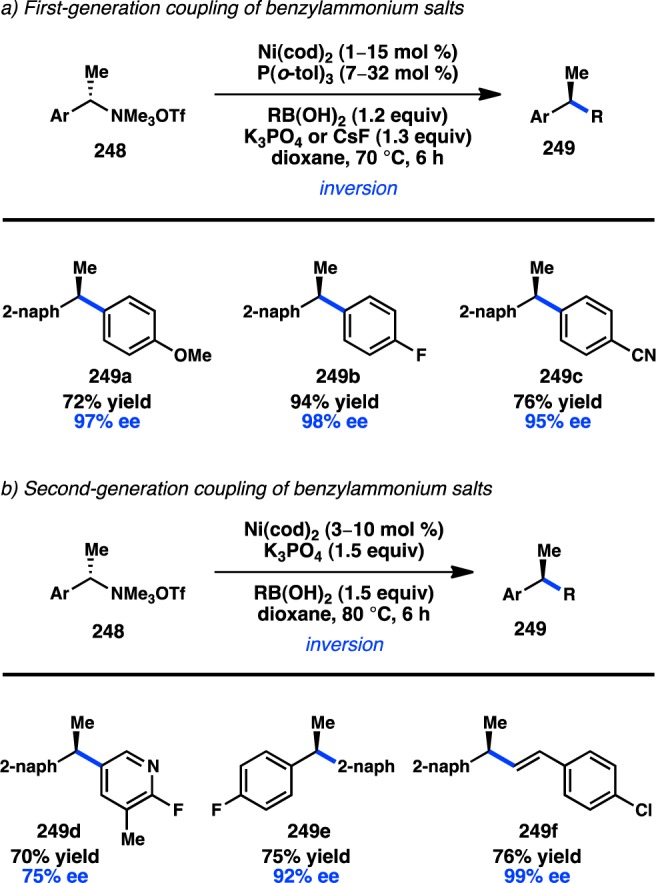
Stereospecific cross-coupling of benzylammonium triflates.
Recently, Takeda, Minakata, and co-workers reported a stereospecific Suzuki–Miyaura cross-coupling of arylaziridines. Ni-catalyzed couplings of aziridines have previously been reported to proceed with stereochemical scrambling,212,213 so the authors instead investigated Pd catalysts for this transformation. Although phosphine ligands tended to promote β-hydride elimination, NHC ligands favored generation of the cross-coupled product, with C–C bond formation occurring at the benzylic position.214 Thus, treatment of aziridine 250 with a boronic acid and Pd-NHC precatalyst 252 furnished 251 in high yield and regioselectivity and with complete inversion of configuration (Figure 57). The authors propose that SN2-type oxidative addition to 250 can form either metallacycle 253 or acyclic species 254. Transmetalation followed by reductive elimination releases product 256 as a single enantiomer and regioisomer.
Figure 57.
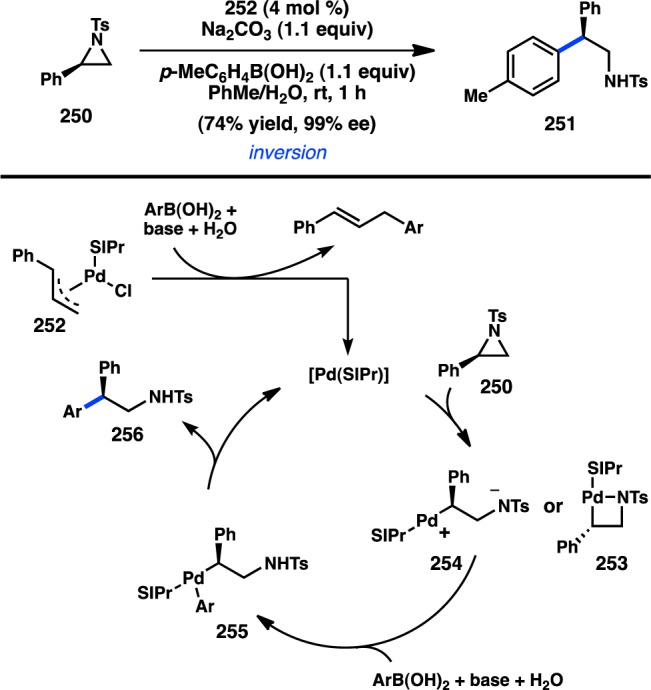
Stereospecific cross-coupling of N-tosylaziridines.
4. Transition-Metal-Catalyzed Enantiocontrolled Allylic Substitution
Over the past several decades, enantiocontrolled allylic substitution has emerged as a fundamental process in organic synthesis, allowing efficient formation of C–C, C–N, and C–O bonds with concomitant production of a versatile alkene functional group.215,216 As a general rule, these transformations proceed through π-allylmetal complexes. With respect to carbon-based nucleophiles, allylic functionalizations can be categorized into two groups (Figure 58): (1) reactions with stabilized or “soft” nucleophiles, defined here as nucleophiles for which the pKa of the conjugate acid is less than 25, and (2) reactions with unstabilized or “hard” nucleophiles, typically comprising organometallic reagents. Whereas soft nucleophiles usually undergo direct attack at the carbon atom of a π-allylmetal complex, hard nucleophiles typically react first with the metal catalyst through a transmetalation mechanism. Following transmetalation, reductive elimination from the metal center forges the C–C bond. Depending on the choice of transition-metal catalyst and ligand, transmetalation may occur either before or after oxidative addition to an allyl electrophile.
Figure 58.
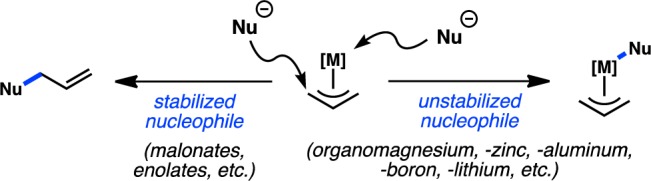
Stabilized vs unstabilized nucleophiles.
In many ways, the principles of stereocontrolled cross-coupling remain relevant when discussing enantioselective or enantiospecific allylic substitution reactions. In a simplified case, allylic substitution with hard nucleophiles can proceed via transmetalation, SN2′ (or γ) oxidative addition (these two steps can also occur in the reverse order), and reductive elimination to deliver the desired product (Figure 59). Controlling the facial selectivity for oxidative addition of allyl electrophile 257 can provide enantioenriched material. A critical complication occurs when the rate of π-allyl formation becomes competitive with reductive elimination: in this case, isomerization from σ-allyl complex 258 to π-allyl complex 260 allows generation of linear product 263. The rate of π-allyl formation is often influenced by the nature of the transition-metal catalyst and ligand.
Figure 59.
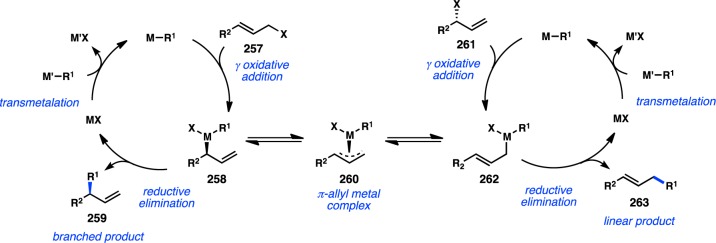
General principles of allylic substitution with hard nucleophiles.
Controlling the regioselectivity of stereocontrolled allylic substitution remains an active area of research. In the case of enantiospecific allylic substitution, transformation of stereogenic starting material 261 to branched product 259 can proceed through an anti SN2′ oxidative addition and π-allylmetal complex 260. Unselective reductive elimination yields a mixture of linear (263) and branched (259) products. Moreover, σ-bond rotation of allylmetal 262 can form a π-allylmetal complex on the opposite enantiotopic face, leading to erosion of enantiospecificity.
Although a number of transition metals have been shown to catalyze allylic substitution reactions, copper217 has received special attention for its ability to promote the coupling of hard nucleophiles and for delivering γ-substituted products with high regiochemical fidelity.4−6,218 With respect to stereospecific and stereoselective reactions, several aspects of Cu-catalyzed allylic substitution are worthy of mention. Electron-deficient copper complexes typically undergo faster reductive elimination to favor the γ-adduct with little stereochemical scrambling. Therefore, the use of electron-rich dialkyl cuprates is usually avoided when γ-selectivity is desired. To establish asymmetric variants of allylic substitution with high regio- and enantiocontrol, the above mechanism has been harnessed and modified in a number of different applications.
This review will cover the transition-metal-catalyzed enantioselective and enantiospecific union of unstabilized, organometallic reagents with allylic and propargylic electrophiles. Copper-catalyzed substitution has been thoroughly reviewed in the past; this paper will only focus on developments in Cu catalysis since 2008.4−6,218 Elegant work in the desymmetrizing ring opening of oxabicyclic electrophiles,3 which typically proceeds via carbometalation and β-oxy elimination, is considered outside the scope of this review.
4.1. Enantioselective Allylic Substitution Reactions
4.1.1. Organomagnesium Reagents
As in the development of transition-metal-catalyzed asymmetric cross-coupling reactions, the first explorations of asymmetric allylic substitution involved the Ni-catalyzed reaction of organomagnesium reagents in the presence of chiral phosphines. In 1980, Consiglio and co-workers showed that Ni in conjunction with bidentate chiral phosphine DIOP (L1) catalyzed the formation of 265 in 16% ee from allylic alcohol 264 (Figure 60).219 Further studies using the allylic phenyl ether determined that use of Chiraphos (L68)220,221 or axially chiral BIPHEMP (L69)222,223 as a ligand provides 265 in improved 51% and 84% ee, respectively. Under similar conditions, low enantioselectivity was obtained for acyclic allylic alcohols.224 In a separate approach, racemic secondary Grignard reagents could be resolved through an enantioselective allylation to deliver products with low ee.21,221
Figure 60.
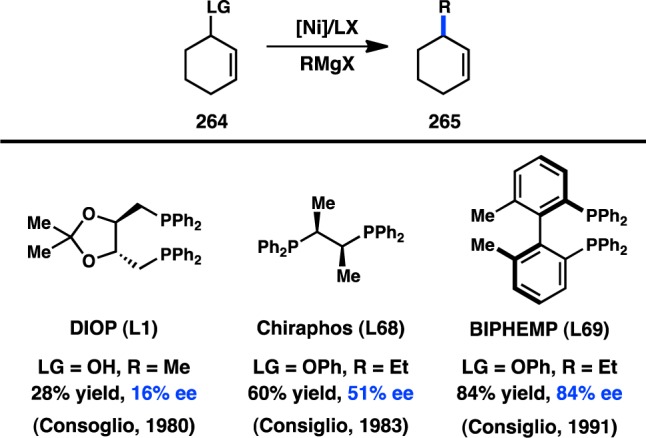
Seminal Ni-catalyzed substitution of cyclic allylic alcohol derivatives.
Shortly after their initial reports of Ni-catalyzed asymmetric allylic substitution reactions with alkyl Grignard reagents, Consiglio and co-workers turned to develop the analogous reactions of arylmagnesium species. However, the decreased nucleophilicity of arylmetallic reagents compared to their alkyl counterparts makes allylic arylation challenging, and the early studies by Consiglio220,221 and Hiyama225 demonstrated both the promise and the challenge of asymmetric allylic arylation. Using Ni as a catalyst, allylic electrophiles and ArMgBr could be coupled to provide a mixture of branched and linear products (Scheme 26a). When acyclic pivalate 266 was utilized, 267 was obtained in a promising 89% ee, but as a mixture with the linear coupling product 268. Similar regioselectivity was observed when diphosphine L70 was employed.226 Symmetrical acyclic electrophiles were instead required to avoid the formation of regioisomers (Scheme 26b). Whereas cyclic substrates reacted with poor enantioselectivity under Consiglio’s conditions, Uemura and co-workers determined that Ni/L29 delivers cyclic product 272 in high ee (Scheme 26c).227 Control experiments revealed that removal of the planar chirality in L29 furnished products with only moderate enantioinduction. Interestingly, Uemura’s method could not be readily extended to acyclic systems.
Scheme 26. Ni-Catalyzed Asymmetric Allylic Arylation with Grignard Reagents.
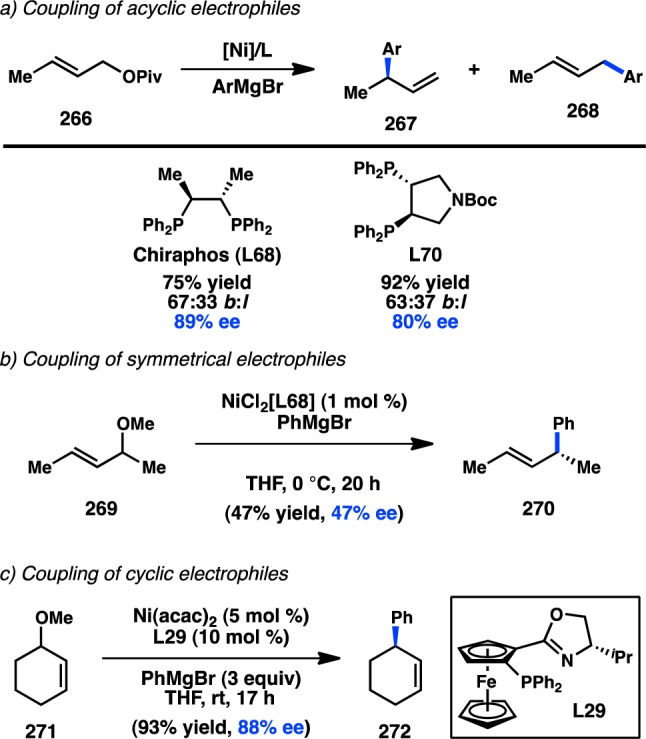
In 1997, RajanBabu and co-workers disclosed an enantioselective Ni/L68-catalyzed alkylation of acyclic diphenylallyl ether 273 (Scheme 27a).228 Methyl ethers were found to provide better results than phenyl ethers, whereas EtMgBr delivered slightly higher ee than MeMgBr. Quenching of the reaction mixture prior to complete conversion led to recovery of enantioenriched 273, implicating a kinetic resolution of the allyl ether in the oxidative addition step. Shortly after these results, Hoveyda and co-workers reported the Ni-catalyzed addition of alkyl Grignard reagents to unsaturated cyclic acetal 275 (Scheme 27b).229 Following acidic workup, ketone 276 was obtained in high yield and good ee. Although Chiraphos (L68) was identified as the optimal chiral ligand, the authors were intrigued to find that the presence of an achiral phosphine, such as PPh3, increased the enantioselectivity of the reaction. Importantly, the reaction conditions permitted both alkyl and aryl Grignard reagents to be incorporated without a difference in enantioinduction.
Scheme 27. Ni-Catalyzed Addition to Allyl Ethers.
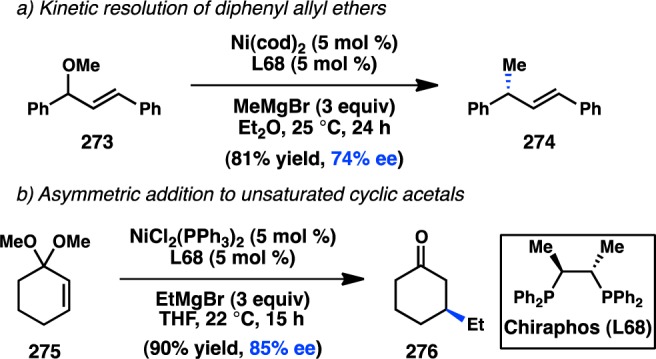
The first Pd-catalyzed asymmetric allylic substitution using Grignard reagents was reported by Buono and co-workers in 1990. The researchers found that a Pd/L71 complex could catalyze the alkenylation of cyclic electrophile 277 in moderate yield and promising ee (Scheme 28).230 Reaction with simple vinylmagnesium bromide only formed reduction products. The related Pd-catalyzed asymmetric allylic arylation with Grignard reagents has yet to be accomplished.
Scheme 28. Pd-Catalyzed Asymmetric Allylic Alkenylation with Grignard Reagents.

Despite the pioneering examples of asymmetric Ni- and Pd-catalyzed coupling of Grignard reagents and allylic electrophiles, the attention of the scientific community has shifted toward Cu catalysis over the past two decades. The Cu-catalyzed addition to linear allylic electrophiles is widely known to proceed with excellent regioselectivity for the branched, stereogenic isomer. In 1995, Bäckvall, van Koten, and co-workers reported the first asymmetric Cu-catalyzed coupling of alkylmagnesium reagents with allylic electrophiles (Scheme 29).231 In successive years, a range of different leaving groups and chiral ligand scaffolds have been exploited to deliver branched products in excellent yield, ee, and regioselectivity; these developments have previously been reviewed by others and will not be discussed here.4−6,218 Since 2008, efforts have focused on diversifying the toolbox of nucleophiles and electrophiles that can deliver high enantioinduction.
Scheme 29. Seminal Example of Cu-Catalyzed Asymmetric Allylic Substitution with Alkyl.
The asymmetric allylic substitution of cinnamyl electrophiles with organomagnesium reagents has served as a model system to test new chiral ligand scaffolds for Cu catalysis (Figure 61). In 2008, Tomioka and co-workers applied chiral amidophosphane L72, which had previously been used in asymmetric conjugate addition reactions, to the allylic substitution of cinnamyl bromide.232 High enantioselectivity was observed for the reaction with ethylmagnesium bromide, but the regioselectivity was poor. Nonetheless, L72 proved to be quite versatile, delivering the branched product in high ee when either alkyl or aryl Grignard reagents were employed.
Figure 61.
Cu-catalyzed asymmetric allylic alkylation of cinnamyl electrophiles.
The laboratories of Schmalz and Zhang independently showed that phosphite or phosphoramidite ligands improve the regioselectivity in the Cu-catalyzed coupling of cinnamyl halides with organomagnesium reagents (Figure 61).233,234 Although L76 was the optimal ligand identified by Schmalz for higher alkyl Grignard reagents, related phosphine–phosphite L77 delivered better results in the coupling of 282 with MeMgBr. Zhang’s novel ligand L75, a rare example of a D2-symmetric ligand being used in asymmetric catalysis, provides the substitution product 283 with good regioselectivity and in good ee. Alexakis subsequently reported phosphite–phosphoramidite L73 in which the ligand chirality is derived from a furanoside core;235L73 imparts excellent regioselectivity favoring 283, but the enantioinduction is modest.
In 2013, Mauduit and co-workers investigated the use of NHC ligands in the Cu-catalyzed substitution of cinnamyl phosphates with alkyl Grignard reagents.236 The chelating, yet relatively simple, NHC L74 produced 283 in high yield and ee and with excellent regioselectivity (Figure 61). Moreover, the reaction conditions could be extended to the formation of all-carbon quaternary centers with little erosion of ee (Scheme 30).
Scheme 30. Preparation of All-Carbon Quaternary Centers by Asymmetric Allylic Alkylation.
In addition to cinnamyl electrophiles, a concerted effort has been made by the Feringa group to develop Cu-catalyzed reactions between organomagnesium reagents and allylic electrophiles bearing synthetically useful functional groups. Halocrotonate 287 reacts with MeMgBr in the presence of Taniaphos (L78) to deliver the desired SN2′ product 288 in excellent ee and without formation of 1,2- or 1,4-addition products (Scheme 31).237 Using the same ligand, Feringa reported the reaction of phosphonate 289 and dienyl bromide 291 with MeMgBr to give products 290 and 291, respectively, in good yields and with excellent enantioinduction.238,239 Recent work by Alexakis and co-workers has demonstrated that skipped enynes can be prepared from enynyl chloride 293 and alkyl Grignard reagents upon exposure to Cu and L79.240 Whereas good selectivity for the SN2′ product is observed for most higher alkylmagnesium reagents, poor regioselection is obtained in the reaction of MeMgBr. A number of recent reports have also demonstrated the utility of tandem asymmetric allylic alkylation/cross-metathesis for the generation of enantioenriched cyclic species.241−244 A tandem asymmetric allylic alkylation/Heck coupling has been realized to form chromene products as well.245
Scheme 31. Expansion of the Scope of Cu-Catalyzed Asymmetric Allylic Alkylation.
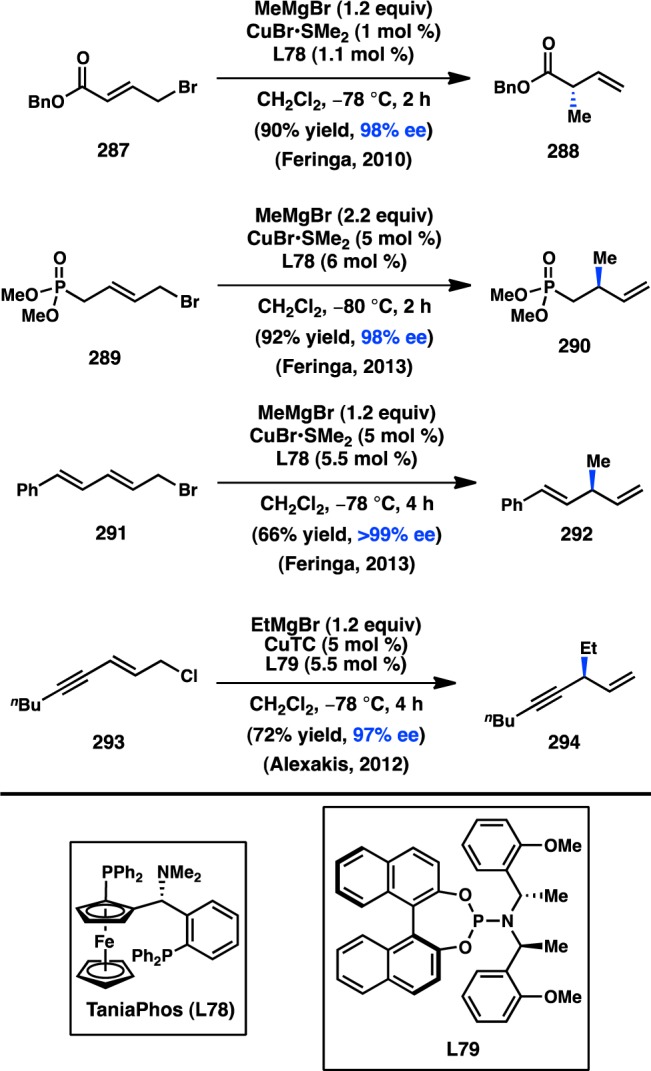
The coupling of more functionalized allylic electrophiles has been extended to include geminal disubstituted electrophiles bearing two leaving groups. In 2010, Feringa and co-workers studied the Cu-catalyzed asymmetric allylic substitution of α-halo acetates (296), which can be prepared in situ from aldehyde 295 (Figure 62a).246 Treatment of 296 with CuTC and L32 delivers 297 with high enantioinduction and exclusive formation of the chloride-displacement product. Higher alkylmagnesium reagents heavily favored SN2′ addition, with good selectivity for the (Z)-alkene in most cases; MeMgBr resulted in decreased regioselectivity. Exposure of 297 to base delivered 298, formally a conjugate addition product. The high Z-selectivity observed in the formation of 297 prompted Feringa and co-workers to investigate the substitution of gem-dichloride 299 (Figure 62b).247 Slightly modified reaction conditions provided chloroalkene 300 with excellent ee and 99:1 Z:E-selectivity. To explain the selectivity for the (Z)-isomer, the authors propose that dative coordination between one of the chlorines of 299 and Cu results in oxidative addition through π-complex 301 to give (Z)-σ-allyl complex 302.
Figure 62.
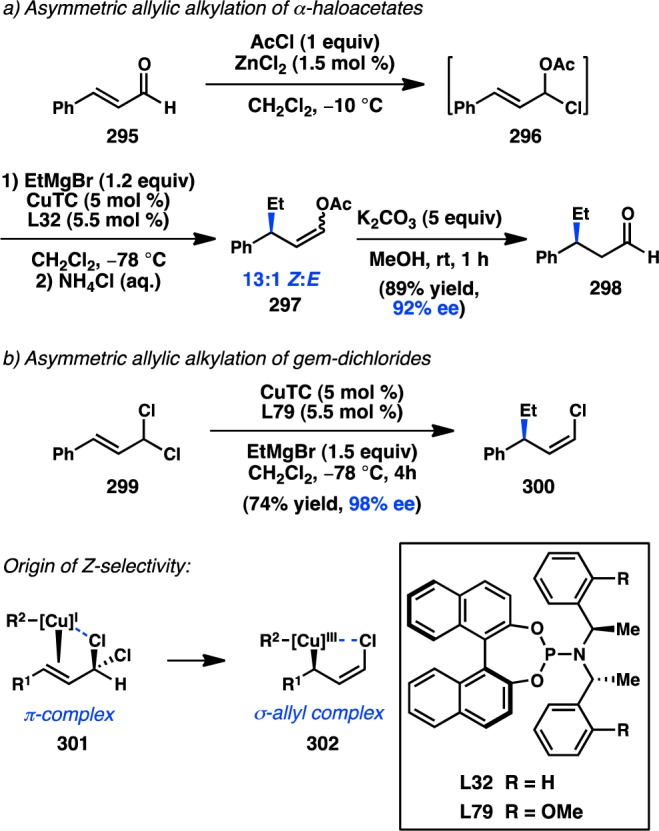
Coupling of allylic electrophiles bearing two leaving groups.
The development of highly enantio- and regioselective Cu-catalyzed allylic substitution reactions with arylmagnesium reagents has proven more challenging than the reactions of the corresponding alkylmagnesium species. In 2009, Tomioka and co-workers were able to improve on their previous studies, which utilized Cu/L72 (see Figure 61), by employing chiral NHC ligands. The preformed copper complex derived from L80 catalyzed the reaction between cinnamyl bromide and PhMgBr to furnish diarylalkene 304 in excellent yield, ee, and branch selectivity (Scheme 32).248 A bulky o-tol group on L80 was hypothesized to increase the steric hindrance of the γ–σ-allylcopper(III) intermediate, thus facilitating a fast reductive elimination and precluding isomerization via π-allyl formation. Using this principle, bulky NHC ligands were applied to the coupling of aliphatic allyl bromides, delivering the SN2′ products in up to 96% branch selectivity and up to 98% ee.249
Scheme 32. Cu-Catalyzed Asymmetric Allylic Arylation with Grignard Reagents.
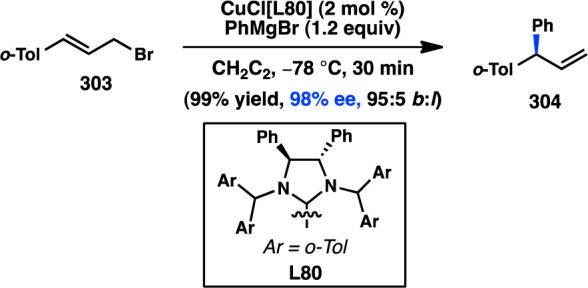
Asymmetric allyl–allyl cross-couplings provide access to versatile intermediates, but they have been slow to develop because of challenges associated with the dual coordination modes of allylmetal species. In 2013, Feringa and co-workers reported an enantioselective Cu-catalyzed coupling of allylmagnesium halides and allyl bromides (Figure 63).250 The use of phosphoramidite (R,S,S)-L32 provided 306 in high ee. A noncoordinating counterion on Cu was necessary to enhance the selectivity for branched product 306; however, the regioselectivity of the reaction was still only modest. The authors propose that oxidative addition of allyl-[Cu]I to an allylic bromide potentially gives rise to a mixture of Cu σ,σ- and σ,π-complexes (308, 309, and 311). Fast reductive elimination from 308 would produce branched product 310, whereas equilibration to π,σ-complex 311 would lead to linear product 312.
Figure 63.
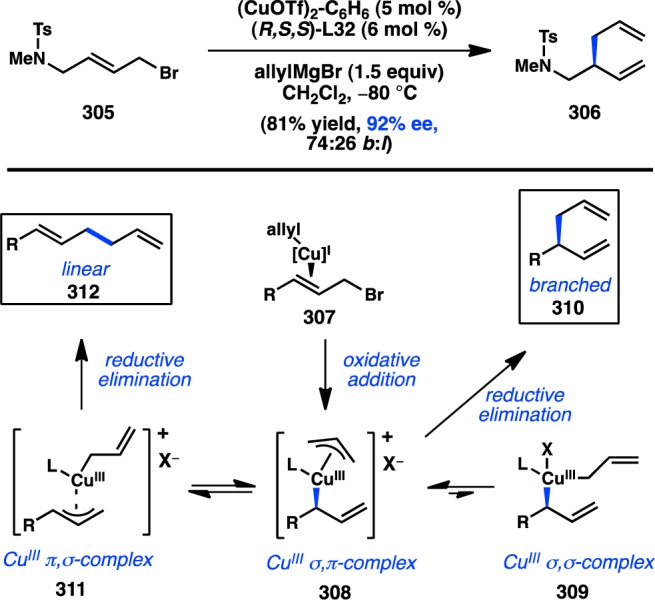
Cu-catalyzed allyl–allyl coupling.
The majority of Cu-catalyzed asymmetric allylic substitution reactions of alkyl Grignard reagents employ nonstereogenic allyl electrophiles. High enantioinduction arises from catalyst differentiation of the pro-chiral faces of the substrate. However, in 2009, Alexakis reported a highly enantioselective allylic substitution of racemic cyclic bromide 313, wherein both enantiomers of starting material converge to a single enantiomer of product (Figure 64).251,252 A subsequent computational study proposed that this reaction is an example of a direct enantioselective transformation (DET). In a DET, each enantiomer of starting material proceeds according to a distinct reaction pathway to provide ultimately the identical enantiomer of product. In the present example, Cu/L32 engages (R)-313 through an SN2′ oxidative addition, whereas (S)-313 reacts by an SN2 oxidative addition, both converging to CuIII complex 315.253
Figure 64.
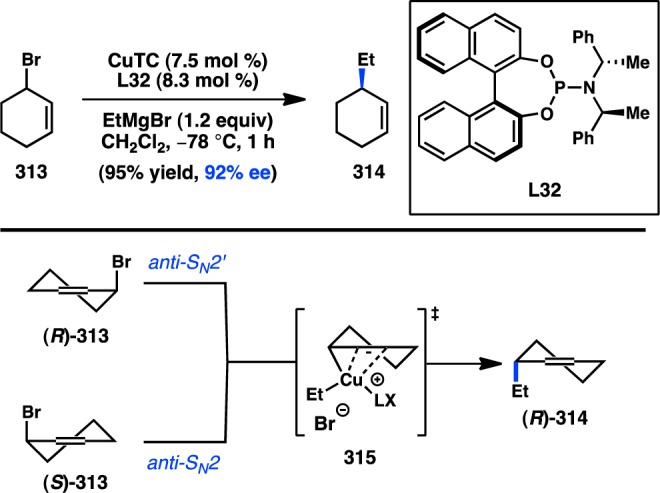
Direct enantioselective transformation (DET) of a racemic allylic electrophile.
When this method was extended to acyclic systems and L81 was employed as the ligand, the authors obtained a ∼1:1 mixture of (R,E)-317 and (R,Z)-317, both of which were formed in high ee (Figure 65).254 To explain this behavior, the authors propose that the reaction proceeds by a stereodivergent kinetic resolution (SKR), in which the chiral catalyst favors an anti SN2′ oxidative addition to the Si face of the alkene. To satisfy these mechanistic conditions, (S)-316 must react in the pro-E-conformation, whereas (R)-316 must react in the pro-Z-conformation.
Figure 65.
Stereodivergent kinetic resolution (SKR) of an acyclic racemic electrophile.
4.1.2. Organozinc Reagents
In 1985, Fiaud and co-workers reported their investigations of the Pd-catalyzed substitution of allylic acetates with arylzinc halides (Scheme 33).255,256 When phosphine L82 was used as the chiral ligand, the reaction between 277 and PhZnCl provided 272 in 60% yield but with poor enantioinduction. Nearly 15 years later, the first enantioselective allylic substitution reaction of organozinc reagents was reported by Knochel and co-workers (Scheme 34).257 Subsequent developments have expanded the scope of leaving groups and the degree of enantioinduction to prepare both tertiary and quaternary centers, primarily using Cu catalysts.4−6,218
Scheme 33. Seminal Enantioselective Pd-Catalyzed Substitution of Cyclic Allylic Acetates.
Scheme 34. Seminal Enantioselective Cu-Catalyzed Substitution of Allylic Chlorides.
Beginning in 2007, researchers began to reinvestigate transition metals other than Cu for the cross-coupling between allylic electrophiles and organozinc reagents. In 2008, Fu and co-workers disclosed a Ni-catalyzed stereoconvergent coupling of alkylzinc halides and branched allylic halides (Figure 66).258 With Pybox L84 as the ligand, a variety of primary alkylzinc halides and unsymmetrical allylic halides could be coupled with high enantio- and regioselectivity, favoring C–C bond formation at the less hindered position. Only in the case of 322b, in which a Me group must be distinguished from an nBu group, was there poor regiodifferentiation. The advantage of this method over many of the Cu-catalyzed reactions lies in its ability to promote stereoconvergent couplings of racemic, stereogenic allylic electrophiles.
Figure 66.
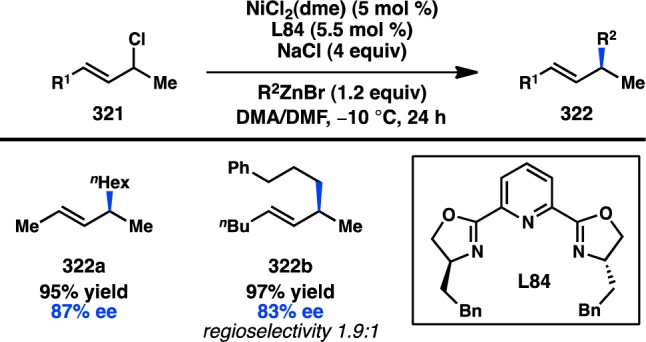
Stereoconvergent Ni-catalyzed coupling of internal allylic electrophiles.
The first highly enantioselective Pd-catalyzed allylic substitution reaction using organozinc reagents was developed by Maulide and co-workers in 2014. This transformation has been challenging to achieve because of the well-precedented umpolung reactivity of allylpalladium complexes in the presence of dialkylzinc reagents.259,260 Upon exposure to R2Zn, allylpalladium species typically acquire nucleophilic character and can react with electrophiles through undesired pathways (Figure 67). Maulide discovered that phosphoramidite L85 imparts unique reactivity to the allylpalladium complex, favoring allylic substitution to provide coupled product 324 in both good yield and good ee.261 Although a variety of cyclic allylic electrophiles were found to be competent reaction partners, only hydrodehalogenation was observed with an acyclic electrophile. The mechanism and origin of enantioinduction have yet to be explored for this intriguing transformation.
Figure 67.
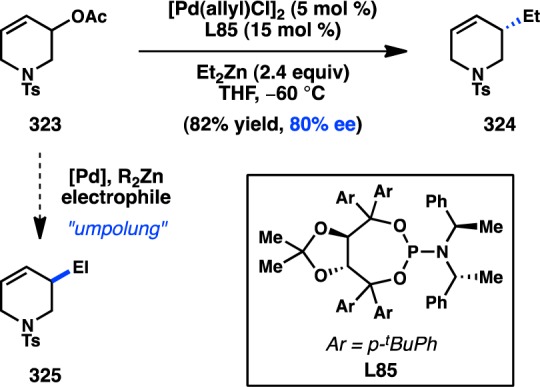
Stereoconvergent Pd-catalyzed coupling of cyclic allylic electrophiles.
Although the Cu-catalyzed asymmetric allylic substitution using alkylmetallic reagents had been extensively developed throughout the early 2000s, examples using arylmetallic reagents were still quite rare. In 2007, Alexakis and co-workers examined the ability of different transition-metal catalysts to promote the branch-selective and enantioselective coupling of aliphatic allylic carbonates and arylmetal reagents.262,263 Iridium was chosen for further study because of its propensity to deliver λ-substituted products in the allylic substitution of stabilized nucleophiles.264,265 Treatment of 326 with PhMgBr in the presence of Ir/L79 as the catalyst led to deprotection of the electrophile but no formation of coupled product. In contrast, transmetalation to the aryl zinc reagent and addition of excess LiBr enabled the desired substitution reaction, providing 327 in good yield and ee, albeit with low selectivity for the branched product (Scheme 35). A variety of phosphoramidite ligands were studied in place of L79, but none delivered enhanced regioselectivity. Modified reaction conditions could be applied to cyclic allylic acetates (277), generating products in as high as 90% ee without any complications from regioisomeric mixtures.
Scheme 35. Ir-Catalyzed Asymmetric Allylic Arylation with Organozinc Reagents.
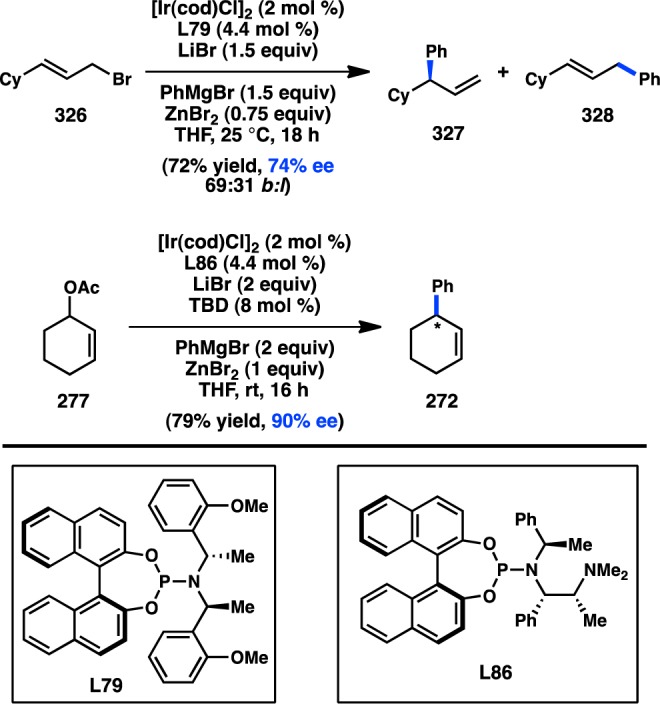
Notwithstanding the recent developments with other transition-metal catalysts, Cu has remained the workhorse of enantioselective allylic substitution reactions of organozinc reagents. Advances in ligand design continue to expand the scope and selectivity of these transformations. In 2009, Nakamura and co-workers disclosed amino hydroxy phosphine L87, with which the Cu-catalyzed reaction between cinnamyl phosphate 329 and dialkylzinc reagents proceeds in modest yield but high ee and regioselectivity (Figure 68).266 The authors suggest that the multiple chelating sites on L87 permit the formation of a Cu/Zn bimetallic complex. Simultaneous π-complexation by Cu and phosphate coordination by Zn forges a closed transition state that can promote an anti SN2′ oxidative addition. The following year, Mauduit and co-workers revealed that hydroxyalkyl NHC L88 can provide 283 in both high yield and high ee.267 The reaction scope was later extended to include the synthesis of skipped dienes as well as the use of Grignard reagents.236
Figure 68.
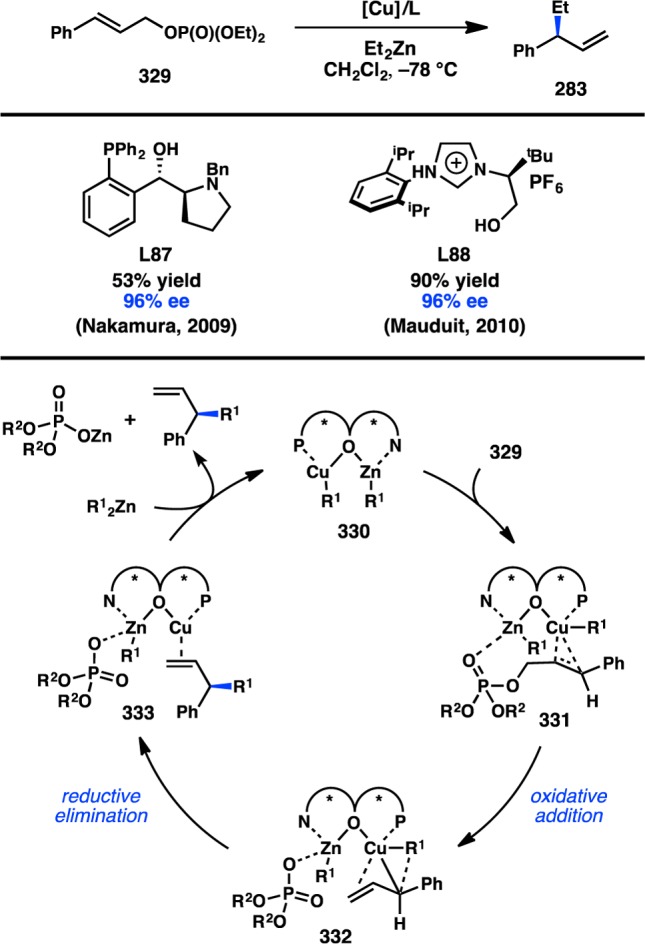
Cu-catalyzed coupling of allylic phosphates.
4.1.3. Organoaluminum Reagents
Trimethylaluminum was first studied in asymmetric allylic alkylation in 2000 by Woodward and co-workers, but both isolated yields and ee’s below 10% were obtained.268 In 2008, Hoveyda and co-workers showed that Cu–NHC complexes catalyze the coupling between allylic phosphates and alkenylaluminum species, which were prepared in situ from an alkyne and DIBAL-H (Scheme 36).269 NHC L89 was unique in delivering both high regioselectivity and high enantioselectivity; furthermore, iBu group transfer was low. If the reaction was conducted using the imidazolium salt precursor to L89, lower conversion was observed. Unfortunately, the reaction scope was limited to alkenylaluminum species that could be prepared by hydroalumination, which excluded cis-alkenylaluminum species, or vinyl groups with aryl substitution. To overcome these issues, Hoveyda investigated the hydroalumination of phenylacetylene derivatives and discovered that trimethyl(phenylethynyl)silane underwent clean hydroalumination with DIBAL-H.270 Moreover, depending on the reaction conditions, either the (E)- or the (Z)-isomer of the vinylmetal could be accessed. Subsequent reaction of 337 and phosphate 329 with Cu/L90 delivered 338 in high yield and excellent ee. The TMS group could be removed under mild conditions to deliver a disubstituted cis-alkene; correspondingly, trans-alkenes could be generated from the (Z)-isomer of 337. Subsequent studies by Hoveyda have established that both alkyl- and aryl-substituted vinylaluminum reagents can also be applied to the synthesis of all-carbon quaternary stereocenters.271
Scheme 36. Cu-Catalyzed Asymmetric Allylic Vinylation with Organoaluminum Reagents.
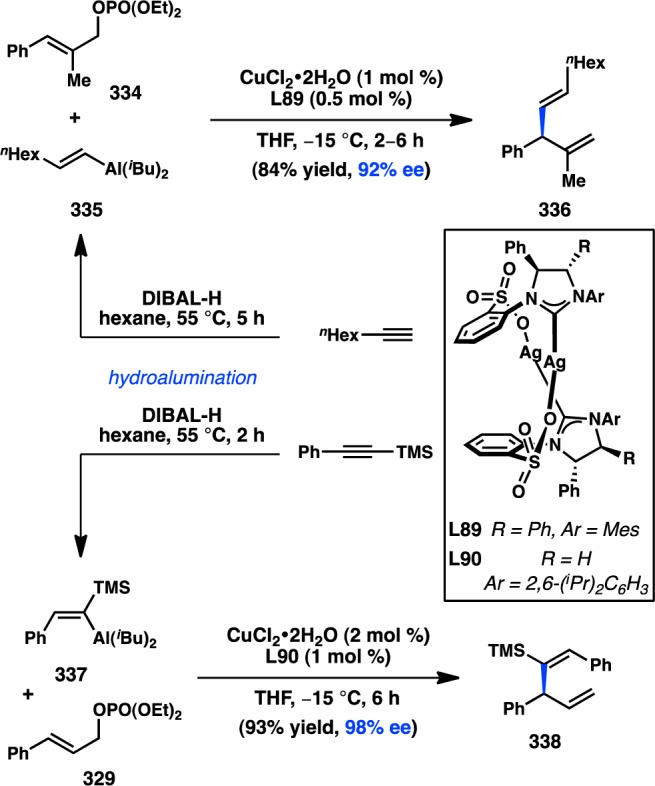
Following their success using vinylaluminum reagents, the Hoveyda laboratory investigated whether arylaluminum reagents could participate in a rare example of transition-metal-catalyzed asymmetric allylic arylation. The requisite organometallic reagents are prepared by treatment of aryllithium species with Et2AlCl (Figure 69).272 The authors expected that the aryl group on Al would be more likely to transmetalate to Cu than the alkyl group.273−276 Cu-catalyzed coupling of an arylaluminum and allylic phosphate 339, using NHC L91 as the ligand, furnished 340 in good yield and high ee. A variety of aryl- and heteroarylaluminum reagents could be used to generate products bearing all-carbon quaternary stereocenters.
Figure 69.
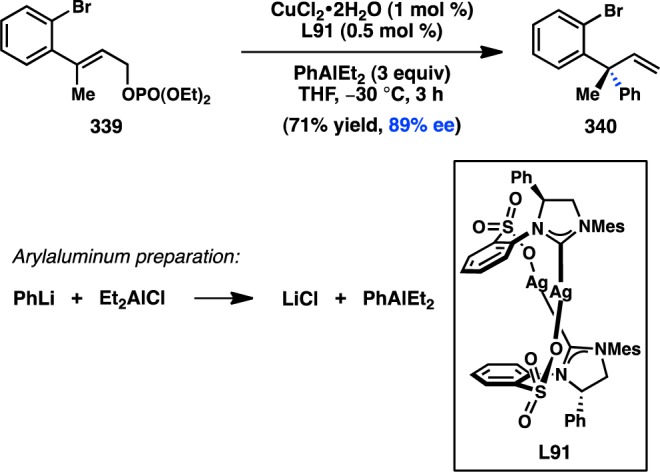
Cu-catalyzed asymmetric allylic arylation with organoaluminum reagents.
In 2011, Hoveyda and co-workers discovered that Cu/L90 catalyzes the asymmetric cross-coupling of alkynylaluminum reagents with allylic phosphates to form all-carbon quaternary stereocenters (Figure 70).277,278 The authors were surprised to see good chemoselectivity for alkynyl transfer, because alkynes are sometimes used as dummy ligands on copper. The organoaluminum reagents could easily be prepared in situ by treatment of an alkyne with DIBAL-H and Et3N.
Figure 70.
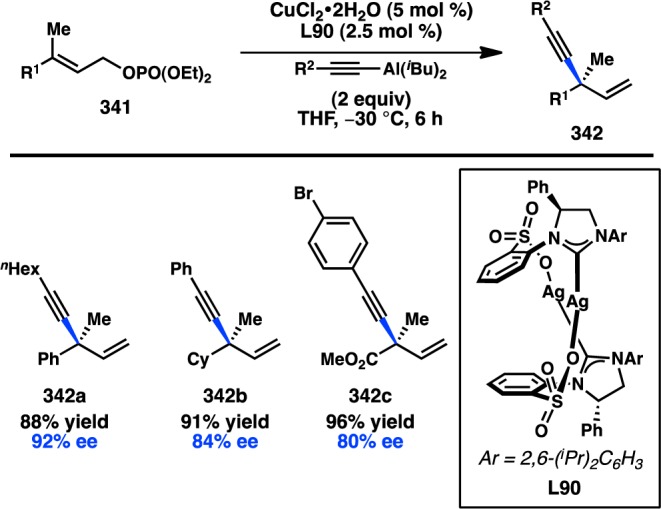
Cu-catalyzed asymmetric allylic alkynylation with alkynylaluminum reagents.
C(sp3) organoaluminum reagents have been less frequently utilized in asymmetric allylic substitution reactions than their organozinc counterparts. However, in 2007, Hoveyda and co-workers showed that AlMe3 provided better results than Me2Zn in Cu/L89-catalyzed SN2′ substitution reactions of allylic phosphates. This reactivity was exploited in an elegant example of a double SN2′ reaction toward a total synthesis of baconipyrone C (Scheme 37a).279 A Ni-catalyzed allylic substitution was also developed by Woodward and co-workers, in which Baylis–Hillman adduct 345 could be coupled with AlMe3 (Scheme 37b).280,281 Axially chiral Ferrocite L92 imparted high enantioinduction, but good control of regioselectivity remains an unsolved obstacle.
Scheme 37. AlMe3 in Ni-Catalyzed Asymmetric Allylic Alkylation.
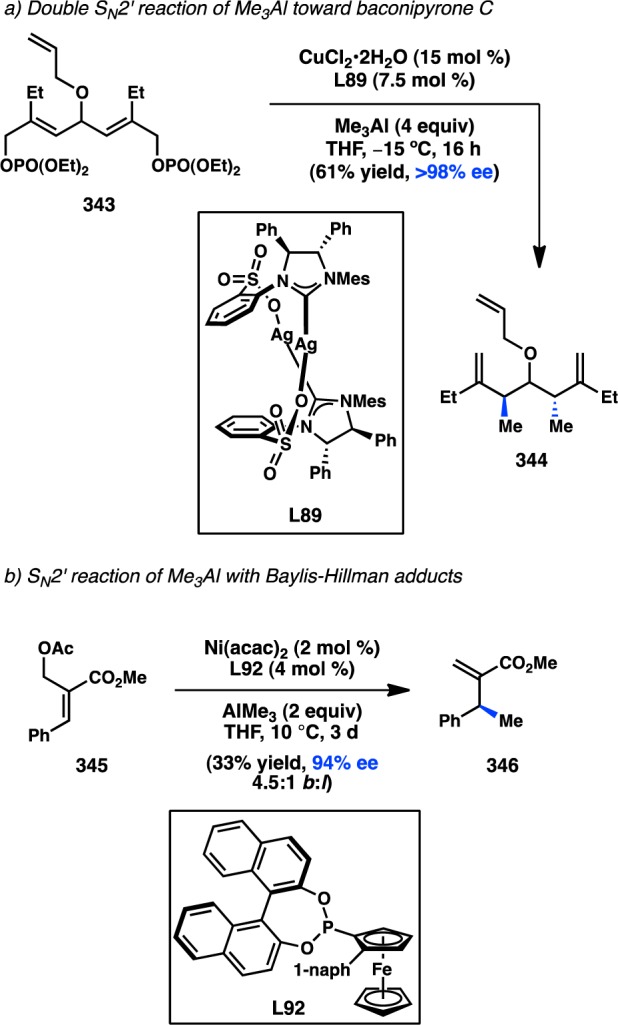
4.1.4. Organoboron Reagents
Of the various organometallic nucleophiles utilized for asymmetric allylic substitution reactions, organoboron reagents have been successfully implemented with the most diverse array of transition-metal catalysts, including Ni, Rh, Ir, Pd, and Cu. In 2000, Uemura studied the reactivity of both organoboron and organomagnesium reagents in the Ni-catalyzed asymmetric allylic arylation of cyclohex-2-en-1-yl acetate (277). The use of (oxazolinylferrocenyl)phosphine L29 enabled formation of 272 in good yield and moderate ee, representing the first example of an arylboron reagent being used in asymmetric allylic arylation (Scheme 38).282 In comparison to Uemura’s related coupling of Grignard reagents (see Scheme 26),227 several points are worth noting: (1) the lower nucleophilicity of organoboron reagents requires the addition of base and for the reaction to be conducted at elevated temperatures, (2) a smaller range of leaving groups participate in the coupling with arylboron species, and (3) lower levels of enantioinduction by L29 are observed. Both methods suffer from reduced ee and poor regioselectivity when applied to acyclic substrates.
Scheme 38. Ni-Catalyzed Asymmetric Allylic Arylation with Organoboron Reagent.
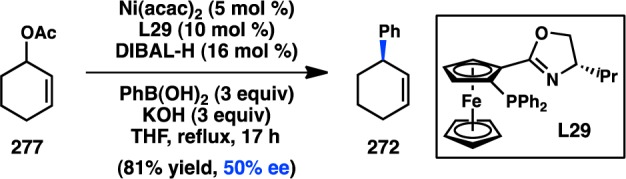
Building on earlier studies of Rh-catalyzed asymmetric 1,4-addition reactions of boronic acids to nitrocyclohexene,283 in 2005, Gong and co-workers reported the Rh-catalyzed substitution of nitroallyl acetate 347 with PhB(OH)2 (Figure 71).284 The authors reasoned that, following insertion into the olefin of 347, a β-elimination of the acetate group might deliver the desired allylic substitution product. Rh-catalyzed coupling of 347 and PhB(OH)2 with BINAP as a chiral ligand furnished 348 in excellent enantioselectivity. Subsequent studies revealed that higher yields but slightly lower enantioselectivity could be obtained when more reactive arylzinc halides were employed as the nucleophile.285
Figure 71.
Rh-catalyzed reactivity of nitroallyl acetates.
Allylic diols have also been studied as electrophiles in Rh-catalyzed allylic substitution reactions with arylboron reagents. Murakami and co-workers discovered that Rh catalyzes the reaction of cis-diol 350 with phenylboroxine to give enantioenriched homoallylic alcohol 351 when chiral diene L93 is employed as the ligand (Figure 72).286 It was observed that allylic cis-diol 350 first reacts with phenylboroxine to generate cyclic arylboronic ester 353. Subsequent carbometalation followed by β-oxygen elimination yields alcohol 351. The production of minor constituent 352 was rationalized by a β-hydride elimination/reinsertion/β-oxygen elimination sequence. A limited scope of arylboroxines was demonstrated for the transformation.
Figure 72.
Allylic substitution by carborhodation.
In 2012, Oi and co-workers reported the Rh/DTBM-SEGPHOS-catalyzed coupling of allyl aryl ethers (355) with arylboronic acids to give allylic substitution product 4 (Figure 73).287 A variety of arylboronic acids were found to react in good yields and with high enantioselectivity. The authors proposed a carborhodation/β-oxygen elimination mechanism analogous to that suggested by Murakami for the reaction of allylic cis-1,4-diols.
Figure 73.
Allylic substitution of allyl aryl ethers.
Lautens and co-workers have studied a closely related reaction, the Rh-catalyzed asymmetric arylation of allylic cis-dicarbonates.288 Rh/L94-catalyzed substitution of dicarbonate 358 with PhB(OH)2 delivered branched product 359 in high yield and ee (Figure 74). The minor product was found to be the isomeric, linear carbonate 360; notably, the alkene corresponding to 352, detected by Murakami in the previously discussed reaction (see Figure 72), was not observed. This finding prompted Lautens to propose instead an oxidative addition/reductive elimination mechanism that proceeds through σ-enyl intermediate 361. Slow isomerization to linear organorhodium complex 362 would explain formation of 360, where the relative rates of reductive elimination from 361 versus isomerization to 362 would determine the product distribution. The Rh-catalyzed desymmetrization of meso-cyclopentene-1,4-diol dicarbonates with arylboron reagents has also been developed by the same group.289,290
Figure 74.
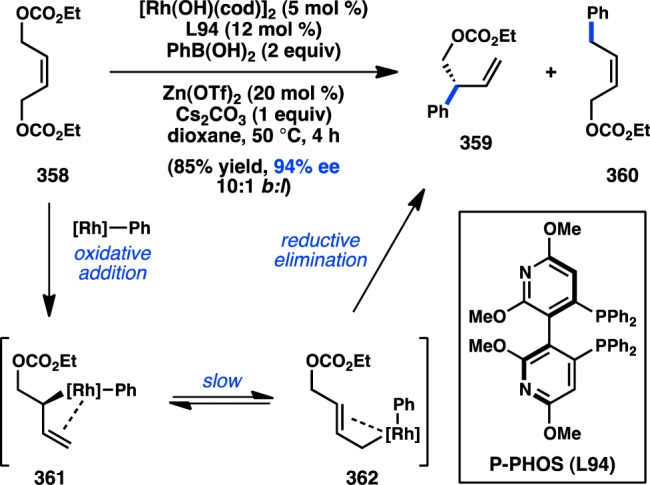
Allylic substitution by oxidative addition of Rh complexes.
Until recently, there have been few reports of Cu-catalyzed stereoselective couplings of allylic electrophiles and C(sp2)-hybridized organoboron reagents. In 2011, Shintani, Hayashi, and co-workers discovered that CuCl, in conjunction with Mauduit-type NHC ligand L95(236,267) bearing a pendant hydroxyl group, catalyzes the union of allylic phosphate 329 and boronate ester 363 (Figure 75).291,292 The use of NaOMe was important to favor formation of the branched product over the linear isomer. A wide scope of arylboron reagents were well tolerated, and trisubstituted allylic phosphates could be employed to deliver all-carbon quaternary stereocenters. Moreover, a cyclohexenylboronate was coupled with only a small erosion of ee. The authors propose a mechanism in which transmetalation proceeds via complex 365, in which the alcohol of L95 undergoes transesterification with the boronic acid. B to Cu transmetalation, followed by oxidative addition to phosphate 368, would generate σ-allylcopper complex 369. Reductive elimination would then provide product 370 and regenerate the Cu catalyst.
Figure 75.
Cu-catalyzed asymmetric allylic arylation with arylboron reagents.
In 2012, Hoveyda and co-workers turned their attention from alkenylaluminum reagents to alkenylboron reagents for Cu-catalyzed asymmetric allylic substitution.293,294 Using NHC ligand L96, allylic phosphate 371 could be coupled with alkenylboronate esters in good yield and high enantio- and regioselectivity (Scheme 39a). Compared to the coupling of vinylaluminum species, these reactions require higher temperatures because of the decreased nucleophilicity of the organoboron. However, the method tolerated functional groups that were not compatible with the vinylaluminum chemistry. Both (Z)-alkenylboron and trisubstituted alkenylboron reagents couple efficiently, readily enabling the synthesis of several natural products. More recently, the Cu/L97-catalyzed coupling of allenylboron reagents with allylic phosphates was disclosed (Scheme 39b).295 These reactions could be conducted at lower temperatures, presumably because of the decreased steric hindrance at the nucleophile. Excellent fidelity for allenylation over propargylation was observed in all cases. A study of ligand effects illustrated the importance of the sulfonate group on L97 for providing γ-selectivity.
Scheme 39. Cu-Catalyzed Asymmetric Allylic Substitution with Other C(sp2)-Hybridized Boron.
Iridium complexes have emerged as privileged catalysts that provide branched products in a wide variety of allylic substitution reactions with heteronucleophiles and stabilized nucleophiles.264,265 Carreira and co-workers were able to extend this reactivity to alkenyl trifluoroborates, finding that Ir/L98 catalyzes the reaction of simple allylic alcohols (374) with alkenyl trifluoroborates to furnish skipped diene products in good yields and remarkable levels of enantioinduction (Figure 76).296 The nBu4HSO4 was proposed to perform two functions: Brønsted acid activation of the allylic alcohol and solubilization of the trifluoroborate salt through phase-transfer catalysis. Both aryl and aliphatic vinyl trifluoroborates are competent in the reaction; however, the scope of electrophiles was limited to aryl- and heteroaryl-substituted allylic alcohols.
Figure 76.
Ir-catalyzed asymmetric allylic vinylation with vinylboron reagents.
Building on their successful Ir-catalyzed asymmetric allylic vinylation, Carreira and co-workers turned to developing the analogous alkynyl coupling reaction. Using the same Ir/L98 catalyst system, allylic alcohol 376 could be coupled to a variety of aryl- and alkyl-substituted alkynyl trifluoroborates (Figure 77).297 Slight modifications to the previously disclosed vinylation conditions were required: instead of using nBu4HSO4 as an additive, the combination of phase transfer catalyst nBu4NBr and Brønsted acid CF3CO2H was used. The ability to form tertiary stereocenters is complementary to Hoveyda’s Cu-catalyzed coupling of alkynylaluminum reagents, a method that was limited to the synthesis of all-carbon quaternary centers.277,278
Figure 77.
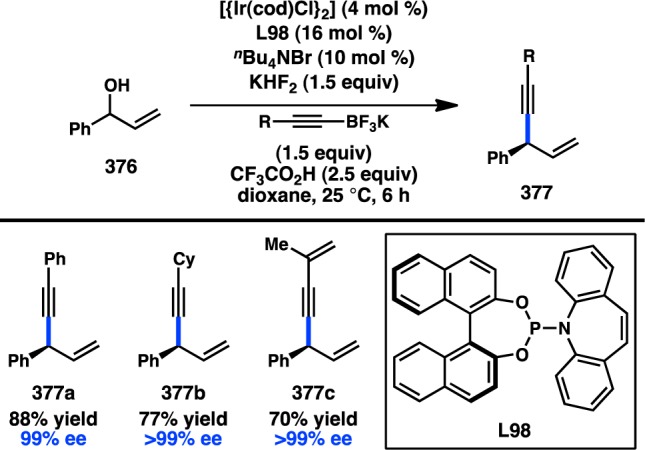
Ir-catalyzed asymmetric allylic alkynylation.
The asymmetric allylic functionalization by C(sp3)-hybridized boron reagents has only recently witnessed success in both Pd- and Cu-catalyzed reactions. In 2010, Morken and co-workers disclosed the use of allylboron reagents in Pd-catalyzed asymmetric allyl–allyl couplings. Using BIPHEP ligand L99, the Pd-catalyzed coupling of allyl carbonate 378 and allylboronate delivered 379 in good yield and high ee; notably, both isomers of the allyl carbonate starting material were found to converge to the same product (Figure 78). The authors propose that bidentate ligand L99 forces the allyl groups of a bisallylpalladium complex 381 to adopt η1 rather than η3 binding modes. Complex 381 is now poised to undergo a 3,3′ reductive elimination to furnish stereogenic product 383.298 The authors hypothesize that equilibration between 381 and 382 should favor the less sterically demanding metal complex. Furthermore, the bite angle of the ligand could affect the ratio of branched (383) to linear (384) products, with small-bite-angle ligands increasing the C1–C1′ separation and disfavoring 1,1′ reductive elimination. Indeed, ligands with small bite angles favored branched products, and those with larger bite angles were unselective.
Figure 78.
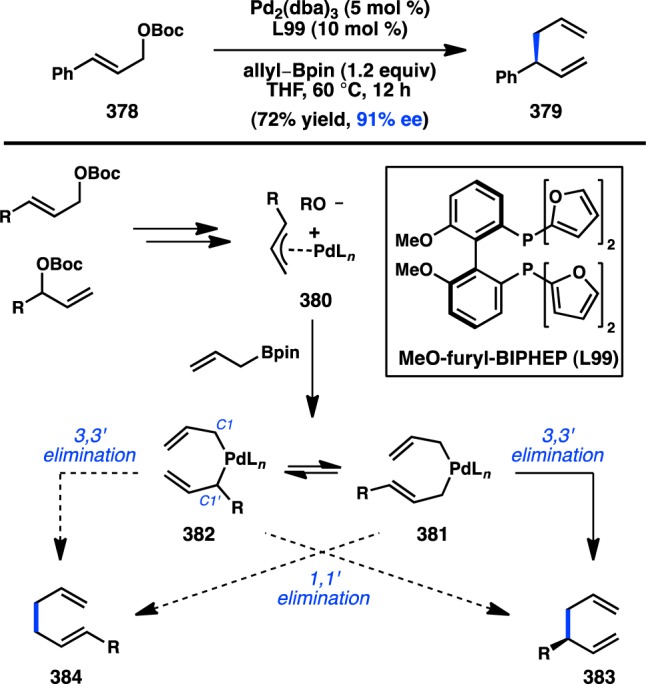
Pd-catalyzed enantioselective allyl–allyl coupling.
Having devised a paradigm for allyl–allyl coupling, Morken and co-workers subsequently expanded the scope of the transformation to form products with all-carbon quaternary or vicinal tertiary centers. Substituted allylboronates were found to react with allylic chlorides (385) to form coupled products (e.g., 388) with both enantio- and diastereocontrol (Figure 79).299 The increased steric hindrance of the boronate nucleophile required the addition of CsF to accelerate transmetalation. Products with all-carbon quaternary centers (388b) could be generated from tertiary allylic carbonates (386) with little erosion of ee.300 Lastly, a bisboronate starting material can be employed, leading to synthetically versatile 388c in high ee.301 Alkyl-substituted allylic electrophiles were also tolerated when a quinoxaline ligand was utilized. A mechanistically distinct Cu-catalyzed allyl–allyl coupling was later reported by Feringa and co-workers.250
Figure 79.
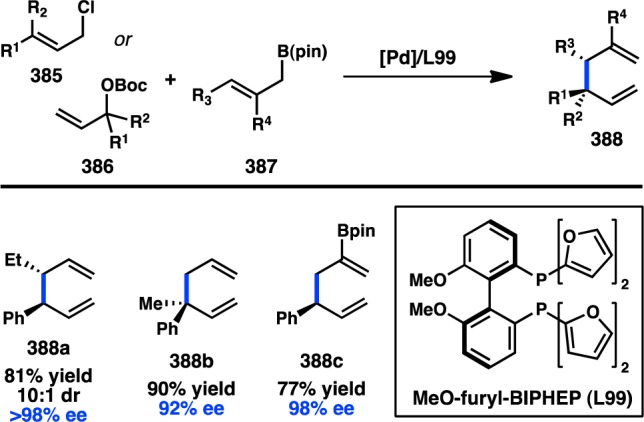
Pd-catalyzed enantioselective allyl–allyl coupling: expansion of the substrate scope.
Morken’s studies on allylboron reagents prompted the discovery of the first enantioselective intramolecular coupling of an allylboronate and an aryl halide. Phosphoramidite L100 was identified as the optimal ligand, providing indane 390 in 80% ee (Figure 80).302 Interestingly, the reversed-polarity intramolecular coupling of an allyl halide and an arylboronate proceeded with similar enantioselectivity. Additional studies revealed that reductive elimination is the stereochemistry-determining step, which occurs after equilibration of transmetalation adducts 392 and 393. Reaction conditions that either slowed reductive elimination or accelerated isomerization delivered higher ee’s of 390. The origin of enantioselectivity stands in stark contrast to the intermolecular coupling of allylboron reagents developed by Yamamoto and Miyaura.107
Figure 80.
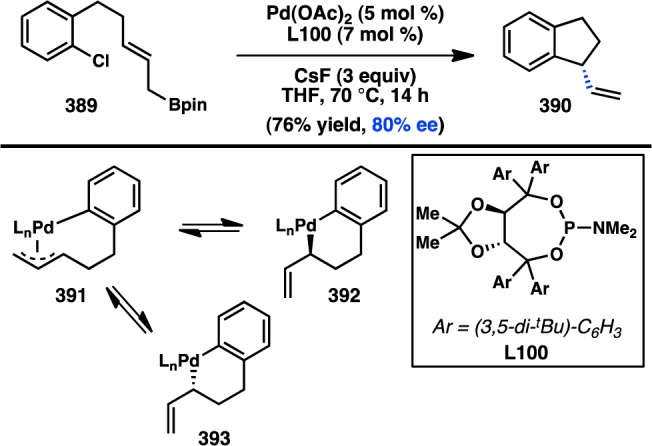
Enantioselective intramolecular coupling of an allylboronate.
In 2012, Sawamura and co-workers reported the first enantioselective allylic alkylation using simple alkylboron reagents. Under Cu-catalyzed conditions derived from their previously reported stereospecific coupling of allylic phosphates and alkylboron reagents,303 the authors were able to couple allylic chloride 395 with alkylborane 394 to deliver 396 in excellent yield with high enantio- and regioselectivity (Scheme 40a).304 The DTBM group on L101 was necessary for both high selectivity and high reactivity; it was proposed this moiety might assist in deaggregation of alkylcopper species, increasing the concentration of active monomeric Cu complexes. Under similar conditions that instead utilize L102, trisubstituted allylic electrophile 398 reacts with alkylboron 397 to form a compound bearing an all-carbon quaternary center in 90% ee (Scheme 40b).305
Scheme 40. Cu-Catalyzed Enantioselective Allylic Alkylation Using Unactivated Alkylborons.
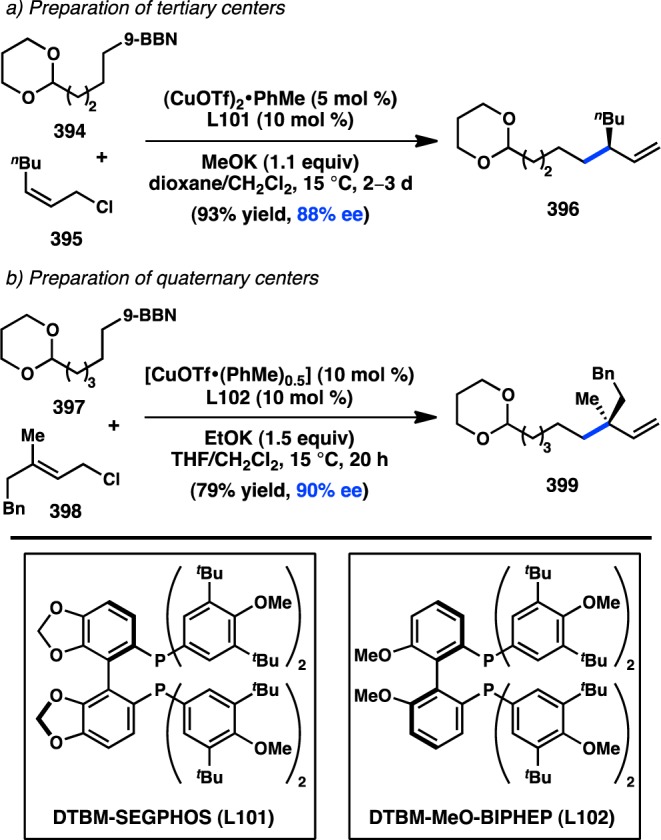
The authors propose that the aforementioned reactions proceed by an addition/elimination mechanism (Figure 81). Transmetalation of the Cu catalyst with the “ate” complex of the alkylboron reagent would give neutral alkylcopper species 400, which would engage allylic chloride 401 through carbocupration, rather than anti SN2′ oxidative addition. Asymmetric induction is thought to arise from differentiation of the two prochiral faces of the olefin by the catalyst. β-Elimination of Cl, possibly assisted by Lewis acidic 9-BBN-OR, would produce the observed product and re-form the active catalyst.
Figure 81.
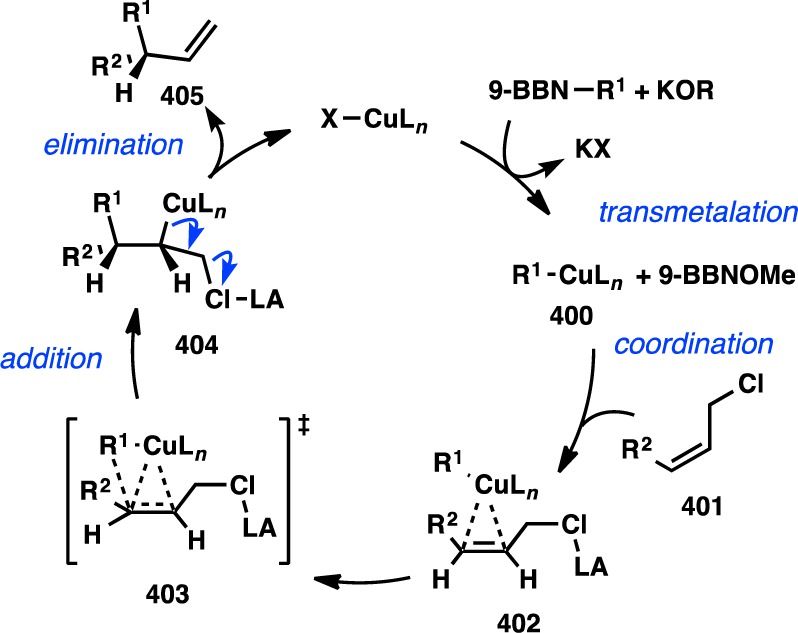
Mechanism of allylic alkylation using alkylboron reagents.
4.1.5. Organolithium Reagents
The high reactivity of organolithium reagents has traditionally prevented their use in asymmetric allylic alkylation reactions: any successful transformations would require conditions that avoid or outcompete unselective background reactivity. By performing spectroscopic studies to understand the parameters that favor catalytically active alkylcopper species and disfavor reactive organolithium aggregates or lithium cuprates, Feringa and co-workers were able to develop the first enantioselective cross-coupling of alkyllithium reagents.306 The key was to exclude ethereal solvents and lower the temperature to −80 °C, under which conditions ligated alkylcopper species could be selectively formed. Thus, slow addition of an organolithium reagent to a dichloromethane solution of allylic halide (406) and Cu/L78 at −80 °C provided alkylated product 407 in high yield with good regioselectivity and excellent enantioinduction (Figure 82).306,307 High chemoselectivity was demonstrated through the coupling of substrates bearing aryl halides and even a free alcohol. The use of phosphoramidite L103 also delivered products with good enantioselectivity: in addition to tertiary centers, the method could be expanded to the synthesis of all-carbon quaternary centers.308 A recent study has shown that addition of a Lewis acid can enable the use of methyl or benzyl ethers as leaving groups, increasing the scope of the electrophile.309
Figure 82.
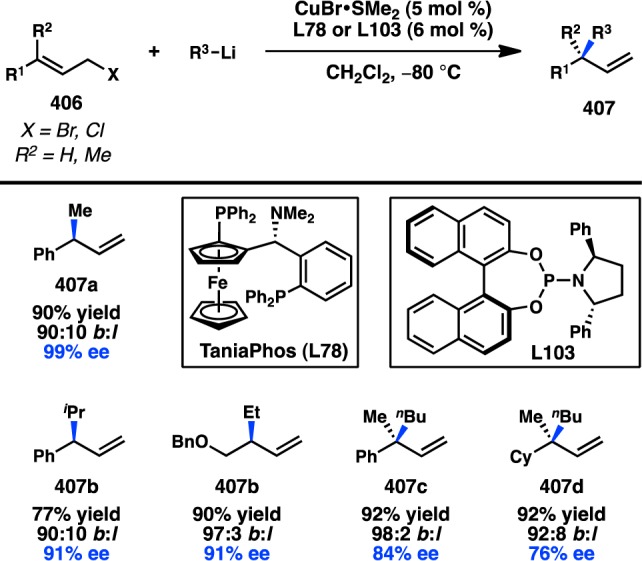
Asymmetric allylic alkylation using organolithium reagents.
4.2. Enantiospecific Allylic Substitutions
Early efforts toward developing stereospecific substitution reactions of allylic electrophiles focused on the use of soft nucleophiles, such as malonates, or hard nucleophiles, such as alkoxides. Whereas enantiospecific allylic substitution reactions using stoichiometric copper nucleophiles have been known for decades and have been exhaustively studied, the corresponding transition-metal-catalyzed reactions of organometallic reagents were much slower to develop.
4.2.1. Organozinc Reagents
The first report of a transition-metal-catalyzed stereospecific allylic substitution reaction using an organometallic nucleophile was disclosed in Hayashi and co-workers’ investigations of Pd-catalyzed reactions of allylic acetates (Scheme 41).310 Pd-catalyzed reaction of acetate 408 with phenylzinc bromide delivered arylation product 410 in good yield and with modest es favoring inversion of stereochemistry. Exposure of isomeric acetate 409 to the same conditions also produced 410 as the major product, albeit with lower es. Although both processes proceed with inversion of stereochemistry, the observation that both 408 and 409 converge to a common product suggests that they proceed through a similar π-allylpalladium intermediate.
Scheme 41. Pd-Catalyzed Stereospecific Allylic Arylation with Arylzinc Reagents.
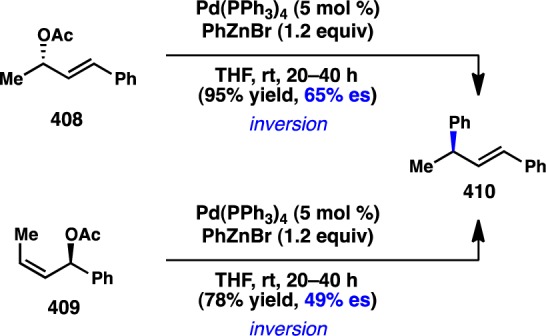
Nearly two decades later, Evans and Uraguchi reported the Rh-catalyzed arylation of allyl carbonates with arylzinc halides to give product 412 in good yield and good regioselectivity (Scheme 42).311 This transformation was found to be completely enantiospecific, giving the product with inversion of configuration. Optimization of additives was critical to achieving good regiospecificity and reproducible yields, with the combined use of LiBr and the exogenous diene ligand dibenzylideneacetone (dba) providing the best results.
Scheme 42. Rh-Catalyzed Stereospecific Allylic Arylation with Arylzinc Reagents.
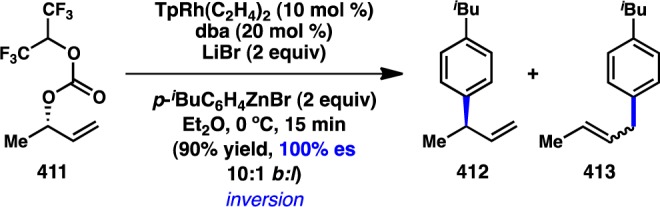
4.2.2. Organomagnesium Reagents
Investigations of transition-metal-catalyzed stereospecific allylic substitution reactions with organomagnesium reagents have focused on rendering the long-established chemistry of stoichiometrically generated cuprate reagents catalytic. The Breit group has focused on developing allylic electrophiles with phosphine-containing leaving groups for γ-selective coupling reactions.312−315 Treatment of enantioenriched o-(diphenylphosphino)benzoyl (o-dppbz) ester 414a with nBuMgBr in the presence of substoichiometric CuBr·SMe2 furnished cyclohexene 415, bearing an all-carbon quaternary center (Scheme 43). The product is formed in high yield with excellent regioselectivity and perfect enantiospecificity for the syn-substitution product. The high selectivity for γ-syn-addition of the nucleophile is proposed to result from the ability of the o-dppbz ester to direct the approach of the copper nucleophile. Consistent with this proposal, (stoichiometric) cuprate addition to the analogous phosphine oxide (414b) provides exclusively the γ-anti-product. The Breit group has employed this method in the synthesis of various polyketide motifs and natural products, including (+)-bourgeanic acid and (R,R,R)-α-tocopherol, to demonstrate its utility.316−319
Scheme 43. o-dppbz-Directed Stereospecific Allylic Alkylation with Grignard Reagents.
Several groups have investigated Cu-catalyzed stereospecific α-selective allylic substitution reactions. In 2010, Bäckvall and co-workers reported their investigations of the CuI-catalyzed coupling of enantioenriched cinnamaldehyde-derived ester 416 with alkyl and aryl Grignard reagents to give styrenyl products 417 and 418 with inversion of stereochemistry (Figure 83a). High levels of α-selectivity and excellent es were reported for a range of nucleophiles, although alkyl Grignard reagents reacted with somewhat lower levels of regioselectivity.320−322 Bäckvall rationalized that the α:γ-selectivity of the transformation is determined by the relative rates of reductive elimination (k1) versus π-allyl formation (k2, k2′) (Figure 83, b). If k1 > k2, then γ-alkylated product 421 is obtained; however, if the rate of reductive elimination is slow, formation of π-allyl 422 can give rise to the thermodynamically stable α-product, 423. The intermediacy of an electron-rich cuprate is expected to slow reductive elimination further, favoring delivery of the α-product. The authors ascribe the erosion of enantiospecificity in some substrates to π-allyl formation via conformation 424, giving rise to undesired 426.
Figure 83.
α-Selective stereospecific allylic substitutions with organomagnesium reagents.
In addition to the allylic esters investigated by the Bäckvall group, other laboratories have begun exploring alternative electrophiles for α-selective Cu-catalyzed allylic substitution reactions with organomagnesium reagents. Tian and co-workers disclosed the Cu-catalyzed arylation of styrenyl sulfonamide 427 with PhMgBr to prepare 410 in good yield, high α-selectivity, and perfect es (Scheme 44).323 Although 427 is the only reported substrate, these preliminary results show promise for the use of sulfonamides as leaving groups.
Scheme 44. α-Selective Allylic Arylation of Enantioenriched Sulfonamides.
Wu and co-workers reported a Cu-catalyzed α-selective allylic substitution of phosphorothioate ester 428 (Scheme 45).324 These studies focused on the use of alkylmagnesium reagents as nucleophiles. The authors propose that reaction through dialkylcopper species bearing an electron-rich ligand favors equilibration to the π-allyl complex, ultimately delivering the thermodynamic product, 429. The phosphorothioate leaving group was critical for imparting both excellent α-selectivity and high yields. It was hypothesized that the phosphorothioate serves as an electron-rich ligand on Cu, further favoring α-selectivity. A single enantioenriched substrate (428) was shown to react with modest es to furnish the stereoinverted product. This transformation is currently limited by a lack of methods to prepare the enantioenriched substrates.
Scheme 45. Stereospecific Allylic Alkylation of Phoshorothioate Esters.
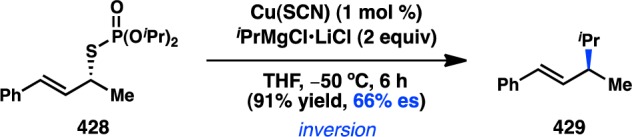
4.2.3. Organoboron Reagents
The Sawamura laboratory has pioneered the development of transition-metal-catalyzed stereospecific allylic substitution reactions using arylboron nucleophiles.325−328 In 2008, they reported that cationic (phen)PdII(OAc)·SbF6 (432) catalyzes the reaction between allylic benzoate 430 and an arylboronic acid to give γ-arylation product 431 with retention of stereochemistry (Figure 84). Product 431 is obtained in good yield and excellent enantiospecificity. The authors propose a mechanism involving ester-directed arylpalladation of the alkene to give 436, followed by preferential β-acetoxy elimination as opposed to β-hydride elimination. The reaction exhibits broad functional group tolerance for both coupling partners. Moreover, the γ-selectivity to form gem-diarylalkanes, which overrides the thermodynamic preference for conjugated products via α-arylation, represents complementary selectivity to many of the Cu-catalyzed methods.
Figure 84.
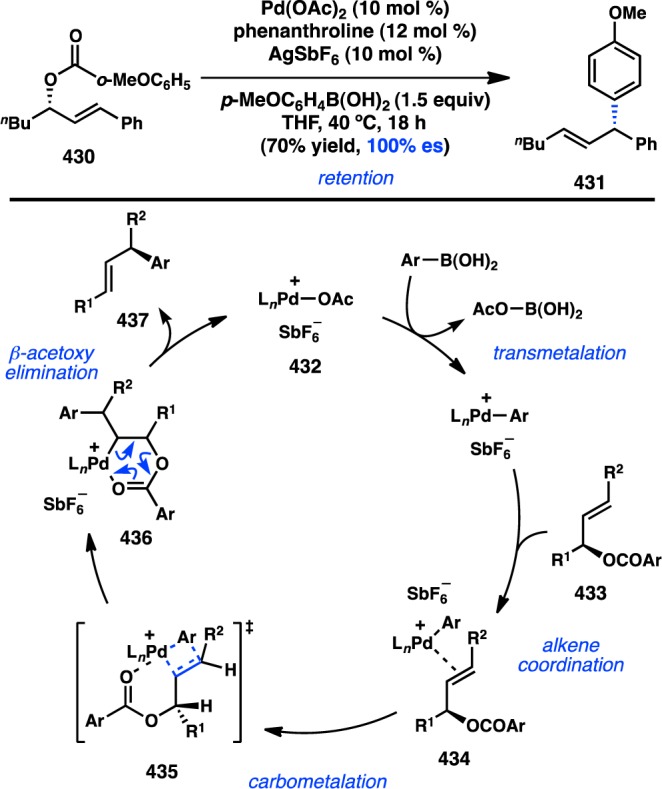
Pd-catalyzed allylic γ-arylation with arylboron nucleophiles.
The Zhang group has investigated α-selective Pd-catalyzed enantiospecific allylic substitution reactions with arylboron nucleophiles. In 2012, they reported the Pd/rac-BINAP-catalyzed reactions of allyl carbonates (438) with arylboronic acids to give α-substitution products (410) in good yield and perfect enantiospecificity (Figure 85a).329 Oxidative addition of Pd to the carbonate electrophile proceeds with extrusion of carbon dioxide to afford π-allyl complex 440 (Figure 85b). This activation mode of the allyl electrophile is distinct from the carbometalation previously invoked by Sawamura.325 Transmetalation of phenylboronic acid delivers intermediate 441, which undergoes reductive elimination to furnish the more thermodynamically favored conjugated α-substitution product 410. This transformation was demonstrated to be general, tolerating a variety of functional groups and sterically encumbered substrates. The authors have subsequently reported that the reaction can be conducted in aqueous conditions and without exogenous ligand.330
Figure 85.
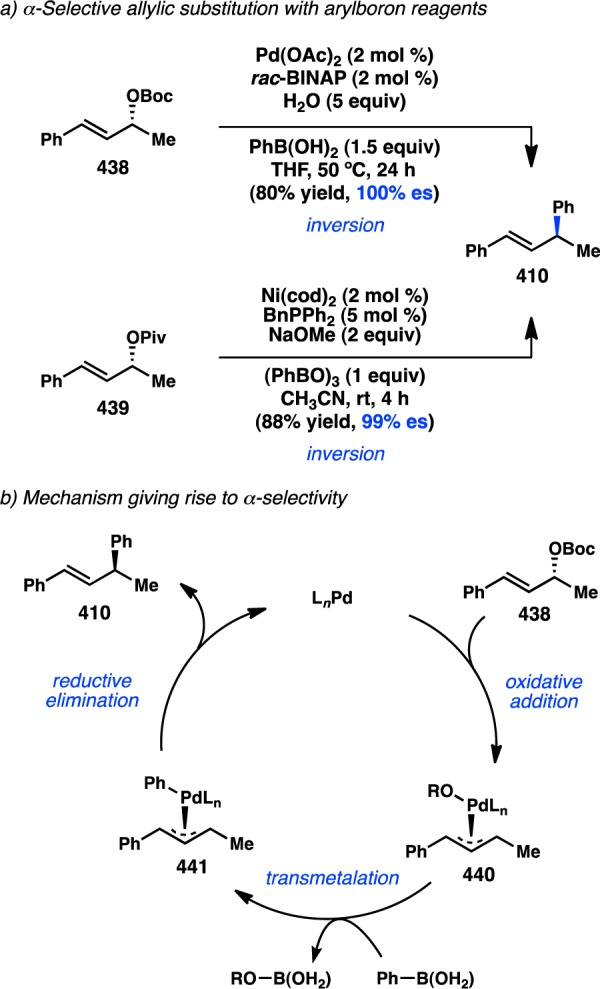
Carbonate leaving groups in allylic arylation.
Recently, the Watson group has reported that the same transformation can be achieved using Ni catalysis. When Ni/BnPPh2 is used as the catalyst, allylic pivalate 439 can be coupled to phenylboroxine in good yield and high es (Figure 85a).331 As in the Pd-catalyzed transformation developed by Zhang, the product is formed with inversion of stereochemistry. The reaction is conducted under mild conditions and exhibits a broad substrate scope.
A limitation of the Pd-catalyzed reactions discussed above is that (Z)-allylic substrates react with poor stereofidelity. To address this shortcoming, Sawamura and co-workers have disclosed a related Cu-catalyzed reaction of (Z)-allylic phosphates (e.g., 442) and arylboronates (Scheme 46).332 Using a simple catalyst system generated from CuCl and acetylacetone, the coupled products are formed in good yield with high γ-selectivity and enantiospecificity. As is often observed in Cu-catalyzed allylic substitution, the products are formed with inversion of stereochemistry.
Scheme 46. Cu-Catalyzed Allylic γ-Arylation with Arylboron Nucleophiles.
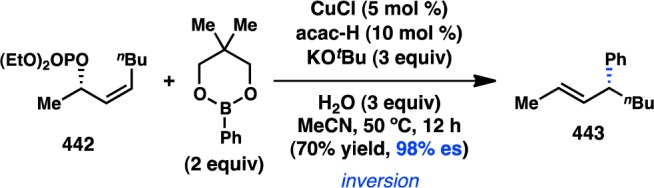
In an effort to simplify substrate preparation and minimize waste, attention has recently turned to the development of γ-selective arylations of electrophiles with Lewis-basic leaving groups. Tian and co-workers reported the arylation of allylic amines (444), whereas both Zhang and Tian have independently disclosed the analogous reaction of free allylic alcohols (445) (Figure 86).333−335 These reactions are proposed to proceed by complexation of the leaving group to the boronic acid nucleophile to form “ate” complex 447, activating the electrophile toward SN2′ oxidative addition. Following transmetalation of π-allyl complex 448 with the arylboronate, reductive elimination furnishes the α-selective products (410). These reactions generally proceed with high enantiospecificity and regioselectivity when bidentate ligands such as TMEDA or rac-BINAP are employed.
Figure 86.
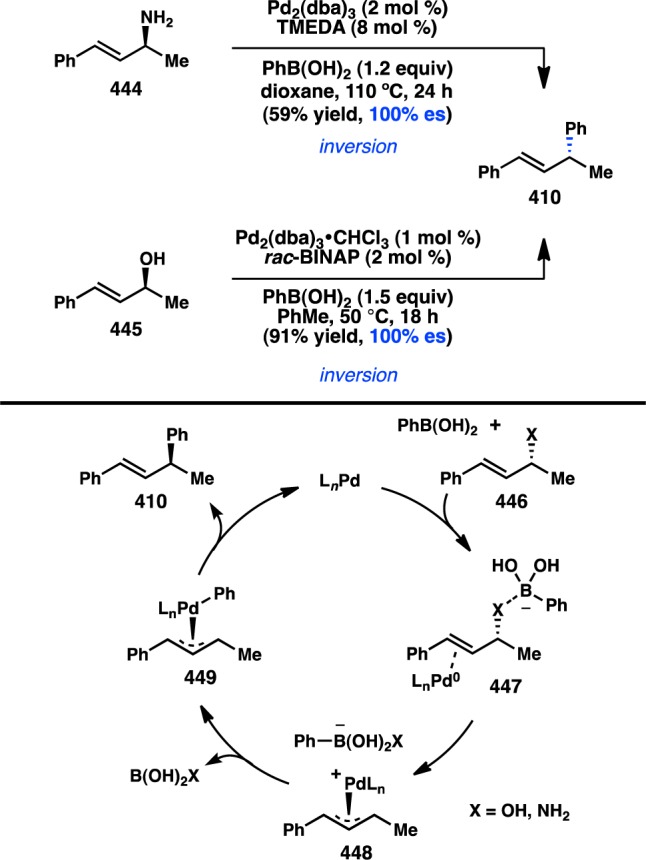
Amine and alcohol leaving groups in allylic arylation.
Building on this mechanism, the Sigman group has developed a novel Pd-catalyzed cross-coupling between homoallylic tosylates and arylboronic acids.336 Using a Pd/L104 complex as the catalyst, tosylate 450 can be coupled with phenylboronic acid to give 451, with inversion of stereochemistry, in good yield and high es (Figure 87). The authors propose that, following oxidative addition, the alkylpalladium intermediate 452 undergoes β-hydride elimination to afford 453, which can reinsert Pd to generate 454 and intercept the standard allylic coupling mechanism. Importantly, the good levels of es observed in this transformation suggest that the olefin remains tightly bound to Pd following β-hydride elimination. It was determined that bulky substituents α to the original position of the tosylate were critical to observe high es in this substitution reaction.
Figure 87.

Stereospecific coupling of homoallylic tosylates.
The use of alkylboron nucleophiles in transition-metal-catalyzed stereospecific allylic substitution reactions has only recently been developed. In 2010, Sawamura and co-workers disclosed a γ-selective Cu-catalyzed reaction of alkyl-9-BBN reagents and enantioenriched (Z)-allylic phosphates (442) (Scheme 47).303 The stereochemical course of the reaction proceeded with overall inversion of configuration and high selectivity for the (E)-alkene, but modest enantiospecificity.
Scheme 47. Initial Report of Enantiospecific Allylic Substitution with Alkylboron Reagents.
The same group subsequently investigated how reaction parameters such as the solvent and base influenced the enantiospecificity of the reaction. Employing silyl-substituted electrophile 457, two sets of conditions that enable stereospecific formation of either (S)-458 or (R)-458 were developed (Figure 88).337 Thus, Cu-catalyzed coupling of (Z)-allylic phosphate 457 and an alkyl-9-BBN in THF with tBuOK as the base furnished (S)-458 in good yield and high es, whereas conducting the same reaction in toluene with MeOK provides (R)-458 with similar levels of es. It is proposed that use of a small base, MeOK, enables coordination of Cu to the leaving group, which directs a syn oxidative addition (i.e., to the same face as the leaving group) via cyclic transition state 459. On the other hand, the bulky base tBuOK precludes coordination of Cu to the “ate” complex, instead favoring anti oxidative addition according to open transition state 460. Along with alkyl-substituted alkenes, this method tolerates both di- and trisubstituted alkenylsilanes, furnishing the corresponding secondary and tertiary chiral silanes, respectively.
Figure 88.
Enantiodivergent allylic alkylation with alkylboron reagents.
Morken and co-workers have reported Pd-catalyzed stereospecific allylation reactions of propargylic and allylic electrophiles, giving rise to 1,5-enynes and -dienes, respectively. In the former case, Pd/rac-BINAP-catalyzed substitution of propargylic acetate 461 with allyl pinacol boronate is proposed to occur by anti SN2′ oxidative addition of Pd0 to 461, followed by transmetalation to generate η1-allenylpalladium species 463 (Figure 89a). Subsequent 3,3′ reductive elimination affords enyne 462 with excellent es and selectivity for formation of the alkyne over the allene.338 Consistent with this mechanism, the reaction proceeds with inversion of stereochemistry. The authors have also disclosed a related kinetic resolution, in which a chiral ligand enables the selective allylation of one enantiomer of the propargylic substrate, allowing recovery of enantioenriched starting material.339 More recently, the Morken laboratory reported the Pd-catalyzed stereospecific substitution of allylic acetate 464 with allyl pinacol boronate to deliver enantioenriched 1,5-diene 465 in good yield and with excellent es (Figure 89b).340 This reaction can also be applied to tertiary acetates, which enables the production of 1,5-dienes bearing all-carbon quaternary centers. This transformation provides a notable alternative to the Cope rearrangement for the synthesis of chiral 1,5-dienes, particularly because there are no enantioselective examples of Cope rearrangements of unfunctionalized substrates (such as those required to produce 465).
Figure 89.
Allylation with allylboronates to form 1,5-dienes and -enynes.
5. Enantiocontrolled Cross-Coupling Reactions To Prepare Molecules with Axial or Planar Chirality
Although centrochirality—arising from a stereogenic C(sp3)-hybridized atom—is most commonly encountered in small organic molecules, other chirality modes include axial and planar chirality. In the case of allenes, axial chirality arises from hindered rotation about a C=C double bond. Alternatively, in atropoisomerism, axial chirality emerges as a result of hindered rotation about a C–C single bond because of either conformational constraints (as in some macrocyclic natural products) or significant steric encumberance about the bonding axis. Most commonly for biaryl systems, ortho substitution prevents rotation about the biaryl axis, allowing isolation of enantiopure atropisomers. Atropisomers maintain a half-life to racemization of at least 1000 s, corresponding to a minimum energy barrier of 93.5 kJ/mol at 300 K.341 Stable atropisomers are expected in biaryls containing four ortho substituents and, in many cases, those with only three substituents. Isolable atropisomers can be found in molecules with only two ortho groups if those groups are sufficiently large.342 The energy of interconversion has been shown to be well-correlated with the van der Waals radii of the blocking groups, demonstrating the steric nature of the effect.343 Similarly, planar chirality may result from the unsymmetrical arrangement of groups about a plane of symmetry. Although a range of elegant diastereoselective and oxidative approaches to control atropochirality have been developed, this discussion will be limited to strategies involving enantioselective and enantiospecific cross-coupling.
5.1. Enantiocontrolled Preparation of Axially Chiral Biaryls
Three general strategies have been developed for the enantioselective preparation of axially chiral biaryl scaffolds using transition-metal-catalyzed cross-coupling: (1) C(sp2)–C(sp2) coupling in which axial chirality is produced in the formation of the biaryl linkage, (2) enantioposition-selective cross-coupling of a prochiral bisfunctionalized biaryl scaffold, usually a biaryl ditriflate, and (3) dynamic kinetic asymmetric transformation by cross-coupling of a racemic, axially chiral starting material.
5.1.1. Organomagnesium Reagents
In 1975, Kumada and co-workers reported the Ni-catalyzed cross-coupling of ortho-substituted aryl electrophiles and ortho-substituted aryl Grignard reagents to make biaryl products.344 Recognizing that this chemistry could provide atroposelective entry to molecules with axial chirality, several chiral ligands were investigated in the coupling of naphthyl partners 466 and 467 (Figure 90). BPPFA (L17) was found to deliver 468 in 5% ee, a low but potentially promising value. Several years later, Kumada reported that NAPHOS (L6) provides 468 in 13% ee,24 whereas Brunner found that phosphine L105 delivers 468 in 50% ee.41 Subsequent to Kumada’s work, Frejd investigated the related Pd-catalyzed cross-coupling which produced 468 in 45% ee when BIPHEMP (L69) was used as the ligand.345
Figure 90.
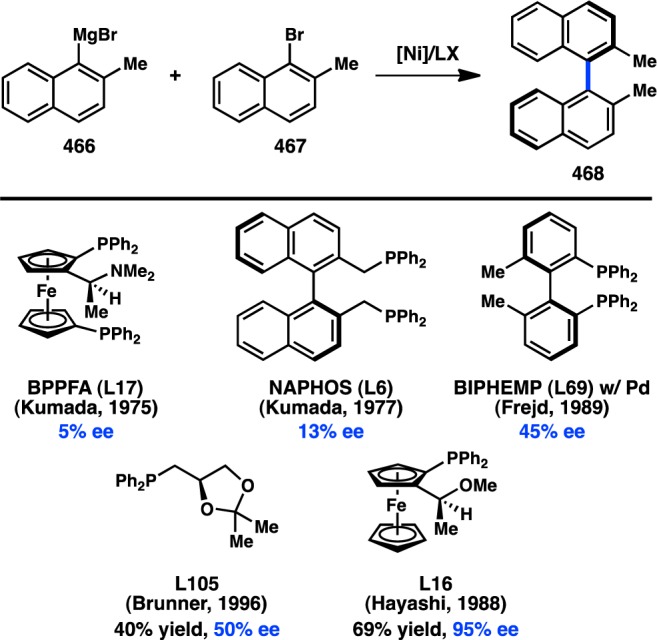
Early ligands for asymmetric C(sp2)–C(sp2) Kumada–Corriu coupling.
The first highly atroposelective examples of Ni-catalyzed biaryl formation emerged from the work of Hayashi, Ito, and co-workers in 1988, when they reinvestigated chiral ferrocenylphosphine ligands for the coupling between 466 and 467.346,347 After observing that monodentate ligands were more catalytically active than bidentate ligands, they identified methoxy-containing ligand L16, which produced binaphthyl 468 in good yield and excellent ee. Critically, a ligand lacking the methoxy group delivered racemic coupling products. The methoxy substituent was proposed to precoordinate the Grignard reagent during a selectivity-determining transmetalation step. A similar proposal was put forth for the DYKAT/cross-coupling of C(sp3) organomagnesium reagents.34
Hayashi and co-workers also investigated transition-metal-catalyzed desymmetrizing cross-coupling reactions to generate enantioenriched biaryl compounds. When chiral Pd complex 471 was used as a catalyst, cross-coupling of prochiral ditriflate 469 with PhMgBr furnished monofunctionalized product 470 in 84% yield and 90% ee (Figure 91).348,349 The major byproduct was diarylated compound 472. The ee of 470 was found to be 85% at low conversion, which increased over the course of the reaction because of a subsequent kinetic resolution of the two monoarylated atropisomers. The minor enantiomer was observed to react approximately 5 times faster than the major enantiomer to form 472, leading to an enhancement in the ee of 470. LiBr was found to increase both the reaction rate and the enantioselectivity.
Figure 91.
Enantioposition-selective Kumada–Corriu coupling.
Following the successful development of the Pd-catalyzed enantioposition-selective arylation of ditriflate 469,348,349 Hayashi and co-workers investigated the analogous atroposelective alkynylation. Using the same chiral Pd catalyst, 471, cross-coupling of biaryl ditriflate 469 and alkynylmagnesium bromide delivered 473 in 92% ee after 6 h (Scheme 48).350 Increasing the reaction time furnished 473 in enantiopure form, albeit in lower yield, because of the kinetic resolution of the monofunctionalized product in a second cross-coupling event.
Scheme 48. Enantioposition-Selective Cross-Coupling of Alkynyl Coupling Partners.
Exploiting the ability of Ni to insert into the C–S bond of benzothiophene, Hayashi and co-workers explored the Ni-catalyzed Kumada–Corriu coupling of 474 with aryl Grignard reagents to give enantioenriched biaryl thiols (Figure 92a).351,352 Dinaphthothiophene (474) is generally considered to be an achiral compound because of the low barrier to flipping about the axial axis of symmetry. When PHOX ligand L52 was employed, the Ni-catalyzed ring-opening/cross-coupling proceeded in high yield and ee. The atroposelectivity was strongly influenced by the identity of the Grignard reagent; alkylmagnesium reagents reacted with poor selectivity. On the basis of this observation, the authors propose that the reaction proceeds by a DYKAT of the rapidly equilibrating oxidative addition products (M)-476 and (P)-476, in which the transmetalation step sets the axial chirality. The reaction could also be extended to dibenzothiophene coupling partners to generate axially chiral biphenyl products. Despite efforts to improve the selectivity of the reaction between 474 and alkyl Grignard reagents, the best conditions identified to date, which utilize chiral phosphine L106, deliver 477 with only moderate enantioselectivity (Figure 92b).
Figure 92.
Atroposelective ring openings of dinaphthothiophene.
5.1.2. Organozinc Reagents
The first atroposelective Negishi cross-coupling to prepare an axially chiral biaryl was reported in 2006 by Espinet and co-workers.353 Using Pd2(dba)3 and chiral ferrocenylphosphine L13, diarylzinc 478 was coupled to bromonaphthalene 467 to furnish 468 in excellent yield and 85% ee (Scheme 49). This result was comparable to that of a related Pd/L13-catalyzed Suzuki–Miyaura coupling to form 468 (see Scheme 50), but a shorter reaction time was achieved.354 The reaction conditions were modified to allow microwave-heating-induced acceleration of the transformation; although the coupling is complete within 1 h, the ee of 468 falls to 60%.355
Scheme 49. Stereoselective Negishi Coupling of Naphthylzinc Reagents.
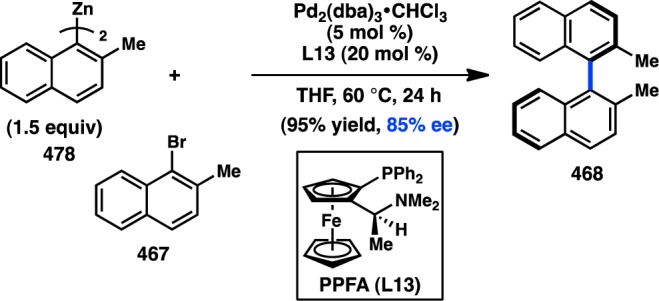
Scheme 50. Cammidge’s Approach to Asymmetric Biaryl Formation.

5.1.3. Organoboron Reagents
Arylboron reagents are attractive partners for cross-coupling reactions because they are easy to handle and store, generate nontoxic byproducts, and are highly functional group tolerant. On the other hand, transition-metal-catalyzed cross-coupling reactions of hindered, ortho-substituted arylboron species often require long reaction times and high temperatures and can suffer from competitive protodeborylation. Likely because of these reasons, atroposelective Suzuki–Miyaura cross-couplings have only emerged in the past 15 years. In contrast, asymmetric variants of the Kumada–Corriu coupling have been known for twice as long. Refinements in ligand design and improved reaction conditions for the racemic formation of hindered biaryls were key to the ultimate discovery of an asymmetric variant.356−358
Following a report from Nicolaou and co-workers describing the use of chiral ligands to enhance the atropodiastereoselectivity of a Pd-catalyzed Suzuki–Miyaura aryl–aryl cross-coupling in the synthesis of vancomycin,359 the laboratories of Cammidge and Buchwald independently disclosed enantioselective reactions to prepare simple biaryl systems. In 2000, Cammidge and co-workers reported the Pd-catalyzed coupling of boronate 479 and iodonaphthalene 480, delivering 468 in 85% ee when L13 was used as a chiral ligand (Scheme 50).354,355,358,360,361 The authors propose that the axial chirality is set in the transmetalation step and that the overall reaction occurs under kinetic control. Consistent with Hayashi’s proposal, the authors hypothesize that the nitrogen atom of L13 can coordinate to the organoboron reagent during or before transmetalation.346
Concurrent with Cammidge’s study, Buchwald and co-workers reported a Pd-catalyzed atroposelective Suzuki–Miyaura cross-coupling between 1-bromonaphthalene 481 and arylboronic acids. When chiral biarylylphosphine L107 was employed, the coupling with (o-methylphenyl)boronic acid provided biaryl 483 in 87% ee (Figure 93).362,363 The presence of the adjacent phosphonate ester was important for achieving high selectivity. A crystal structure of an oxidative addition product revealed a potential interaction between Pd and the oxygen atom of the phosphorus substituent.364 The authors proposed that this dative interaction might guide a selectivity-determining reductive elimination. Qiu and co-workers have subsequently developed bridged, axially chiral ligands (L108 and L109) to effect the same transformation in similar ee.365,366 Likewise, Suginome and co-workers have synthesized chiral helical polymers capable of catalyzing the formation of phosphonate-bearing products in the presence of Pd.367−369 In addition to accomplishing high enantioselectivity, Suginome showed that the sense of chiral induction can be reversed simply through a change of solvent.
Figure 93.
Buchwald’s approach to asymmetric biaryl formation.
On the basis of the hypothesis that the phosphonate of 481 coordinates to Pd and enhances the atroposelectivity of biaryl bond formation, Buchwald and co-workers investigated the asymmetric Suzuki–Miyaura cross-coupling of other substrates bearing proximal coordinating groups, including amide and nitro functional groups (Scheme 51).364,370,362,371 A more recent report by Blakemore and co-workers disclosed that biquinoline-derived phosphine L110 provides biaryl 491 in good yield but modest ee (Scheme 52). Iwasawa and Lang have prepared new ligands for the coupling of substrates 489 and 492, respectively, but with varied results.372−374 In collaboration with the process group at Boehringer-Ingelheim, Tang and co-workers have identified P-chiral ligand L113 as providing high atroposelectivity in the Pd-catalyzed cross-coupling of carbonylbenzoxazolidinone 496 with arylboronic acids. Excellent yields and ee’s are obtained for several different boronic acid substrates;375 optimization of the reaction enabled its application in the efficient total synthesis of several biaryl natural products.376
Scheme 51. Coordinating Groups Developed by Buchwald for Asymmetric Biaryl Couplings.

Scheme 52. Other Coordinating Functional Groups in Asymmetric Suzuki–Miyaura Couplings.

Despite advances in the asymmetric Suzuki–Miyaura cross-coupling of aryl halides bearing proximal coordinating groups, the development of highly atroposelective cross-coupling reactions of simple substrates has remained challenging. The coupling between 1-naphthylboronic acid 487 and 1-halonaphthalene 498 has emerged as a test reaction for new chiral ligands (Figure 94). Often, o-Me groups in the starting materials provide higher ee’s, but their presence can also decrease the yield of the coupling. High temperatures and long reaction times are typical of these methods. Nonetheless, in 2009, Uozumi and co-workers identified an imidazoindole phosphine that facilitates production of 468 in high yield and ee.377 Tethering the ligand to a polymer support, as in L115, permits 468 to be made in water and under heterogeneous conditions without loss of enantioinduction. Additional experiments confirmed that the polymer-supported ligand could be recycled through multiple reaction runs. Notably, ferrocenylphosphine L114, lacking the dimethylamino group in L13, delivers the desired product in moderate enantioselectivity;378 similar enantioinduction has been demonstrated by Guiry379 and Putala.380
Figure 94.
Other ligands for the formation of tetrasubstituted biaryls.
With respect to the formation of trisubstituted biaryl compounds lacking polar directing groups, Lin and co-workers have developed a chiral diene to promote generation of 500 in up to 90% ee and good yield (Figure 95).381 Kündig and co-workers have recently demonstrated that NHC L120 can be used as part of a chiral PEPPSI complex to forge 500 in good ee.382 Phebox L119(383) and phosphine–NHC L118(384,385) deliver more modest atroposelectivity.
Figure 95.
Other ligands for the formation of trisubstituted biaryls.
The Pd-catalyzed Suzuki–Miyaura cross-coupling between 2-methoxyhalonaphthalenes (501) and 1-naphthylboronic acid (499) is another cross-coupling reaction that serves as a testing ground for new ligands (Figure 96). Binaphthyl 502 can be obtained in high yields and moderate ee when BINAP ligands L122 and L123 are employed.386−388 Lassaletta achieved higher atroposelectivity in the Pd-catalyzed coupling of 499 and 501 through the use of bishydrazone L124 and phosphino hydrazone L125: the improved result with L124 was proposed to arise from π-stacking interactions between the substrate and the ligand.389,390 Notably, Pd/L124-catalyzed biaryl formation exhibited a relatively broad substrate scope, enabling the atroposelective cross-coupling of 1-bromonaphthalene or 1-bromo-2-methylnaphthalene with a variety of arylboronic acids. Several additional ligands that have proven less successful are shown in Figure 96.391−398 Enantioenriched dialkoxybinaphthalenes have also been targeted through an asymmetric Suzuki–Miyaura coupling (Scheme 53).
Figure 96.
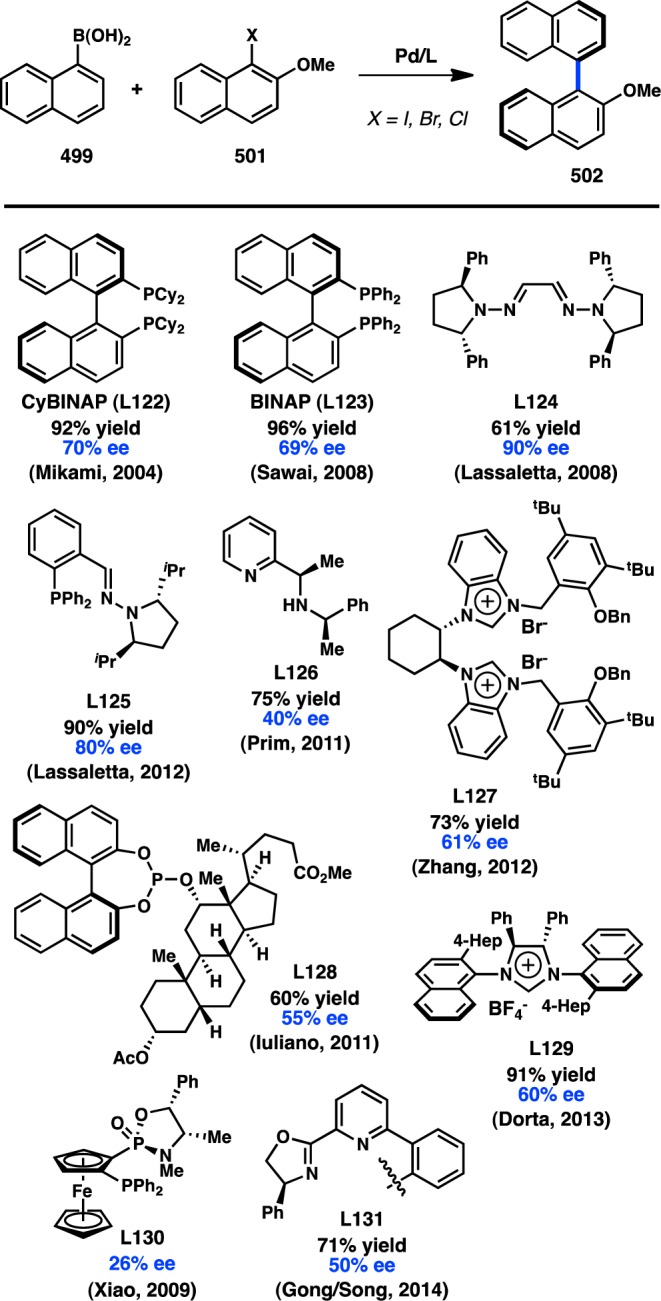
Other ligands for the asymmetric Suzuki–Miyaura coupling of alkoxynaphthyls.
Scheme 53. Preparation of Dialkoxybinaphthyls.
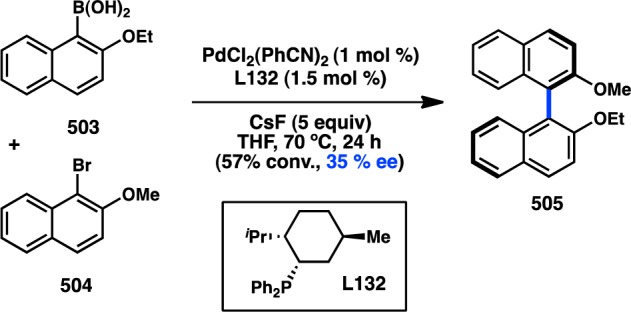
Pd-catalyzed oxidative cross-coupling of unfunctionalized aryl substrates with arylboronic acids can provide direct access to enantioenriched biaryl compounds without the need to prepare the intermediate organo(pseudo)halide. In 2012, Yamaguchi, Itami, and co-workers examined the Pd-catalyzed oxidative cross-coupling of thiophenes with ortho-substituted naphthylboronic acids. Using TEMPO as the stoichiometric oxidant and chiral bioxazoline ligand L133, the C–H activation/cross-coupling between 506 and 507 proceeds in poor yield but with promising atroposelectivity (Scheme 54).399 In a second-generation approach, an aerobic oxidation using iron phthalocyanine (FePc) as a cocatalyst was developed.400 Realizing that DMSO can coordinate to Pd under the reaction conditions, the authors developed a bidentate sulfoxide–oxazoline ligand (L134) that was capable of furnishing 508 in a better yield than the first-generation strategy, although the enantioinduction was still modest.
Scheme 54. Atroposelective Oxidative Cross-Coupling.
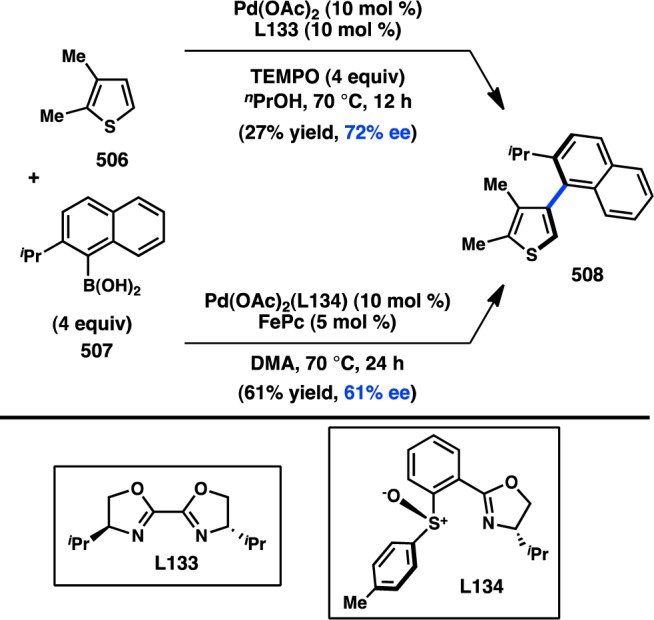
The majority of research toward atropoenantioselective Suzuki–Miyaura cross-couplings has focused on reactions in which the axial chirality is produced in the formation of the biaryl linkage. Alternatively, Lassaletta and co-workers recently reported a Pd-catalyzed arylation of triflate 509 that is an example of a DYKAT (Figure 97).401 When TADDOL-based phosphoramidite L135 is used as a ligand, coupling of triflate 509 with arylboroxines occurs in good yields and with a high level of enantioinduction. Control experiments confirmed the inability of 509 to racemize on the time scale of the reaction, ruling out a dynamic kinetic resolution of the starting material. Instead, the authors hypothesized that oxidative addition of 509 to Pd would be accompanied by chelation of the proximal isoquinoline. This bidentate coordination to Pd was proposed to distort the biaryl C–C bond, widening the angle φ, and decreasing the configurational stability of the complex. In this scenario, fast racemization about the axis of chirality, followed by selective transmetalation of one atropisomer ((P)-511), would ultimately lead to high atroposelectivity. A similar DYKAT approach has been disclosed by Virgil, Stoltz, and co-workers for the preparation of C–P bonds on QUINAP scaffolds.402
Figure 97.
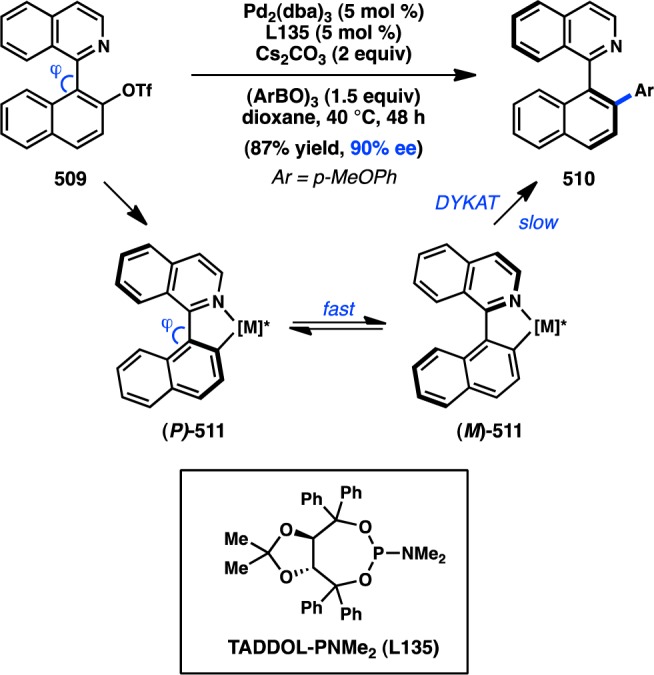
DYKAT approach to enantioenriched biaryls.
5.1.4. Organoindium Reagents
Organoindium reagents are also viable nucleophiles in transition-metal-catalyzed biaryl formation, prompting the group of Sarandeses to investigate atroposelective variants of these reactions.403 Surprisingly, low ee was observed when the chiral Buchwald ligand KenPhos (L107) was employed (Figure 98). On the other hand, PPFA (L13), which is known to promote similar atroposelective cross-coupling reactions of organozinc reagents,353 delivers 500 in good ee. Further studies are needed to assess the full scope and synthetic utility of these organoindium cross-coupling reactions.
Figure 98.
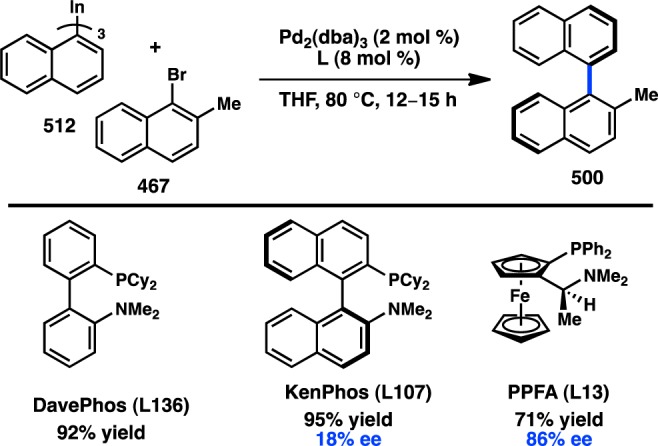
Atroposelective cross-coupling of organoindium reagents.
5.2. Enantiocontrolled Preparation of Allenes
The synthesis of axially chiral allenes has garnered attention because of their presence in bioactive natural products and the intriguing chiroptical properties of cumulenes in general.404 Both enantioselective and enantiospecific transition-metal-catalyzed reactions have been developed for the synthesis of chiral allenes. For each approach, as in the case of allylic substitution, oxidative addition to allenyl or propargyl electrophiles can occur either by an SN2 mechanism or by an SN2′ mechanism, resulting in a potential mixture of regio- and stereochemical outcomes. A second key challenge arises from the possible equilibrium between the allenylmetal and its isomeric propargylmetal species prior to reductive elimination.
5.2.1. Enantioselective Preparation of Allenes
Boersma, Elsevier, and co-workers have investigated a Pd-catalyzed asymmetric Negishi reaction for the synthesis of enantioenriched allenes. When a chiral Pd complex was used as the catalyst, the coupling of racemic allenylzinc iodide 513 with iodobenzene furnished chiral allene 514 in 26% ee (Scheme 55).405 Other allenylmetal species, such as organomagnesium and -copper reagents, resulted in lower enantioselectivity. The reversed-polarity cross-coupling of an arylzinc halide and a haloallene was simultaneously investigated, but enantioinduction higher than 9% ee could not be accomplished.
Scheme 55. Stereoselective Negishi Coupling of Allenylzinc Halides.
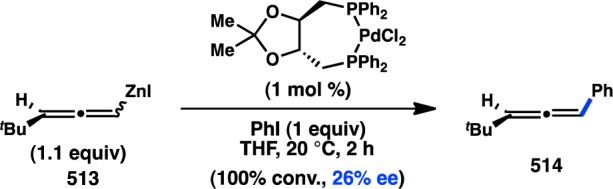
Transition-metal-catalyzed coupling of an organometallic reagent to a propargyl electrophile represents a direct and powerful method for the preparation of chiral allenes. Alexakis and co-workers recently reported the Cu-catalyzed asymmetric addition of alkyl Grignard reagents to propargylic dichloride 515 (Figure 99).406 The complex generated from CuBr and phosphoramidite L137 catalyzed the enantioselective reaction to provide chloroallenes 516 in high yield and ee. Both primary and secondary organomagnesium reagents were well tolerated; however, tBuMgBr only delivered racemic product. The authors demonstrated that the newly formed chiral chloroallene could undergo a Cu-catalyzed enantiospecific cross-coupling with aryl Grignard reagents to afford cross-coupled products without loss of stereochemical information.
Figure 99.
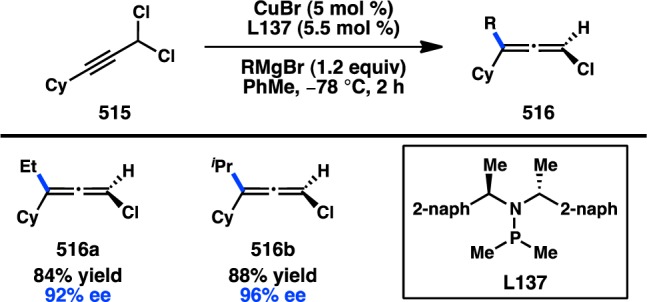
Cu-catalyzed enantioselective propargylic alkylation.
5.2.2. Enantiospecific Preparation of Allenes
In 1984, Vermeer and co-workers investigated the stereochemical outcome of Pd-catalyzed cross-coupling reactions of enantioenriched allenyl halide 517 (Figure 100).407,408 The authors initially anticipated haloallenes would behave like haloalkenes and react with retention of configuration in the presence of Pd0. To the contrary, the coupling of either 517a or 517b delivered 518 with good enantiospecificity and inversion of configuration. Iodoallene 517c furnished the desired product with stereoretention, albeit the degree of es was lower. The nature of the organozinc reagent and the catalyst had smaller effects on the stereospecificity.
Figure 100.
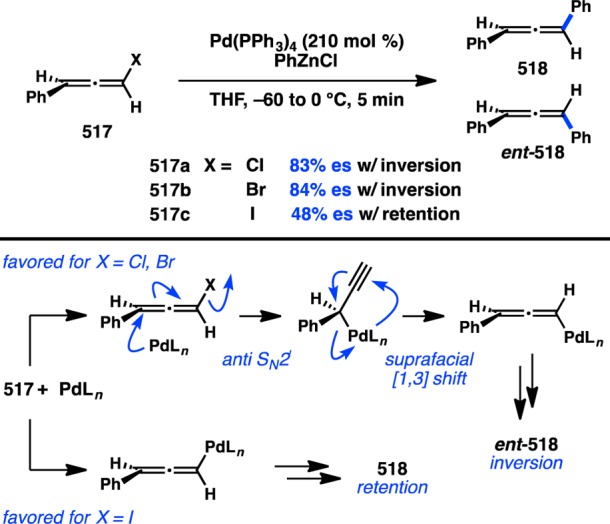
Stereochemical outcome of the coupling of enantioenriched allenyl halides.
The authors rationalize this behavior by proposing two competing mechanisms for the cross-coupling. In the case of 517a and 517b, Pd precoordinates to the allene, resulting in oxidative addition by an anti SN2′ mechanism to give a propargylmetal species. A [1,3]-suprafacial Pd shift would furnish an allenylmetal complex with net inversion of configuration, which following reductive elmination would provide product 518. For 517c, the weaker C–I bond could result in direct oxidative addition of Pd, which upon reductive elimination would furnish the product with retention of stereochemistry.
In 2010, Burke and co-workers performed a similar analysis for the Suzuki–Miyaura coupling of haloallenes and PhB(OH)2 (Scheme 56).409 In analogy to Negishi couplings discussed above, allenyl bromides and chlorides delivered products with net inversion. However, increasing steric profiles of the substituents at C3 and employing large supporting ligands, such as XPhos (L63), can disfavor the anti SN2′ pathway, as demonstrated in the coupling of allenyl iodide 519 to produce 514 exclusively with stereoretention. A DFT study on the Stille coupling of enantiopure haloallenes has supported a similar conclusion.410
Scheme 56. Suzuki–Miyaura Cross-Coupling of Enantioenriched Haloallenes.

Enantiospecific allylic functionalization is a versatile tool for the preparation of chiral allenes from propargylic electrophiles. Readily prepared enantiopure propargylic electrophiles make stereospecific transformations especially attractive. Whereas Pd-catalyzed substitutions with organozinc reagents have been known for many years, only recently have Cu-catalyzed transformations, as well as reactions that use organoboron nucleophiles, emerged.
In the early 1980s, Vermeer and co-workers examined the stereochemical outcome for the Pd-catalyzed coupling of arylzinc reagents and enantioenriched propargylic acetates. Initial studies revealed that allene 518 could be generated in modest enantiospecificity and with inversion of configuration (Figure 101).411,412 The authors hypothesized that acetate 520 undergoes a stereoinvertive SN2′ oxidative addition to form η1-allenylpalladium complex 521. Transmetalation with PhZnCl and a stereoretentive reductive elimination would deliver allene 518 with overall inversion. Isomerization of 521 to propargyl complex 523 is one potential mechanism for the racemization of the product. Vermeer’s protocol continues to be a robust method for the synthesis of enantioenriched aryl-substituted allenes.413,414
Figure 101.
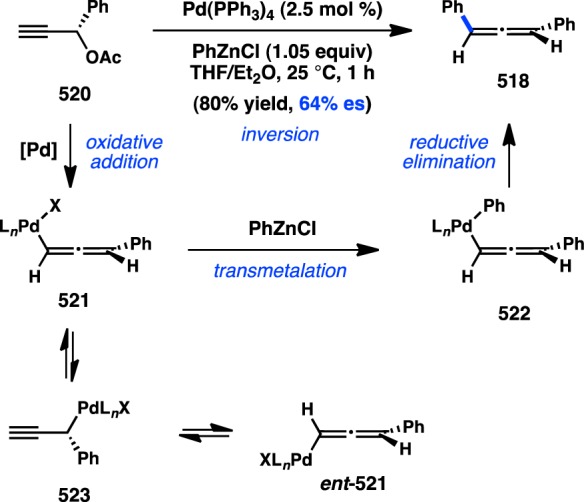
Pd-catalyzed enantiospecific functionalization with arylzinc reagents.
Yoshida, Ihara, and co-workers reported the Pd-catalyzed enantiospecific arylations of propargylic electrophiles using arylboronic acid nucleophiles. Using Pd(PPh3)4 as the catalyst, the reaction of carbonate 524 with arylboronic acids proceeds with perfect invertive transfer of chirality to furnish allene 525 (Scheme 57).415,416 In comparison to arylzinc reagents, relatively high temperatures are required to obtain good yields. The identity of the leaving group influenced the degree of enantiospecificity, with the parent propargyl alcohols delivering nearly racemic products. In an interesting extension of the reaction, propargylic oxiranes can also be coupled with arylboronic acids to form products in high yield and with good es.417
Scheme 57. Pd-Catalyzed Enantiospecific Functionalization with Arylboronic Acids.
In 2006, Molander reported the Pd-catalyzed vinylation of propargylic phosphates to prepare enylallenes. Using Pd(PPh3)4 as the catalyst, propargylic phosphate 526 could be coupled with vinyl trifluoroborate under mild conditions at room temperature to give 527 with good levels of enantiospecifity (Scheme 58). The use of less reactive electrophiles, such as propargylic carbonates, or an increase in the reaction time notably decreased the ee of the product through an off-pathway racemization process. Unfortunately, a limited scope of vinylboron nucleophiles was reported.
Scheme 58. Pd-Catalyzed Enantiospecific Functionalization with Vinyl-BF3K.
Sawamura and co-workers reported a Cu-catalyzed enantiospecific synthesis of chiral allenes in 2011. Building off their prior investigations of stereospecific coupling reactions between allylic phosphates and alkylboron nucleophiles,303 it was found that CuOAc catalyzed the reaction of propargyl phosphate 528 with alkyl-9-BBN reagents to deliver allene 529 with excellent γ-selectivity and high point-to-axial chirality transfer (Figure 102).418,419 The authors propose that the reaction proceeds via initial transmetalation between Cu and the “ate” complex of the nucleophile to form neutral CuI complex 530, which is only mildly nucleophilic. π-Complexation of the alkyne followed by carbometalation would provide 533. anti-Elimination from 533 would deliver the desired allene without stereochemical scrambling. A similar transformation was developed by Lalic and co-workers the following year, in which a Cu–NHC complex catalyzes the stereospecific substitution of a propargylic phosphate with both alkyl- and arylboron nucleophiles.420
Figure 102.
Cu-catalyzed enantiospecific functionalization of propargyl phosphates.
A year later, Sawamura and Lalic independently reported the Cu-catalyzed stereospecific coupling of propargylic phosphates 528 with either aryl- or vinylboronate reagents (Figure 103a).421 The Sawamura coupling employed either CuCl or CuCl2 as the catalyst, under conditions similar to those utilized in the previously reported enantiospecific arylation of allylic phosphates.332 On the other hand, Lalic and co-workers identified a Cu–NHC catalyst that provided the enantioenriched allene in good yield and with excellent point-to-axial chirality transfer (Figure 103b).420 Under Lalic’s conditions, both aryl- and alkylboron reagents were found to react with high stereochemical fidelity.
Figure 103.
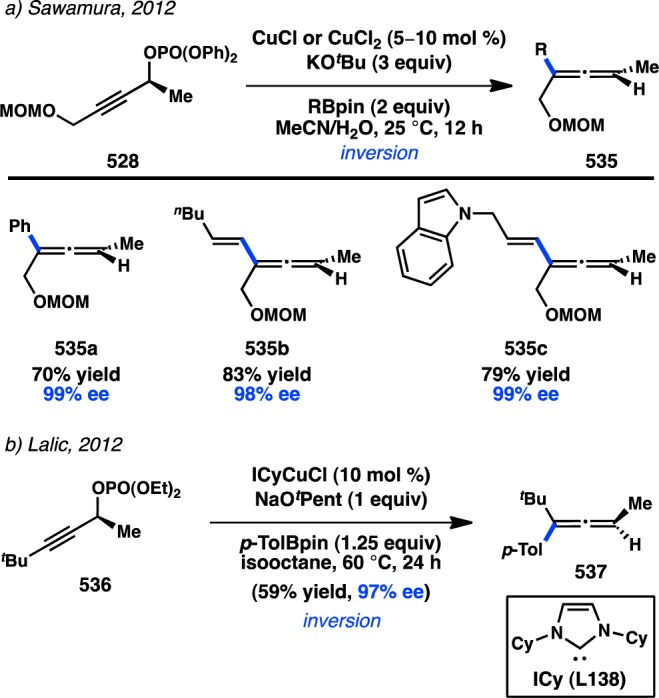
Cu-catalyzed propargylic arylation and vinylation with organoboronates.
5.3. Enantioselective Preparation of Planar Chiral Compounds
Few catalytic asymmetric methods have been developed for the synthesis of molecules with planar or helical chirality; most routes focus on classical resolutions or chiral auxiliary approaches.422 Tricarbonyl(η6-arene)chromium complexes have found utility in the synthesis of planar chiral compounds because the complexes can exist in two enantiomeric forms when the arene is not symmetrically substituted. The first enantioselective cross-coupling to generate a planar chiral compound was reported by Uemera, Hayashi, and co-workers in 1993, in which meso-dichloride 538 was desymmetrized to give 539 (Figure 104a).423 Screening an array of chiral ligands and alkenylmetallic species while using [PdCl-π-allyl]2 as the palladium source revealed that the combination of PPFA (L13) as the ligand and prop-1-en-2-ylboronic acid as the coupling partner provides product 539a in 61% yield and 44% ee. Poor enantioinduction was observed when organotin and organomagnesium reagents were employed. Under similar conditions, arylboronic acids can be coupled with 538 to afford 539b in up to 69% ee.424 In 2011, Kündig and co-workers reported a similar strategy for the desymmetrization of 1,4-dibromonaphthalene complexes. Upon exposure to Pd(dba)2 and phosphoramidite L139, 540 and phenylboronic acid can be coupled to prepare 541 with excellent enantioselectivity (Figure 104b).425 Additional experiments confirmed that the ee increased at longer reaction times, suggesting a kinetic resolution of the product enantiomers takes place during a second cross-coupling to give the meso-compound. Oxidative addition was presumed to be the enantio-determining step. In a separate report, meso-cobalt metallocenes could be desymmetrized in up to 24% ee through an asymmetric Suzuki–Miyaura cross-coupling.426
Figure 104.
Desymmetrization of tricarbonyl(η6-arene)chromium complexes.
In 2013, Gu, You, and co-workers reported an asymmetric C–H activation approach to planar chiral ferrocenes. Building on reports from Yu and co-workers that chiral monoprotected amino acids were competent ligands for asymmetric C–H activation/cross-coupling techniques,133,134 the authors subjected 542 and PhB(OH)2 to Pd(OAc)2 and a series of different amino acid-derived ligands (Scheme 59).427 Boc-l-Val-OH was found to effect formation of 543 with excellent enantioinduction and a minimal amount of the bisarylated product. The authors propose that the Lewis basic dimethylamino group acts as a directing group to facilitate the C–H activation event.
Scheme 59. Synthesis of Planar Chiral Ferrocenes by C–H Activation.

6. Conclusion and Future Outlook
The past several decades have seen a profound growth in cross-coupling methods, and the wealth of new tools for enantioselective and enantiospecific C–C bond formation has revolutionized synthetic planning and practice. As this review illustrates, more and more stereogenic structural motifs have become accessibile through transition-metal-catalyzed cross-coupling, allowing chemists to leverage the flexibility of cross-coupling to streamline the synthesis of complex molecules. However, several challenges remain at the forefront of this field. Whereas enantiocontrolled cross-couplings of sec-alkyl partners with n-alkyl or C(sp2) partners have been well-explored, the analogous asymmetric cross-coupling reactions of tert-alkyl partners represent a largely undeveloped area of great synthetic promise. Similarly, the completely stereocontrolled cross-coupling of two sec-alkyl partners is yet to be realized. These reactions would provide entry to molecules with all-carbon quaternary centers or vicinal tertiary centers, respectively—motifs that are present in many bioactive small molecules and natural product targets. Stereospecific cross-couplings of enantiopure C(sp3) boron and tin nucleophiles show great promise but are currently limited by the difficulty of preparing such substrates. Despite considerable research in the area of allylic substitution, reactions using aryl nucleophiles to prepare all-carbon quaternary centers remain underdeveloped yet valuable. Finally, although atroposelective methods to provide axially chiral products have blossomed in recent years, few ligands have been shown to support a broad substrate scope with high selectivity. More general methods would be of great utility in accessing these atropoisomeric products. The promise of enantiocontrolled cross-coupling methods has been made clear in the years since their inception. We anticipate that their development and application will continue to address long-standing challenges in the field of organic synthesis.
Acknowledgments
We are grateful for financial support from the National Institutes of Health (Grant GM111805-01), Novartis, and Eli Lilly. Fellowship support from Bristol-Myers Squibb (A.H.C.), the American Chemical Society Division of Organic Chemistry, with sponsorship by Amgen (A.H.C.), and the National Science Foundation (A.H.C., Grant No. DGE-1144469) is gratefully acknowledged.
Glossary
Abbreviations
- 9-BBN
9-borabicyclo[3.3.1]nonane
- acac
acetylacetonate
- Boc
tert-butoxycarbonyl
- Bz
benzoyl
- CDR
catalytic dynamic resolution
- cod
1,5-cyclooctadiene
- CPME
cyclopentyl methyl ether
- dan
1,8-diaminonaphthalene
- dba
dibenzylideneacetone
- DCC
N,N′-dicyclohexylcarbodiimide
- DET
direct enantioselective transformation
- DFT
density functional theory
- DIBAL-H
diisobutylaluminum hydride
- DKR
dynamic kinetic resolution
- DMA
N,N-dimethylacetamide
- DMAP
p-(dimethylamino)pyridine
- DME
1,2-dimethoxyethane
- DMF
N,N-dimethylformamide
- dppb
1,4-bis(diphenylphosphino)butane
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- dppo
1,8-bis(diphenylphosphino)octane
- DTBM
3,5-di-tert-butyl-4-methoxyphenyl
- DTR
dynamic thermodynamic resolution
- DYKAT
dynamic kinetic asymmetric transformation
- ee
enantiomeric excess
- es
enantiospecificity
- HMPA
hexamethylphosphoramide
- MS
molecular sieves
- MTBE
methyl tert-butyl ether
- nbd
norbornadiene
- Neop
neopentanediol, 2,2-dimethyl-1,3-propanediol
- NHC
N-heterocyclic carbene
- PEPPSI
pyridine-enhanced precatalyst preparation stabilization and initiation
- phen
1,10-phenanthroline
- Pin
pinacol, 2,3-dimethyl-2,3-butanediol
- PMB
p-methoxybenzyl
- SiMes
1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene
- SKR
stereodivergent kinetic resolution
- TADDOLs
α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanols
- TASF
tris(dimethylamino)sulfonium difluorotrimethylsilicate
- TBAB
tetrabutylammonium bromide
- TBAF
tetrabutylammonium fluoride
- TBAT
tetrabutylammonium difluorotriphenylsilicate
- TBD
1,5,7-triazabicyclo[4.4.0]dec-5-ene
- TC
thiophene-2-carboxylate
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- THF
tetrahydrofuran
- TMEDA
N,N,N′,N′-tetramethylethylenediamine
- Ts
tosyl, p-toluenesulfonyl
Biographies

Alan H. Cherney was born and raised in Chicago, IL. Alan earned his B.S. in chemical and biomolecular engineering from the University of Illinois at Urbana-Champaign in 2010. In 2015, he completed his Ph.D. in chemistry at the California Institute of Technology under the direction of Professor Sarah Reisman.

Nathaniel T. Kadunce was born in Beaver, PA, in 1988. He received his B.A. in chemistry and biochemistry from Oberlin College in 2011. Nathaniel is currently pursuing his Ph.D. in chemistry with Prof. Sarah Reisman at the California Institute of Technology.

Sarah E. Reisman received her B.A. in chemistry in 2001 from Connecticut College. In 2006, she earned her Ph.D. in chemistry from Yale University under the direction of Prof. John L. Wood. As an NIH NRSA (National Institutes of Health National Research Service Award) fellow, Sarah conducted postdoctoral studies in the area of asymmetric catalysis with Prof. Eric Jacobsen at Harvard University. In 2008, Sarah joined the faculty at the California Institute of Technology, where her laboratory seeks to discover, develop, and study new chemical reactions within the context of natural product total synthesis.
The authors declare no competing financial interest.
References
- Mc Cartney D.; Guiry P. J. The asymmetric Heck and related reactions. Chem. Soc. Rev. 2011, 40, 5122–5150 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]
- Karimi B.; Behzadnia H.; Elhamifar D.; Akhavan P. F.; Esfahani F. K.; Zamani A. Transition-metal-catalyzed oxidative Heck reactions. Synthesis 2010, 2010, 1399–1427 10.1055/s-0029-1218748. [DOI] [Google Scholar]
- Lautens M.; Fagnou K.; Hiebert S. Transition metal-catalyzed enantioselective ring-opening reactions of oxabicyclic alkenes. Acc. Chem. Res. 2003, 36, 48–58 10.1021/ar010112a. [DOI] [PubMed] [Google Scholar]
- Alexakis A.; Bäckvall J.-E.; Krause N.; Pàmies O.; Diéguez M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 2008, 108, 2796–2823 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]
- Falciola C. A.; Alexakis A. Copper-catalyzed asymmetric allylic alkylation. Eur. J. Org. Chem. 2008, 2008, 3765–3780 10.1002/ejoc.200800025. [DOI] [Google Scholar]
- Harutyunyan S. R.; den Hartog T.; Geurts K.; Minnaard A. J.; Feringa B. L. Catalytic asymmetric conjugate addition and allylic alkylation with Grignard reagents. Chem. Rev. 2008, 108, 2824–2852 10.1021/cr068424k. [DOI] [PubMed] [Google Scholar]
- Jana R.; Pathak T. P.; Sigman M. S. Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 2011, 111, 1417–1492 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph A.; Lautens M. Secondary alkyl halides in transition-metal-catalyzed cross-coupling reactions. Angew. Chem., Int. Ed. 2009, 48, 2656–2670 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- Whitesides G. M.; Roberts J. D. Nuclear magnetic resonance spectroscopy. The configurational stability of primary Grignard reagents. Structure and medium effects. J. Am. Chem. Soc. 1965, 87, 4878–4888 10.1021/ja00949a035. [DOI] [Google Scholar]
- Whitesides G. M.; Witanowski M.; Roberts J. D. Magnetic resonance spectroscopy. The configurational stability of primary Grignard reagents. 3,3-Dimethylbutylmagnesium chloride. J. Am. Chem. Soc. 1965, 87, 2854–2862 10.1021/ja01091a014. [DOI] [Google Scholar]
- Witanowski M.; Roberts J. D. Proton magnetic resonance spectroscopy. Configurational stability of neohexyl(3,3-dimethylbutyl) organometallic compounds. J. Am. Chem. Soc. 1966, 88, 737–741 10.1021/ja00956a023. [DOI] [Google Scholar]
- Hoffmann R. W.; Hölzer B.; Knopff O.; Harms K. Asymmetric synthesis of a chiral secondary Grignard reagent. Angew. Chem., Int. Ed. 2000, 39, 3072–3074. [DOI] [PubMed] [Google Scholar]
- Corriu R. J. P.; Masse J. P. Activation of Grignard reagents by transition-metal complexes - new and simple synthesis of trans-stilbenes and polyphenyls. J. Chem. Soc., Chem. Commun. 1972, 144a. 10.1039/c3972000144a. [DOI] [Google Scholar]
- Tamao K.; Sumitani K.; Kumada M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376 10.1021/ja00767a075. [DOI] [Google Scholar]
- Tamao K. Discovery of the cross-coupling reaction between Grignard reagents and C(sp2) halides catalyzed by nickel-phosphine complexes. J. Organomet. Chem. 2002, 653, 23–26 10.1016/S0022-328X(02)01159-2. [DOI] [Google Scholar]
- Hayashi T. Catalytic asymmetric cross-coupling. J. Organomet. Chem. 2002, 653, 41–45 10.1016/S0022-328X(02)01272-X. [DOI] [Google Scholar]
- Consiglio G.; Botteghi C. Stereoselective formation of C-C bond in coupling reaction between secondary alkylmagnesium bromides and unsaturated halides - asymmetrical induction by optically-active diphosphine nickel-complex. Helv. Chim. Acta 1973, 56, 460–463 10.1002/hlca.19730560137. [DOI] [Google Scholar]
- Kiso Y.; Tamao K.; Miyake N.; Yamamoto K.; Kumada M. Asymmetric cross-coupling reaction of sec-alkyl Grignard reagents with organic halides in the presence of a chiral phosphine-nickel complex as a catalyst. Tetrahedron Lett. 1974, 15, 3–6 10.1016/S0040-4039(01)82121-3. [DOI] [Google Scholar]
- Consiglio G.; Piccolo O.; Morandini F. Nickel catalyzed asymmetric cross-coupling reaction between C6H5X and s-BuMgX. The influence of the nature of the halogens of the two reagents. J. Organomet. Chem. 1979, 177, C13–C15 10.1016/S0022-328X(00)94089-0. [DOI] [Google Scholar]
- Consiglio G.; Morandini F.; Piccolo O. Asymmetric nickel catalysis in the cross-coupling reaction of aryl halides with Grignard reagents. A study of the factors influencing asymmetric induction. Tetrahedron 1983, 39, 2699–2707 10.1016/S0040-4020(01)91980-5. [DOI] [Google Scholar]
- Consiglio G.; Indolese A. Enantioselective cross-coupling of vinyl-, aryl- and allyl-electrophiles catalyzed by nickel complexes containing (R,R)-1,2-cyclopentanediylbis(diphenylphosphine) and related ligands. J. Organomet. Chem. 1991, 417, C36–C40 10.1016/0022-328X(91)80189-Q. [DOI] [Google Scholar]
- Iida A.; Yamashita M. Asymmetric Grignard cross-coupling reaction using chiral ligands derived from carbohydrates. Bull. Chem. Soc. Jpn. 1988, 61, 2365–2367 10.1246/bcsj.61.2365. [DOI] [Google Scholar]
- Czaplik W. M.; Mayer M.; Jacobi von Wangelin A. Direct cobalt-catalyzed cross-coupling between aryl and alkyl halides. Synlett 2009, 2009, 2931–2934 10.1055/s-0029-1218012. [DOI] [Google Scholar]
- Tamao K.; Yamamoto H.; Matsumoto H.; Miyake N.; Hayashi T.; Kumada M. Optically active 2,2′-bis(diphenylphosphinomethyl)-1,1′-binaphthyl: a new chiral bidentate phosphine ligand for transition-metal complex catalyzed asymmetric reactions. Tetrahedron Lett. 1977, 18, 1389–1392 10.1016/S0040-4039(01)93051-5. [DOI] [Google Scholar]
- Brunner H.; Pröbster M. Asymmetric catalyzes. 6. NiCl2(−)Norphos catalyzed cross-coupling reaction of 1-phenylethyl-Grignard with vinylbromide. J. Organomet. Chem. 1981, 209, C1–C3 10.1016/S0022-328X(00)88984-6. [DOI] [Google Scholar]
- Hayashi T.; Fukushima M.; Konishi M.; Kumada M. Chiral β-dimethylaminoalkylphosphines. Highly efficient ligands for a nickel-complex catalyzed asymmetric Grignard cross-coupling reaction. Tetrahedron Lett. 1980, 21, 79–82 10.1016/S0040-4039(00)93629-3. [DOI] [Google Scholar]
- Hayashi T.; Nagashima N.; Kumada M. Asymmetric Grignard cross-coupling by means of polymer-supported optically-active β-dimethylaminoalkylphosphine-nickel catalysts. Tetrahedron Lett. 1980, 21, 4623–4626 10.1016/0040-4039(80)80090-6. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Fukushima M.; Kanehira K.; Hioki T.; Kumada M. Chiral (β-aminoalkyl)phosphines. Highly efficient phosphine-ligands for catalytic asymmetric Grignard cross-coupling. J. Org. Chem. 1983, 48, 2195–2202 10.1021/jo00161a013. [DOI] [Google Scholar]
- Baker K. V.; Brown J. M.; Cooley N. A.; Hughes G. D.; Taylor R. J. Reactive intermediates in asymmetric cross-coupling catalyzed by palladium P-N chelates. J. Organomet. Chem. 1989, 370, 397–406 10.1016/0022-328X(89)87301-2. [DOI] [Google Scholar]
- Cross G. A.; Kellogg R. M. Zinc halide induced switch in enantioselection in nickel and palladium catalyzed cross coupling reactions. J. Chem. Soc., Chem. Commun. 1987, 1746–1747 10.1039/c39870001746. [DOI] [Google Scholar]
- Cross G.; Vriesema B. K.; Boven G.; Kellogg R. M.; van Bolhuis F. Transition metal catalyzed asymmetric cross coupling reactions. New ligands and the effects of added salts. Crystal structures of [Ph2PCH2CH((CH2)3SMe)NMe2]•PdCl2 and [Ph2PCH2CH((CH2)2SMe)NMe2]•PdCl2. J. Organomet. Chem. 1989, 370, 357–381 10.1016/0022-328X(89)87299-7. [DOI] [Google Scholar]
- Hayashi T.; Tajika M.; Tamao K.; Kumada M. High stereoselectivity in asymmetric Grignard cross-coupling catalyzed by nickel complexes of chiral(aminoalkylferrocenyl)phosphines. J. Am. Chem. Soc. 1976, 98, 3718–3719 10.1021/ja00428a061. [DOI] [Google Scholar]
- Tamao K.; Hayashi T.; Matsumoto H.; Yamamoto H.; Kumada M. Asymmetric total synthesis of optically-active α-curcumene. Tetrahedron Lett. 1979, 20, 2155–2156 10.1016/S0040-4039(01)86288-2. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Fukushima M.; Mise T.; Kagotani M.; Tajika M.; Kumada M. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition metal complexes 0.2. Nickel- and palladium-catalyzed asymmetric Grignard cross-coupling. J. Am. Chem. Soc. 1982, 104, 180–186 10.1021/ja00365a033. [DOI] [Google Scholar]
- Hayashi T.; Kumada M. Asymmetric synthesis catalyzed by transition-metal complexes with functionalized chiral ferrocenylphosphine ligands. Acc. Chem. Res. 1982, 15, 395–401 10.1021/ar00084a003. [DOI] [Google Scholar]
- Indolese A.; Consiglio G. Influence of the extent of conversion on enantioselectivity in the nickel-catalyzed cross-coupling reaction. J. Organomet. Chem. 1993, 463, 23–27 10.1016/0022-328X(93)83393-A. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Hioki T.; Kumada M.; Ratajczak A.; Niedbala H. Preparation of (R)-N,N-dimethyl-1-[2-(diphenylphosphino)ferrocenyl]-2-propanamines and asymmetric Grignard cross-coupling catalyzed by nickel complexes with the phosphine ligands. Bull. Chem. Soc. Jpn. 1981, 54, 3615–3616 10.1246/bcsj.54.3615. [DOI] [Google Scholar]
- Lemaire M.; Buter J.; Vriesema B. K.; Kellogg R. M. Chiral (macrocyclic) sulfides as ligands for nickel catalyzed carbon–carbon bond formation. J. Chem. Soc., Chem. Commun. 1984, 309–310 10.1039/c39840000309. [DOI] [Google Scholar]
- Vriesema B. K.; Lemaire M.; Buter J.; Kellogg R. M. Chiral (macrocyclic) sulfide- and sulfide/alkylamino-containing ligands for nickel-catalyzed Grignard cross-coupling reactions. J. Org. Chem. 1986, 51, 5169–5177 10.1021/jo00376a022. [DOI] [Google Scholar]
- Brunner H.; Li W.; Weber H. Asymmetrical catalysts 0.24. Cross-coupling of 1-phenylethyl Grignard and vinylbromide with Ni-catalysts of optically-active P/N-ligands. J. Organomet. Chem. 1985, 288, 359–363 10.1016/0022-328X(85)80130-3. [DOI] [Google Scholar]
- Terfort A.; Brunner H. Phosphane ligands with two binding sites of differing hardness for enantioselective Grignard cross coupling. J. Chem. Soc., Perkin Trans. 1 1996, 1467–1479 10.1039/p19960001467. [DOI] [Google Scholar]
- Jedlicka B.; Kratky C.; Weissensteiner W.; Widhalm M. Palladium-catalyzed enantioselective Grignard cross-coupling with use of a new ferrocenylaminophosphine ligand. J. Chem. Soc., Chem. Commun. 1993, 1329–1330 10.1039/c39930001329. [DOI] [Google Scholar]
- Pellet-Rostaing S.; Saluzzo C.; Ter Halle R.; Breuzard J.; Vial L.; Le Guyader F.; Lemaire M. Grignard cross-coupling catalyzed by chiral phosphino-quincorine and phosphino-quincoridine derivatives. Tetrahedron: Asymmetry 2001, 12, 1983–1985 10.1016/S0957-4166(01)00342-1. [DOI] [Google Scholar]
- Hayashi M.; Takaoki K.; Hashimoto Y.; Saigo K. Synthesis of a new chiral (β-aminoalkyl)phosphine ligand and its application to a catalytic asymmetric Grignard cross-coupling reaction. Enantiomer 1997, 2, 293–296. [Google Scholar]
- Yamago S.; Yanagawa M.; Nakamura E. Tertiary phosphines and P-chiral phosphinites bearing a fullerene substituent. J. Chem. Soc., Chem. Commun. 1994, 2093–2094 10.1039/c39940002093. [DOI] [Google Scholar]
- Yamago S.; Yanagawa M.; Mukai H.; Nakamura E. Tertiary phosphines, P-chiral phosphinites and phosphonic acid esters bearing fullerene substituent. Metal complexes and redox properties. Tetrahedron 1996, 52, 5091–5102 10.1016/0040-4020(96)00116-0. [DOI] [Google Scholar]
- Kreuzfeld H. J.; Döbler C.; Abicht H. P. Asymmetric Grignard cross-coupling reaction between (E)-β-bromostyrene and 1-phenylethylmagnesium chloride. J. Organomet. Chem. 1987, 336, 287–292 10.1016/0022-328X(87)85186-0. [DOI] [Google Scholar]
- Döbler C.; Kreuzfeld H. J. Asymmetric Grignard cross-coupling reaction between (E)-β-bromostyrene and 1-phenylethylmagnesium chloride. J. Organomet. Chem. 1988, 344, 249–252 10.1016/0022-328X(88)80483-2. [DOI] [Google Scholar]
- Horibe H.; Kazuta K.; Kotoku M.; Kondo K.; Okuno H.; Murakami Y.; Aoyama T. Further application of an N-Ar axially chiral mimetic-type ligand: Asymmetric Grignard cross-coupling reaction. Synlett 2003, 2047–2051 10.1055/s-2003-41485. [DOI] [Google Scholar]
- Horibe H.; Fukuda Y.; Kondo K.; Okuno H.; Murakami Y.; Aoyama T. Asymmetric Kumada-Corriu cross-coupling reaction with Pd2(dba)3 and an N-Ar axially chiral mimetic-type ligand catalyst. Tetrahedron 2004, 60, 10701–10709 10.1016/j.tet.2004.09.010. [DOI] [Google Scholar]
- Richards C. J.; Hibbs D. E.; Hursthouse M. B. PdCl2-complexes containing phosphinoferrocenyloxazoline ligands. X-ray crystal-structure analysis and application to Grignard cross-coupling. Tetrahedron Lett. 1995, 36, 3745–3748 10.1016/0040-4039(95)00583-X. [DOI] [Google Scholar]
- Lloyd-Jones G. C.; Butts C. P. Nickel(II) complexes bearing phosphinoaryl oxazoline ligands as pro-catalysts for Grignard cross-coupling. Tetrahedron 1998, 54, 901–914 10.1016/S0040-4020(97)10332-5. [DOI] [Google Scholar]
- Schwink L.; Knochel P. Enantioselective preparation of C2-symmetrical ferrocenyl ligands for asymmetric catalysis. Chem. - Eur. J. 1998, 4, 950–968. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Ito H.; Kumada M. Optically active allylsilanes. 1. Preparation by palladium-catalyzed asymmetric Grignard cross-coupling and anti stereochemistry in electrophilic substitution reactions. J. Am. Chem. Soc. 1982, 104, 4962–4963 10.1021/ja00382a045. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Okamoto Y.; Kabeta K.; Kumada M. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine transition-metal complexes. 3. Preparation of optically active allylsilanes by palladium-catalyzed asymmetric Grignard cross-coupling. J. Org. Chem. 1986, 51, 3772–3781 10.1021/jo00370a006. [DOI] [Google Scholar]
- Hayashi T.; Kanehira K.; Hioki T.; Kumada M. Kinetic resolution of racemic Grignard reagents by nickel-catalyzed asymmetric Grignard cross-coupling. Tetrahedron Lett. 1981, 22, 137–140 10.1016/0040-4039(81)80169-4. [DOI] [Google Scholar]
- Hayashi T.; Okamoto Y.; Kumada M. An optically active propargylsilane: preparation by asymmetric Grignard cross-coupling and stereochemistry in an electrophilic substitution. Tetrahedron Lett. 1983, 24, 807–808 10.1016/S0040-4039(00)81533-6. [DOI] [Google Scholar]
- Hayashi T.; Hagihara T.; Katsuro Y.; Kumada M. Asymmetric cross-coupling of organozinc reagents with alkenyl bromides catalyzed by a chiral ferrocenylphosphine palladium complex. Bull. Chem. Soc. Jpn. 1983, 56, 363–364 10.1246/bcsj.56.363. [DOI] [Google Scholar]
- Hayashi T.; Yamamoto A.; Hojo M.; Ito Y. A new chiral ferrocenylphosphine ligand with C2 symmetry: preparation and use for palladium-catalyzed asymmetric cross-coupling. J. Chem. Soc., Chem. Commun. 1989, 495–496 10.1039/c39890000495. [DOI] [Google Scholar]
- Cherney A. H.; Reisman S. E. Pd-catalyzed Fukuyama cross-coupling of secondary organozinc reagents for the direct synthesis of unsymmetrical ketones. Tetrahedron 2014, 70, 3259–3265 10.1016/j.tet.2013.11.104. [DOI] [Google Scholar]
- Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C.-y. Enantioselective, palladium-catalyzed α-arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2006, 128, 3538–3539 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- Cordier C. J.; Lundgren R. J.; Fu G. C. Enantioconvergent cross-couplings of racemic alkylmetal reagents with unactivated secondary alkyl electrophiles: Catalytic asymmetric Negishi α-alkylations of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2013, 135, 10946–10949 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zembayashi M.; Tamao K.; Hayashi T.; Mise T.; Kumada M. New asymmetric Grignard cross-coupling reaction via an alkyl group isomerization catalyzed by chiral phosphine nickel complexes. Tetrahedron Lett. 1977, 18, 1799–1802 10.1016/S0040-4039(01)83607-8. [DOI] [Google Scholar]
- Binder J. T.; Cordier C. J.; Fu G. C. Catalytic enantioselective cross-couplings of secondary alkyl electrophiles with secondary alkylmetal nucleophiles: Negishi reactions of racemic benzylic bromides with achiral alkylzinc reagents. J. Am. Chem. Soc. 2012, 134, 17003–17006 10.1021/ja308460z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoroafor M. O.; Ward D. L.; Brubaker C. H. Synthesis of new chiral ferrocenyl amine sulfide complexes and their applications as asymmetric catalysts. The structure of (R,S)- C5H5FeC5H3[CHMeNMe2][SCH3][PdCl2]. Organometallics 1988, 7, 1504–1511 10.1021/om00097a009. [DOI] [Google Scholar]
- Naiini A. A.; Lai C.-K.; Ward D. L.; Brubaker C. H. Synthesis of new ferrocenyl amine sulfide and selenide complexes of group 10 metals and their catalytic activities toward selective hydrogenation, isomerization, and asymmetric Grignard cross-coupling reactions. J. Organomet. Chem. 1990, 390, 73–90 10.1016/0022-328X(90)85083-B. [DOI] [Google Scholar]
- Ali H. M.; Brubaker C. H. Syntheses of new chiral ferrocenylamine sulfide and selenide complexes of group X metals and their application to enantioselective cross-coupling reactions and selective hydrogenation. J. Mol. Catal. 1990, 60, 331–342 10.1016/0304-5102(90)85256-H. [DOI] [Google Scholar]
- Lou S.; Fu G. C. Nickel/bis(oxazoline)-catalyzed asymmetric Kumada reactions of alkyl electrophiles: Cross-couplings of racemic α-bromoketones. J. Am. Chem. Soc. 2010, 132, 1264–1266 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin P. M.; Esquivias J.; Fu G. C. Catalytic asymmetric cross-couplings of racemic α-bromoketones with arylzinc reagents. Angew. Chem., Int. Ed. 2009, 48, 154–156 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer C.; Fu G. C. Asymmetric nickel-catalyzed Negishi cross-couplings of secondary α-bromo amides with organozinc reagents. J. Am. Chem. Soc. 2005, 127, 4594–4595 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- Arp F. O.; Fu G. C. Catalytic enantioselective Negishi reactions of racemic secondary benzylic halides. J. Am. Chem. Soc. 2005, 127, 10482–10483 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- Lin X.; Sun J.; Xi Y.; Lin D. How racemic secondary alkyl electrophiles proceed to enantioselective products in Negishi cross-coupling reactions. Organometallics 2011, 30, 3284–3292 10.1021/om1012049. [DOI] [Google Scholar]
- Smith S. W.; Fu G. C. Nickel-catalyzed asymmetric cross-couplings of racemic propargylic halides with arylzinc reagents. J. Am. Chem. Soc. 2008, 130, 12645–12647 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schley N. D.; Fu G. C. Nickel-catalyzed Negishi arylations of propargylic bromides: A mechanistic investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge B. M.; Chausset-Boissarie L.; Burns M.; Pulis A. P.; Aggarwal V. K. Enantioselective synthesis and cross-coupling of tertiary propargylic boronic esters using lithiation-borylation of propargylic carbamates. Angew. Chem., Int. Ed. 2012, 51, 11795–11799 10.1002/anie.201203198. [DOI] [PubMed] [Google Scholar]
- Oelke A. J.; Sun J.; Fu G. C. Nickel-catalyzed enantioselective cross-couplings of racemic secondary electrophiles that bear an oxygen leaving group. J. Am. Chem. Soc. 2012, 134, 2966–2969 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H.-Q.; Chandrashekar E. R. R.; Fu G. C. Nickel/bis(oxazoline)-catalyzed asymmetric Negishi arylations of racemic secondary benzylic electrophiles to generate enantioenriched 1,1-diarylalkanes. J. Am. Chem. Soc. 2013, 135, 16288–16291 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imao D.; Glasspoole B. W.; Laberge V. S.; Crudden C. M. Cross coupling reactions of chiral secondary organoboronic esters with retention of configuration. J. Am. Chem. Soc. 2009, 131, 5024–5025 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- Taylor B. L. H.; Swift E. C.; Waetzig J. D.; Jarvo E. R. Stereospecific nickel-catalyzed cross-coupling reactions of alkyl ethers: Enantioselective synthesis of diarylethanes. J. Am. Chem. Soc. 2011, 133, 389–391 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]
- Greene M. A.; Yonova I. M.; Williams F. J.; Jarvo E. R. Traceless directing group for stereospecific nickel-catalyzed alkyl-alkyl cross-coupling reactions. Org. Lett. 2012, 14, 4293–4296 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]
- Yonova I. M.; Johnson A. G.; Osborne C. A.; Moore C. E.; Morrissette N. S.; Jarvo E. R. Stereospecific nickel-catalyzed cross-coupling reactions of alkyl Grignard reagents and identification of selective anti-breast-cancer agents. Angew. Chem., Int. Ed. 2014, 53, 2422–2427 10.1002/anie.201308666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Srinivas H. D.; Dasgupta S.; Watson M. P. Nickel-catalyzed cross-couplings of benzylic pivalates with arylboroxines: Stereospecific formation of diarylalkanes and triarylmethanes. J. Am. Chem. Soc. 2013, 135, 3307–3310 10.1021/ja312087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity P.; Shacklady-McAtee D. M.; Yap G. P. A.; Sirianni E. R.; Watson M. P. Nickel-catalyzed cross couplings of benzylic ammonium salts and boronic acids: Stereospecific formation of diarylethanes via C-N bond activation. J. Am. Chem. Soc. 2013, 135, 280–285 10.1021/ja3089422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacklady-McAtee D. M.; Roberts K. M.; Basch C. H.; Song Y.-G.; Watson M. P. A general, simple catalyst for enantiospecific cross couplings of benzylic ammonium triflates and boronic acids: No phosphine ligand required. Tetrahedron 2014, 70, 4257–4263 10.1016/j.tet.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Fu G. C. Catalytic asymmetric synthesis of tertiary alkyl fluorides: Negishi cross-couplings of racemic α,α-dihaloketones. J. Am. Chem. Soc. 2014, 136, 5520–5524 10.1021/ja501815p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A.; Falck J. R. Stereospecific Suzuki cross-coupling of alkyl α-cyanohydrin triflates. J. Am. Chem. Soc. 2010, 132, 2524–2525 10.1021/ja910582n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Fu G. C. Catalytic asymmetric synthesis of secondary nitriles via stereoconvergent Negishi arylations and alkenylations of racemic α-bromonitriles. J. Am. Chem. Soc. 2012, 134, 9102–9105 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D. A.; Maki T.; Fu G. C. Stille cross-couplings of unactivated secondary alkyl halides using monoorganotin reagents. J. Am. Chem. Soc. 2005, 127, 510–511 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Bobes F.; Fu G. C. Amino alcohols as ligands for nickel-catalyzed Suzuki reactions of unactivated alkyl halides, including secondary alkyl chlorides, with arylboronic acids. J. Am. Chem. Soc. 2006, 128, 5360–5361 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]
- Choi J.; Martín-Gago P.; Fu G. C. Stereoconvergent arylations and alkenylations of unactivated alkyl electrophiles: Catalytic enantioselective synthesis of secondary sulfonamides and sulfones. J. Am. Chem. Soc. 2014, 136, 12161–12165 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito B.; Fu G. C. Alkyl-alkyl Suzuki cross-couplings of unactivated secondary alkyl halides at room temperature. J. Am. Chem. Soc. 2007, 129, 9602–9603 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito B.; Fu G. C. Enantioselective alkyl-alkyl Suzuki cross-couplings of unactivated homobenzylic halides. J. Am. Chem. Soc. 2008, 130, 6694–6695 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- Owston N. A.; Fu G. C. Asymmetric alkyl-alkyl cross-couplings of unactivated secondary alkyl electrophiles: Stereoconvergent Suzuki reactions of racemic acylated halohydrins. J. Am. Chem. Soc. 2010, 132, 11908–11909 10.1021/ja105924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Wilsily A.; Fu G. C. Stereoconvergent amine-directed alkyl-alkyl Suzuki reactions of unactivated secondary alkyl chlorides. J. Am. Chem. Soc. 2011, 133, 8154–8157 10.1021/ja203560q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zultanski S. L.; Fu G. C. Catalytic asymmetric γ-alkylation of carbonyl compounds via stereoconvergent Suzuki cross-couplings. J. Am. Chem. Soc. 2011, 133, 15362–15364 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsily A.; Tramutola F.; Owston N. A.; Fu G. C. New directing groups for metal-catalyzed asymmetric carbon-carbon bond-forming processes: Stereoconvergent alkyl-alkyl Suzuki cross-couplings of unactivated electrophiles. J. Am. Chem. Soc. 2012, 134, 5794–5797 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Sakthivel S.; Kulbitski K.; Nisnevich G.; Gandelman M. Efficient synthesis of secondary alkyl fluorides via Suzuki cross-coupling reaction of 1-halo-1-fluoroalkanes. J. Am. Chem. Soc. 2014, 136, 9548–9551 10.1021/ja504089y. [DOI] [PubMed] [Google Scholar]
- Lundin P. M.; Fu G. C. Asymmetric Suzuki cross-couplings of activated secondary alkyl electrophiles: Arylations of racemic α-chloroamides. J. Am. Chem. Soc. 2010, 132, 11027–11029 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong H.; Fu G. C. Catalytic enantioselective cyclization/cross-coupling with alkyl electrophiles. J. Am. Chem. Soc. 2014, 136, 3788–3791 10.1021/ja500706v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T. J. A.; Shields J. D.; Doyle A. G. Transition metal-catalyzed cross coupling with N-acyliminium ions derived from quinolines and isoquinolines. Chem. Sci. 2011, 2, 980–984 10.1039/C1SC00026H. [DOI] [Google Scholar]
- Sylvester K. T.; Wu K.; Doyle A. G. Mechanistic investigation of the nickel-catalyzed Suzuki reaction of N,O-acetals: Evidence for boronic acid assisted oxidative addition and an iminium activation pathway. J. Am. Chem. Soc. 2012, 134, 16967–16970 10.1021/ja3079362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields J. D.; Ahneman D. T.; Graham T. J. A.; Doyle A. G. Enantioselective, nickel-catalyzed Suzuki cross-coupling of quinolinium ions. Org. Lett. 2014, 16, 142–145 10.1021/ol4031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau S. T.; Lutz J. P.; Wu K.; Doyle A. G. Nickel-catalyzed enantioselective arylation of pyridinium ions: Harnessing an iminium ion activation mode. Angew. Chem., Int. Ed. 2013, 52, 9153–9156 10.1002/anie.201303994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y.; Takada S.; Miyaura N. γ-Selective cross-coupling of potassium allyltrifluoroborates with aryl and 1-alkenyl bromides catalyzed by a Pd(OAc)2/D-t-BPF complex. Chem. Lett. 2006, 35, 704–705 10.1246/cl.2006.704. [DOI] [Google Scholar]
- Yamamoto Y.; Takada S.; Miyaura N. Asymmetric cross-coupling of potassium 2-butenyltrifluoroborates with aryl and 1-alkenyl bromides catalyzed by a Pd(OAc)2/Josiphos complex. Chem. Lett. 2006, 35, 1368–1369 10.1246/cl.2006.1368. [DOI] [Google Scholar]
- Yamamoto Y.; Takada S.; Miyaura N.; Iyama T.; Tachikawa H. γ-Selective cross-coupling reactions of potassium allyltrifluoroborates with haloarenes catalyzed by a Pd(0)/D-t-BPF or Pd(0)/Josiphos ((R,S)-CyPF-t-Bu) complex: Mechanistic studies on transmetalation and enantioselection. Organometallics 2009, 28, 152–160 10.1021/om800832r. [DOI] [Google Scholar]
- Chausset-Boissarie L.; Ghozati K.; LaBine E.; Chen J. L.-Y.; Aggarwal V. K.; Crudden C. M. Enantiospecific, regioselective cross-coupling reactions of secondary allylic boronic esters. Chem. - Eur. J. 2013, 19, 17698–17701 10.1002/chem.201303683. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y.; Goda K.-I.; Hiyama T. Regio- and stereoselective cross-coupling reaction of optically active allylsilanes: Stereocontrol of palladium-mediated SE′ reactions. Tetrahedron Lett. 1994, 35, 1279–1282 10.1016/0040-4039(94)88043-3. [DOI] [Google Scholar]
- Denmark S. E.; Werner N. S. On the stereochemical course of palladium-catalyzed cross-coupling of allylic silanolate salts with aromatic bromides. J. Am. Chem. Soc. 2010, 132, 3612–3620 10.1021/ja910804u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X.; Strotman N. A.; Fu G. C. Catalytic asymmetric Hiyama cross-couplings of racemic α-bromo esters. J. Am. Chem. Soc. 2008, 130, 3302–3303 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- Lou S.; Fu G. C. Enantioselective alkenylation via nickel-catalyzed cross-coupling with organozirconium reagents. J. Am. Chem. Soc. 2010, 132, 5010–5011 10.1021/ja1017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caeiro J.; Pérez Sestelo J.; Sarandeses L. A. Enantioselective nickel-catalyzed cross-coupling reactions of trialkynylindium reagents with racemic secondary benzyl bromides. Chem. - Eur. J. 2008, 14, 741–746 10.1002/chem.200701035. [DOI] [PubMed] [Google Scholar]
- Willis M. C. Enantioselective desymmetrisation. J. Chem. Soc., Perkin Trans. 1 1999, 1765–1784 10.1039/a906269b. [DOI] [Google Scholar]
- Chen Y.; McDaid P.; Deng L. Asymmetric alcoholysis of cyclic anhydrides. Chem. Rev. 2003, 103, 2965–2983 10.1021/cr020037x. [DOI] [PubMed] [Google Scholar]
- Díaz de Villegas M. D.; Gálvez J. A.; Etayo P.; Badorrey R.; López-Ram-de-Víu P. Recent advances in enantioselective organocatalyzed anhydride desymmetrization and its application to the synthesis of valuable enantiopure compounds. Chem. Soc. Rev. 2011, 40, 5564–5587 10.1039/c1cs15120g. [DOI] [PubMed] [Google Scholar]
- Johnson J. B.; Rovis T. Enantioselective cross-coupling of anhydrides with organozinc reagents: The controlled formation of carbon-carbon bonds through the nucleophilic interception of metalacycles. Acc. Chem. Res. 2008, 41, 327–338 10.1021/ar700176t. [DOI] [PubMed] [Google Scholar]
- Bercot E. A.; Rovis T. Highly efficient nickel-catalyzed cross-coupling of succinic and glutaric anhydrides with organozinc reagents. J. Am. Chem. Soc. 2005, 127, 247–254 10.1021/ja044588b. [DOI] [PubMed] [Google Scholar]
- Giovannini R.; Stüdemann T.; Dussin G.; Knochel P. An efficient nickel-catalyzed cross-coupling between sp3 carbon centers. Angew. Chem., Int. Ed. 1998, 37, 2387–2390. [DOI] [PubMed] [Google Scholar]
- Giovannini R.; Stüdemann T.; Devasagayaraj A.; Dussin G.; Knochel P. New efficient nickel-catalyzed cross-coupling reaction between two Csp3 centers. J. Org. Chem. 1999, 64, 3544–3553 10.1021/jo982317b. [DOI] [PubMed] [Google Scholar]
- Bercot E. A.; Rovis T. A mild and efficient catalytic alkylative monofunctionalization of cyclic anhydrides. J. Am. Chem. Soc. 2002, 124, 174–175 10.1021/ja017086w. [DOI] [PubMed] [Google Scholar]
- Johnson J. B.; Bercot E. A.; Rowley J. M.; Coates G. W.; Rovis T. Ligand-dependent catalytic cycle and role of styrene in nickel-catalyzed anhydride cross-coupling: Evidence for turnover-limiting reductive elimination. J. Am. Chem. Soc. 2007, 129, 2718–2725 10.1021/ja067845g. [DOI] [PubMed] [Google Scholar]
- Bercot E. A.; Rovis T. A palladium-catalyzed enantioselective alkylative desymmetrization of meso-succinic anhydrides. J. Am. Chem. Soc. 2004, 126, 10248–10249 10.1021/ja046528b. [DOI] [PubMed] [Google Scholar]
- Cook M. J.; Rovis T. Rhodium-catalyzed enantioselective desymmetrization of meso-3,5-dimethyl glutaric anhydride: A general strategy to syn-deoxypolypropionate synthons. J. Am. Chem. Soc. 2007, 129, 9302–9303 10.1021/ja073269s. [DOI] [PubMed] [Google Scholar]
- Johnson J. B.; Cook M. J.; Rovis T. Ligand differentiated complementary Rh-catalyst systems for the enantioselective desymmetrization of meso-cyclic anhydrides. Tetrahedron 2009, 65, 3202–3210 10.1016/j.tet.2008.10.075. [DOI] [Google Scholar]
- Cook M. J.; Rovis T. Enantioselective rhodium-catalyzed alkylative desymmetrization of 3,5-dimethylglutaric anhydride. Synthesis 2009, 2009, 335–338 10.1055/s-0028-1083275. [DOI] [Google Scholar]
- Brand G. J.; Studte C.; Breit B. Iterative synthesis of (oligo)deoxypropionates via zinc-catalyzed enantiospecific sp3-sp3 cross-coupling. Org. Lett. 2009, 11, 4668–4670 10.1021/ol901944b. [DOI] [PubMed] [Google Scholar]
- Johnson J. B.; Bercot E. A.; Williams C. M.; Rovis T. A concise synthesis of eupomatilones 4, 6, and 7 by rhodium-catalyzed enantioselective desymmetrization of cyclic meso anhydrides with organozinc reagents generated in situ. Angew. Chem., Int. Ed. 2007, 46, 4514–4518 10.1002/anie.200700816. [DOI] [PubMed] [Google Scholar]
- Cho S. Y.; Shibasaki M. The asymmetric synthesis of cyclopentane derivatives by palladium-catalyzed coupling of prochiral alkylboron compounds. Tetrahedron: Asymmetry 1998, 9, 3751–3754 10.1016/S0957-4166(98)00386-3. [DOI] [Google Scholar]
- Sun C.; Potter B.; Morken J. P. A catalytic enantiotopic-group-selective Suzuki reaction for the construction of chiral organoboronates. J. Am. Chem. Soc. 2014, 136, 6534–6537 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo K.; Ohkubo T.; Hirokami M.; Shibata T. Chemoselective and regiospecific Suzuki coupling on a multisubstituted sp3-carbon in 1,1-diborylalkanes at room temperature. J. Am. Chem. Soc. 2010, 132, 11033–11035 10.1021/ja105176v. [DOI] [PubMed] [Google Scholar]
- Willis M. C.; Powell L. H. W.; Claverie C. K.; Watson S. J. Enantioselective Suzuki reactions: Catalytic asymmetric synthesis of compounds containing quaternary carbon centers. Angew. Chem., Int. Ed. 2004, 43, 1249–1251 10.1002/anie.200352648. [DOI] [PubMed] [Google Scholar]
- Shi B.-F.; Maugel N.; Zhang Y.-H.; Yu J.-Q. Pd(II)-catalyzed enantioselective activation of C(sp2)-H and C(sp3)-H bonds using monoprotected amino acids as chiral ligands. Angew. Chem., Int. Ed. 2008, 47, 4882–4886 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- Wasa M.; Engle K. M.; Lin D. W.; Yoo E. J.; Yu J.-Q. Pd(II)-catalyzed enantioselective C-H activation of cyclopropanes. J. Am. Chem. Soc. 2011, 133, 19598–19601 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K.-J.; Lin D. W.; Miura M.; Zhu R.-Y.; Gong W.; Wasa M.; Yu J.-Q. Palladium(II)-catalyzed enantioselective C(sp3)-H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc. 2014, 136, 8138–8142 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Tamao K.; Katsuro Y.; Nakae I.; Kumada M. Asymmetric hydrosilylation of olefins catalyzed by a chiral ferrocenylphosphine-palladium complex. Asymmetric synthesis of optically active alcohols and bromides from olefins. Tetrahedron Lett. 1980, 21, 1871–1874 10.1016/S0040-4039(00)92802-8. [DOI] [Google Scholar]
- Hatanaka Y.; Hiyama T. Stereochemistry of the cross-coupling reaction of chiral alkylsilanes with aryl triflates: A novel approach to optically active compounds. J. Am. Chem. Soc. 1990, 112, 7793–7794 10.1021/ja00177a048. [DOI] [Google Scholar]
- Ye J.; Bhatt R. K.; Falck J. R. Stereospecific α-alkoxystannane couplings with acyl chlorides: Total synthesis of (+)-goniofufurone. Tetrahedron Lett. 1993, 34, 8007–8010 10.1016/S0040-4039(00)61436-3. [DOI] [Google Scholar]
- Ye J.; Bhatt R. K.; Falck J. R. Stereospecific palladium/copper cocatalyzed cross-coupling of α-alkoxystannes and α-aminostannanes with acyl chlorides. J. Am. Chem. Soc. 1994, 116, 1–5 10.1021/ja00080a001. [DOI] [Google Scholar]
- Itami K.; Kamei T.; Yoshida J.-i. Unusually accelerated silylmethyl transfer from tin in Stille coupling: Implication of coordination-driven transmetalation. J. Am. Chem. Soc. 2001, 123, 8773–8779 10.1021/ja0160593. [DOI] [PubMed] [Google Scholar]
- Falck J. R.; Bhatt R. K.; Ye J. Tin-copper transmetalation: Cross-coupling of α-heteroatom-substituted alkyltributylstannanes with organohalides. J. Am. Chem. Soc. 1995, 117, 5973–5982 10.1021/ja00127a010. [DOI] [Google Scholar]
- Lange H.; Fröhlich R.; Hoppe D. Cu(I)-catalyzed stereospecific coupling reactions of enantioenriched α-stannylated benzyl carbamates and their application. Tetrahedron 2008, 64, 9123–9135 10.1016/j.tet.2008.06.092. [DOI] [Google Scholar]
- Mohapatra S.; Bandyopadhyay A.; Barma D. K.; Capdevila J. H.; Falck J. R. Chiral α,β-dialkoxy- and α-alkoxy-β-aminostannanes: Preparation and copper-mediated cross-coupling. Org. Lett. 2003, 5, 4759–4762 10.1021/ol035458v. [DOI] [PubMed] [Google Scholar]
- Falck J. R.; Barma D. K.; Mohapatra S.; Bandyopadhyay A.; Reddy K. M.; Qi J.; Campbell W. B. Asymmetric synthesis of the stereoisomers of 11,12,15(S)-trihydroxyeicosa-5(Z),8(Z),13(E)-trienoic acid, a potent endothelium-derived vasodilator. Bioorg. Med. Chem. Lett. 2004, 14, 4987–4990 10.1016/j.bmcl.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Li H.; He A.; Falck J. R.; Liebeskind L. S. Stereocontrolled synthesis of α-amino-α′-alkoxy ketones by a copper-catalyzed cross-coupling of peptidic thiol esters and α-alkoxyalkylstannanes. Org. Lett. 2011, 13, 3682–3685 10.1021/ol201330j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goli M.; He A.; Falck J. R. Pd-catalyzed cross-coupling of α-(acyloxy)-tri-N-butylstannanes with alkenyl, aryl, and heteroaryl electrophiles. Org. Lett. 2011, 13, 344–346 10.1021/ol102863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells K. W.; Chong J. M. Stille coupling of stereochemically defined α-sulfonamidoorganostannanes. J. Am. Chem. Soc. 2004, 126, 15666–15667 10.1021/ja044354s. [DOI] [PubMed] [Google Scholar]
- Kalkofen R.; Hoppe D. First example of an enantiospecific sp3-sp2 Stille coupling of a chiral allylstannane with aryl halides. Synlett 2006, 2006, 1959–1961 10.1055/s-2006-947353. [DOI] [Google Scholar]
- Vedejs E.; Haight A. R.; Moss W. O. Internal coordination at tin promotes selective alkyl transfer in the Stille coupling reaction. J. Am. Chem. Soc. 1992, 114, 6556–6558 10.1021/ja00042a044. [DOI] [Google Scholar]
- Li L.; Wang C.-Y.; Huang R.; Biscoe M. R. Stereoretentive Pd-catalysed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl halides. Nat. Chem. 2013, 5, 607–612 10.1038/nchem.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Z.; Deng M.-Z. Cross-coupling reaction of cyclopropylboronic acid with bromoarenes. J. Chem. Soc., Perkin Trans. 1 1996, 2663–2664 10.1039/p19960002663. [DOI] [Google Scholar]
- Fang G. H.; Yan Z. J.; Deng M. Z. Palladium-catalyzed cross-coupling of stereospecific potassium cyclopropyl trifluoroborates with aryl bromides. Org. Lett. 2004, 6, 357–360 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]
- Charette A. B.; Mathieu S.; Fournier J.-F. Diastereoselective synthesis of 1,2,3-substituted potassium cyclopropyl trifluoroborates via an unusual zinc-boron exchange. Synlett 2005, 1779–1782 10.1055/s-2005-871557. [DOI] [Google Scholar]
- Zhou S.-M.; Deng M.-Z.; Xia L.-J.; Tang M.-H. Efficient Suzuki-type cross-coupling of enantiomerically pure cyclopropylboronic acids. Angew. Chem., Int. Ed. 1998, 37, 2845–2847. [DOI] [PubMed] [Google Scholar]
- Luithle J. E. A.; Pietruszka J. Synthesis of enantiomerically pure cyclopropanes from cyclopropylboronic acids. J. Org. Chem. 1999, 64, 8287–8297 10.1021/jo9910278. [DOI] [PubMed] [Google Scholar]
- Luithle J. E. A.; Pietruszka J. (2R,3R)-1,4-dimethoxy-1,1,4,4-tetraphenyl-2,3-butanediol: Chiral auxiliary and efficient protecting group for boronic acids. J. Org. Chem. 2000, 65, 9194–9200 10.1021/jo0056601. [DOI] [PubMed] [Google Scholar]
- Rubina M.; Rubin M.; Gevorgyan V. Catalytic enantioselective hydroboration of cyclopropenes. J. Am. Chem. Soc. 2003, 125, 7198–7199 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]
- Hohn E.; Pietruszka J.; Solduga G. Synthesis of enantiomerically pure cyclopropyl trifluoroborates. Synlett 2006, 2006, 1531–1534 10.1055/s-2006-944185. [DOI] [Google Scholar]
- Yao M.-L.; Deng M.-Z. A practical approach to stereodefined cyclopropyl-substituted heteroarenes using a Suzuki-type reaction. New J. Chem. 2000, 24, 425–428 10.1039/b001051k. [DOI] [Google Scholar]
- Yao M.-L.; Deng M.-Z. Palladium-catalyzed cross-coupling reaction of cyclopropylboronic acids with aryl triflates. Synthesis 2000, 2000, 1095–1100 10.1055/s-2000-6313. [DOI] [PubMed] [Google Scholar]
- Chen H.; Deng M.-Z. A novel stereocontrolled synthesis of 1,2-trans cyclopropyl ketones via Suzuki-type coupling of acid chlorides with cyclopropylboronic acids. Org. Lett. 2000, 2, 1649–1651 10.1021/ol000013h. [DOI] [PubMed] [Google Scholar]
- Edwards D. R.; Hleba Y. B.; Lata C. J.; Calhoun L. A.; Crudden C. M. Regioselectivity of the rhodium-catalyzed hydroboration of vinyl arenes: Electronic twists and mechanistic shifts. Angew. Chem., Int. Ed. 2007, 46, 7799–7802 10.1002/anie.200702563. [DOI] [PubMed] [Google Scholar]
- Glasspoole B. W.; Oderinde M. S.; Moore B. D.; Antoft-Finch A.; Crudden C. M. Highly chemoselective and enantiospecific Suzuki-Miyaura cross-couplings of benzylic organoboronic esters. Synthesis 2013, 45, 1759–1763 10.1055/s-0033-1338875. [DOI] [Google Scholar]
- Glasspoole B. W.; Ghozati K.; Moir J. W.; Crudden C. M. Suzuki-Miyaura cross-couplings of secondary allylic boronic esters. Chem. Commun. 2012, 48, 1230–1232 10.1039/C2CC16076E. [DOI] [PubMed] [Google Scholar]
- Matthew S. C.; Glasspoole B. W.; Eisenberger P.; Crudden C. M. Synthesis of enantiomerically enriched triarylmethanes by enantiospecific Suzuki-Miyaura cross-coupling reactions. J. Am. Chem. Soc. 2014, 136, 5828–5831 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]
- Stymiest J. L.; Bagutski V.; French R. M.; Aggarwal V. K. Enantiodivergent conversion of chiral secondary alcohols into tertiary alcohols. Nature 2008, 456, 778–783 10.1038/nature07592. [DOI] [PubMed] [Google Scholar]
- Daini M.; Suginome M. Palladium-catalyzed, stereoselective, cyclizative alkenylboration of carbon-carbon double bonds through activation of a boron-chlorine bond. J. Am. Chem. Soc. 2011, 133, 4758–4761 10.1021/ja200856t. [DOI] [PubMed] [Google Scholar]
- Ohmura T.; Awano T.; Suginome M. Stereospecific Suzuki-Miyaura coupling of chiral α-(acylamino)benzylboronic esters with inversion of configuration. J. Am. Chem. Soc. 2010, 132, 13191–13193 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- Awano T.; Ohmura T.; Suginome M. Inversion or retention? Effects of acidic additives on the stereochemical course in enantiospecific Suzuki-Miyaura coupling of α-(acetylamino)benzylboronic esters. J. Am. Chem. Soc. 2011, 133, 20738–20741 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- Chea H.; Sim H.-S.; Yun J. Copper-catalyzed conjugate addition of diboron reagents to α,β-unsaturated amides: Highly reactive copper-1,2-bis(diphenylphosphino)benzene catalyst system. Adv. Synth. Catal. 2009, 351, 855–858 10.1002/adsc.200900040. [DOI] [Google Scholar]
- Sandrock D. L.; Jean-Gérard L.; Chen C.-y.; Dreher S. D.; Molander G. A. Stereospecific cross-coupling of secondary alkyl β-trifluoroboratoamides. J. Am. Chem. Soc. 2010, 132, 17108–17110 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; Wisniewski S. R.; Hosseini-Sarvari M. Synthesis and Suzuki-Miyaura cross-coupling of enantioenriched secondary potassium β-trifluoroboratoamides: Catalytic, asymmetric conjugate addition of bisboronic acid and tetrakis(dimethylamino) diboron to α,β-unsaturated carbonyl compounds. Adv. Synth. Catal. 2013, 355, 3037–3057 10.1002/adsc.201300640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudasaini B.; Janesko B. G. Computational investigation of selectivity in Suzuki–Miyaura coupling of secondary alkyl boranes. Organometallics 2011, 30, 4564–4571 10.1021/om200305r. [DOI] [Google Scholar]
- Molander G. A.; Wisniewski S. R. Stereospecific cross-coupling of secondary organotrifluoroborates: Potassium 1-(benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc. 2012, 134, 16856–16868 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C. H.; McDonald R.; Hall D. G. Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem. 2011, 3, 894–899 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jeon H.; Yun J. Regio- and enantioselective copper(I)-catalyzed hydroboration of borylalkenes: Asymmetric synthesis of 1,1-diborylalkanes. Angew. Chem., Int. Ed. 2013, 52, 3989–3992 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]
- Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. Efficient cross-coupling of secondary alkyltrifluoroborates with aryl chlorides-reaction discovery using parallel microscale experimentation. J. Am. Chem. Soc. 2008, 130, 9257–9259 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogenband A.; Lange J. H. M.; Terpstra J. W.; Koch M.; Visser G. M.; Visser M.; Korstanje T. J.; Jastrzebski J. T. B. H. Ruphos-mediated Suzuki cross-coupling of secondary alkyl trifluoroborates. Tetrahedron Lett. 2008, 49, 4122–4124 10.1016/j.tetlet.2008.04.129. [DOI] [Google Scholar]
- Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. Stereospecific Pd-catalyzed cross-coupling reactions of secondary alkylboron nucleophiles and aryl chlorides. J. Am. Chem. Soc. 2014, 136, 14027–14030 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin D. Y.; Koehl W. J. Effect of solvent on the steric stability of lithium reagents. J. Am. Chem. Soc. 1962, 84, 1967–1973 10.1021/ja00869a039. [DOI] [Google Scholar]
- Hoffmann R. W. The quest for chiral Grignard reagents. Chem. Soc. Rev. 2003, 32, 225–230 10.1039/b300840c. [DOI] [PubMed] [Google Scholar]
- Hölzer B.; Hoffmann R. W. Kumada-Corriu coupling of Grignard reagents, probed with a chiral Grignard reagent. Chem. Commun. 2003, 732–733 10.1039/b300033h. [DOI] [PubMed] [Google Scholar]
- Boudier A.; Knochel P. Palladium catalyzed stereoselective cross-couplings and acylations of chiral secondary diorganozincs. Tetrahedron Lett. 1999, 40, 687–690 10.1016/S0040-4039(98)02581-7. [DOI] [Google Scholar]
- Boudier A.; Darcel C.; Flachsmann F.; Micouin L.; Oestreich M.; Knochel P. Stereoselective preparation and reactions of configurationally defined dialkylzinc compounds. Chem. - Eur. J. 2000, 6, 2748–2761. [DOI] [PubMed] [Google Scholar]
- Boudier A.; Hupe E.; Knochel P. Highly diastereoselective synthesis of monocyclic and bicyclic secondary diorganozinc reagents with defined configuration. Angew. Chem., Int. Ed. 2000, 39, 2294–2297. [DOI] [PubMed] [Google Scholar]
- Hupe E.; Calaza M. I.; Knochel P. Synthesis and reaction of secondary and primary diorganozinc reagents using a boron-zinc exchange reaction - a useful method for the stereo- and regioselective formation of new carbon-carbon bonds. J. Organomet. Chem. 2003, 680, 136–142 10.1016/S0022-328X(03)00237-7. [DOI] [Google Scholar]
- Beak P.; Basu A.; Gallagher D. J.; Park Y. S.; Thayumanavan S. Regioselective, diastereoselective, and enantioselective lithiation-substitution sequences: Reaction pathways and synthetic applications. Acc. Chem. Res. 1996, 29, 552–560 10.1021/ar950142b. [DOI] [Google Scholar]
- Gelardi G.; Barker G.; O’Brien P.; Blakemore D. C. Asymmetric lithiation trapping of N-Boc heterocycles at temperatures above −78 °C. Org. Lett. 2013, 15, 5424–5427 10.1021/ol402395j. [DOI] [PubMed] [Google Scholar]
- Klapars A.; Campos K. R.; Waldman J. H.; Zewge D.; Dormer P. G.; Chen C.-y. Enantioselective Pd-catalyzed α-arylation of N-Boc-pyrrolidine: The key to an efficient and practical synthesis of a glucokinase activator. J. Org. Chem. 2008, 73, 4986–4993 10.1021/jo8006804. [DOI] [PubMed] [Google Scholar]
- Barker G.; McGrath J. L.; Klapars A.; Stead D.; Zhou G.; Campos K. R.; O’Brien P. Enantioselective, palladium-catalyzed α-arylation of N-Boc pyrrolidine: In situ react IR spectroscopic monitoring, scope, and synthetic applications. J. Org. Chem. 2011, 76, 5936–5953 10.1021/jo2011347. [DOI] [PubMed] [Google Scholar]
- Bailey W. F.; Beak P.; Kerrick S. T.; Ma S.; Wiberg K. B. An experimental and computational investigation of the enantioselective deprotonation of Boc-piperidine. J. Am. Chem. Soc. 2002, 124, 1889–1896 10.1021/ja012169y. [DOI] [PubMed] [Google Scholar]
- Beak P.; Anderson D. R.; Curtis M. D.; Laumer J. M.; Pippel D. J.; Weisenburger G. A. Dynamic thermodynamic resolution: Control of enantioselectivity through diastereomeric equilibration. Acc. Chem. Res. 2000, 33, 715–727 10.1021/ar000077s. [DOI] [PubMed] [Google Scholar]
- Coldham I.; Raimbault S.; Whittaker D. T. E.; Chovatia P. T.; Leonori D.; Patel J. J.; Sheikh N. S. Asymmetric substitutions of 2-lithiated N-Boc-piperidine and N-Boc-azepine by dynamic resolution. Chem. - Eur. J. 2010, 16, 4082–4090 10.1002/chem.200903059. [DOI] [PubMed] [Google Scholar]
- Beng T. K.; Yousaf T. I.; Coldham I.; Gawley R. E. Enantiomerization dynamics and a catalytic dynamic resolution of N-trimethylallyl-2-lithiopyrrolidine. J. Am. Chem. Soc. 2009, 131, 6908–6909 10.1021/ja900755a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beng T. K.; Gawley R. E. Highly enantioselective catalytic dynamic resolution of N-Boc-2-lithiopiperidine: Synthesis of (R)-(+)-N-Boc-pipecolic acid, (S)-(−)-coniine, (S)-(+)-pelletierine, (+)-β-conhydrine, and (S)-(−)-ropivacaine and formal synthesis of (−)-lasubine II and (+)-cermizine C. J. Am. Chem. Soc. 2010, 132, 12216–12217 10.1021/ja105772z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beng T. K.; Woo J. S.; Gawley R. E. Synthetic applications and inversion dynamics of configurationally stable 2-lithio-2-arylpyrrolidines and -piperidines. J. Am. Chem. Soc. 2012, 134, 14764–14771 10.1021/ja306276w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beng T. K.; Gawley R. E. Application of catalytic dynamic resolution of N-Boc-2-lithiopiperidine to the asymmetric synthesis of 2-aryl and 2-vinyl piperidines. Org. Lett. 2011, 13, 394–397 10.1021/ol102682r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beng T. K.; Tyree W. S.; Parker T.; Su C.; Williard P. G.; Gawley R. E. Dynamics of catalytic resolution of 2-lithio-N-Boc-piperidine by ligand exchange. J. Am. Chem. Soc. 2012, 134, 16845–16855 10.1021/ja307796e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit Y.; Sanner C.; Larchevêque M. Stereoselective synthesis of optically active α-methyl esters. Tetrahedron Lett. 1990, 31, 2149–2152 10.1016/0040-4039(90)80095-4. [DOI] [Google Scholar]
- Studte C.; Breit B. Zinc-catalyzed enantiospecific sp3-sp3 cross-coupling of α-hydroxy ester triflates with Grignard reagents. Angew. Chem., Int. Ed. 2008, 47, 5451–5455 10.1002/anie.200800733. [DOI] [PubMed] [Google Scholar]
- López-Pérez A.; Adrio J.; Carretero J. C. Palladium-catalyzed cross-coupling reaction of secondary benzylic bromides with Grignard reagents. Org. Lett. 2009, 11, 5514–5517 10.1021/ol902335c. [DOI] [PubMed] [Google Scholar]
- Taylor B. L. H.; Harris M. R.; Jarvo E. R. Synthesis of enantioenriched triarylmethanes by stereospecific cross-coupling reactions. Angew. Chem., Int. Ed. 2012, 51, 7790–7793 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- Yang C.-T.; Zhang Z.-Q.; Liang J.; Liu J.-H.; Lu X.-Y.; Chen H.-H.; Liu L. Copper-catalyzed cross-coupling of nonactivated secondary alkyl halides and tosylates with secondary alkyl Grignard reagents. J. Am. Chem. Soc. 2012, 134, 11124–11127 10.1021/ja304848n. [DOI] [PubMed] [Google Scholar]
- Terao J.; Todo H.; Begum S. A.; Kuniyasu H.; Kambe N. Copper-catalyzed cross-coupling reaction of Grignard reagents with primary alkyl halides: Remarkable effect of 1-phenylpropyne. Angew. Chem., Int. Ed. 2007, 46, 2086–2089 10.1002/anie.200603451. [DOI] [PubMed] [Google Scholar]
- Malosh C. F.; Ready J. M. Catalytic cross-coupling of alkylzinc halides with α-chloroketones. J. Am. Chem. Soc. 2004, 126, 10240–10241 10.1021/ja0467768. [DOI] [PubMed] [Google Scholar]
- Wisniewska H. M.; Swift E. C.; Jarvo E. R. Functional-group-tolerant, nickel-catalyzed cross-coupling reaction for enantioselective construction of tertiary methyl-bearing stereocenters. J. Am. Chem. Soc. 2013, 135, 9083–9090 10.1021/ja4034999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefson E. J.; Dawson D. D.; Osborne C. A.; Jarvo E. R. Stereospecific cross-coupling reactions of aryl-substituted tetrahydrofurans, tetrahydropyrans, and lactones. J. Am. Chem. Soc. 2014, 136, 14951–14958 10.1021/ja5076426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez N.; Ramírez de Arellano C.; Asensio G.; Medio-Simón M. Palladium-catalyzed Suzuki-Miyaura reaction involving a secondary sp3 carbon: Studies of stereochemistry and scope of the reaction. Chem. - Eur. J. 2007, 13, 4223–4229 10.1002/chem.200601488. [DOI] [PubMed] [Google Scholar]
- Gourlaouen C.; Ujaque G.; Lledós A.; Medio-Simon M.; Asensio G.; Maseras F. Why is the Suzuki-Miyaura cross-coupling of sp3 carbons in α-bromo sulfoxide systems fast and stereoselective? A DFT study on the mechanism. J. Org. Chem. 2009, 74, 4049–4054 10.1021/jo900178c. [DOI] [PubMed] [Google Scholar]
- Pudasaini B.; Janesko B. G. Computational mechanistic study of stereoselective Suzuki coupling of an α-cyano-activated secondary alkyl. Organometallics 2012, 31, 4610–4618 10.1021/om300455g. [DOI] [Google Scholar]
- Harris M. R.; Hanna L. E.; Greene M. A.; Moore C. E.; Jarvo E. R. Retention or inversion in stereospecific nickel-catalyzed cross-coupling of benzylic carbamates with arylboronic esters: Control of absolute stereochemistry with an achiral catalyst. J. Am. Chem. Soc. 2013, 135, 3303–3306 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-Y.; Doyle A. G. Nickel-catalyzed Negishi alkylations of styrenyl aziridines. J. Am. Chem. Soc. 2012, 134, 9541–9544 10.1021/ja3013825. [DOI] [PubMed] [Google Scholar]
- Nielsen D. K.; Huang C.-Y.; Doyle A. G. Directed nickel-catalyzed Negishi cross coupling of alkyl aziridines. J. Am. Chem. Soc. 2013, 135, 13605–13609 10.1021/ja4076716. [DOI] [PubMed] [Google Scholar]
- Takeda Y.; Ikeda Y.; Kuroda A.; Tanaka S.; Minakata S. Pd/NHC-catalyzed enantiospecific and regioselective Suzuki-Miyaura arylation of 2-arylaziridines: Synthesis of enantioenriched 2-arylphenethylamine derivatives. J. Am. Chem. Soc. 2014, 136, 8544–8547 10.1021/ja5039616. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Van Vranken D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem. Rev. 2003, 103, 2921–2943 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Yoshikai N.; Nakamura E. Mechanisms of nucleophilic organocopper(I) reactions. Chem. Rev. 2012, 112, 2339–2372 10.1021/cr200241f. [DOI] [PubMed] [Google Scholar]
- Geurts K.; Fletcher S. P.; van Zijl A. W.; Minnaard A. J.; Feringa B. L. Copper-catalyzed asymmetric allylic substitution reactions with organozinc and Grignard reagents. Pure Appl. Chem. 2008, 80, 1025–1037 10.1351/pac200880051025. [DOI] [Google Scholar]
- Consiglio G.; Morandini F.; Piccolo O. Nickel-catalyzed asymmetric alkylation of some chiral and achiral allylic alcohols. Helv. Chim. Acta 1980, 63, 987–989 10.1002/hlca.19800630427. [DOI] [Google Scholar]
- Consiglio G.; Morandini F.; Piccolo O. Nickel catalysed asymmetric coupling reaction between allyl phenyl ethers and Grignard reagents. J. Chem. Soc., Chem. Commun. 1983, 112–114 10.1039/c39830000112. [DOI] [Google Scholar]
- Consiglio G.; Piccolo O.; Roncetti L.; Morandini F. Asymmetric nickel-catalyzed cross-coupling reaction of allylic substrates with grignard-reagents. Tetrahedron 1986, 42, 2043–2053 10.1016/S0040-4020(01)87621-3. [DOI] [Google Scholar]
- Indolese A. F.; Consiglio G. Enantioselective nickel-catalyzed Grignard cross-coupling of allyl electrophiles. The influence of the alkyl group of the Grignard reagent. Organometallics 1994, 13, 2230–2234 10.1021/om00018a016. [DOI] [Google Scholar]
- Consiglio G.; Indolese A. Enantioselective allylation of Grignard reagents with nickel-diphosphine catalysts. Organometallics 1991, 10, 3425–3427 10.1021/om00056a007. [DOI] [Google Scholar]
- Chérest M.; Felkin H.; Umpleby J. D.; Davies S. G. Asymmetric synthesis in the nickel-complex-catalyzed formation of olefins from allyl alcohols and Grignard reagents. J. Chem. Soc., Chem. Commun. 1981, 681–682 10.1039/c39810000681. [DOI] [Google Scholar]
- Hiyama T.; Wakasa N. Asymmetric coupling of arylmagnesium bromides with allylic esters. Tetrahedron Lett. 1985, 26, 3259–3262 10.1016/S0040-4039(00)98166-8. [DOI] [Google Scholar]
- Nagel U.; Nedden H. G. Enantioselective catalysis XVI: Regio- and enantioselectivity in nickel-catalysed cross-coupling reactions of allylic substrates with Grignard reagents. Inorg. Chim. Acta 1998, 269, 34–42 10.1016/S0020-1693(97)05770-8. [DOI] [Google Scholar]
- Chung K.-G.; Miyake Y.; Uemura S. Nickel(0)-catalysed asymmetric cross-coupling reactions of allylic compounds with Grignard reagents using optically active oxazolinylferrocenylphosphines as ligands. J. Chem. Soc., Perkin Trans. 1 2000, 2725–2729 10.1039/b002547j. [DOI] [Google Scholar]
- Nomura N.; RajanBabu T. V. Nickel-catalyzed asymmetric allylation of alkyl Grignard reagents. Effect of ligands, leaving groups and a kinetic resolution with a hard nucleophile. Tetrahedron Lett. 1997, 38, 1713–1716 10.1016/S0040-4039(97)00178-0. [DOI] [Google Scholar]
- Gomez-Bengoa E.; Heron N. M.; Didiuk M. T.; Luchaco C. A.; Hoveyda A. H. Ni-catalyzed asymmetric addition of Grignard reagents to unsaturated cyclic acetals. The influence of added phosphine on enantioselectivity. J. Am. Chem. Soc. 1998, 120, 7649–7650 10.1021/ja980499l. [DOI] [Google Scholar]
- Fotiadu F.; Cros P.; Faure B.; Buono G. Application of AMPP-Pd catalysts in an unusual asymmetric allylic coupling reaction of 1-trimethylsilyl vinyl magnesium bromide. Tetrahedron Lett. 1990, 31, 77–80 10.1016/S0040-4039(00)94338-7. [DOI] [Google Scholar]
- van Klaveren M.; Persson E. S. M.; del Villar A.; Grove D. M.; Bäckvall J.-E.; van Koten G. Chiral arenethiolatocopper(I) catalyzed substitution reactions of acyclic allylic substrates with Grignard reagents. Tetrahedron Lett. 1995, 36, 3059–3062 10.1016/0040-4039(95)00426-D. [DOI] [Google Scholar]
- Selim K. B.; Yamada K.; Tomioka K. Copper-catalyzed asymmetric allylic substitution with aryl and ethyl Grignard reagents. Chem. Commun. 2008, 5140–5142 10.1039/b809140d. [DOI] [PubMed] [Google Scholar]
- Lölsberg W.; Ye S.; Schmalz H.-G. Enantioselective copper-catalysed allylic alkylation of cinnamyl chlorides by Grignard reagents using chiral phosphine-phosphite ligands. Adv. Synth. Catal. 2010, 352, 2023–2031 10.1002/adsc.201000213. [DOI] [Google Scholar]
- Fang F.; Zhang H.; Xie F.; Yang G.; Zhang W. B. Highly enantioselective copper-catalyzed allylic alkylation with atropos phosphoramidites bearing a D2-symmetric biphenyl backbone. Tetrahedron 2010, 66, 3593–3598 10.1016/j.tet.2010.03.031. [DOI] [Google Scholar]
- Magre M.; Mazuela J.; Diéguez M.; Pàmies O.; Alexakis A. Furanoside phosphite-phosphoroamidite and diphosphoroamidite ligands applied to asymmetric Cu-catalyzed allylic substitution reactions. Tetrahedron: Asymmetry 2012, 23, 67–71 10.1016/j.tetasy.2012.01.003. [DOI] [Google Scholar]
- Magrez M.; Le Guen Y.; Baslé O.; Crévisy C.; Mauduit M. Bidentate hydroxyalkyl NHC ligands for the copper-catalyzed asymmetric allylic substitution of allyl phosphates with Grignard reagents. Chem. - Eur. J. 2013, 19, 1199–1203 10.1002/chem.201203969. [DOI] [PubMed] [Google Scholar]
- den Hartog T.; Maciá B.; Minnaard A. J.; Feringa B. Copper-catalyzed asymmetric allylic alkylation of halocrotonates: Efficient synthesis of versatile chiral multifunctional building blocks. Adv. Synth. Catal. 2010, 352, 999–1013 10.1002/adsc.201000109. [DOI] [Google Scholar]
- Hornillos V.; Pérez M.; Fañanás-Mastral M.; Feringa B. L. Cu-catalyzed asymmetric allylic alkylation of phosphonates and phosphine oxides with Grignard reagents. Chem. - Eur. J. 2013, 19, 5432–5441 10.1002/chem.201204364. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Fañanás-Mastral M.; Minnaard A. J.; Feringa B. L. A novel catalytic asymmetric route towards skipped dienes with a methyl-substituted central stereogenic carbon. Chem. Commun. 2013, 49, 3309–3311 10.1039/c3cc41021h. [DOI] [PubMed] [Google Scholar]
- Li H.; Alexakis A. Enyne chlorides: Substrates for copper-catalyzed asymmetric allylic alkylation. Angew. Chem., Int. Ed. 2012, 51, 1055–1058 10.1002/anie.201107129. [DOI] [PubMed] [Google Scholar]
- Giacomina F.; Riat D.; Alexakis A. ω-Ethylenic allylic substrates as alternatives to cyclic substrates in copper- and iridium-catalyzed asymmetric allylic alkylation. Org. Lett. 2010, 12, 1156–1159 10.1021/ol100162y. [DOI] [PubMed] [Google Scholar]
- Teichert J. F.; Zhang S.; van Zijl A. W.; Slaa J. W.; Minnaard A. J.; Feringa B. L. Asymmetric allylic alkylation in combination with ring-closing metathesis for the preparation of chiral N-heterocycles. Org. Lett. 2010, 12, 4658–4660 10.1021/ol101944j. [DOI] [PubMed] [Google Scholar]
- Mao B.; Geurts K.; Fañanás-Mastral M.; van Zijl A. W.; Fletcher S. P.; Minnaard A. J.; Feringa B. L. Catalytic enantioselective synthesis of naturally occurring butenolides via hetero-allylic alkylation and ring closing metathesis. Org. Lett. 2011, 13, 948–951 10.1021/ol102994q. [DOI] [PubMed] [Google Scholar]
- Giacomina F.; Alexakis A. Construction of enantioenriched cyclic compounds by asymmetric allylic alkylation and ring-closing metathesis. Eur. J. Org. Chem. 2013, 2013, 6710–6721 10.1002/ejoc.201300971. [DOI] [Google Scholar]
- Hornillos V.; van Zijl A. W.; Feringa B. L. Catalytic asymmetric synthesis of chromenes and tetrahydroquinolines via sequential allylic alkylation and intramolecular Heck coupling. Chem. Commun. 2012, 48, 3712–3714 10.1039/c2cc30395g. [DOI] [PubMed] [Google Scholar]
- Fañanás-Mastral M.; Feringa B. L. Copper-catalyzed regio- and enantioselective synthesis of chiral enol acetates and β-substituted aldehydes. J. Am. Chem. Soc. 2010, 132, 13152–13153 10.1021/ja105585y. [DOI] [PubMed] [Google Scholar]
- Giannerini M.; Fañanás-Mastral M.; Feringa B. L. Z-selective copper-catalyzed asymmetric allylic alkylation with Grignard reagents. J. Am. Chem. Soc. 2012, 134, 4108–4111 10.1021/ja300743t. [DOI] [PubMed] [Google Scholar]
- Selim K. B.; Matsumoto Y.; Yamada K.; Tomioka K. Efficient chiral N-heterocyclic carbene/copper(I)-catalyzed asymmetric allylic arylation with aryl Grignard reagents. Angew. Chem., Int. Ed. 2009, 48, 8733–8735 10.1002/anie.200904676. [DOI] [PubMed] [Google Scholar]
- Selim K. B.; Nakanishi H.; Matsumoto Y.; Yamamoto Y.; Yamada K.; Tomioka K. Chiral N-heterocyclic carbene-copper(I)-catalyzed asymmetric allylic arylation of aliphatic allylic bromides: Steric and electronic effects on γ-selectivity. J. Org. Chem. 2011, 76, 1398–1408 10.1021/jo102386s. [DOI] [PubMed] [Google Scholar]
- Hornillos V.; Pérez M.; Fañanás-Mastral M.; Feringa B. L. Copper-catalyzed enantioselective allyl-allyl cross-coupling. J. Am. Chem. Soc. 2013, 135, 2140–2143 10.1021/ja312487r. [DOI] [PubMed] [Google Scholar]
- Langlois J.-B.; Alexakis A. Dynamic kinetic asymmetric transformation in copper catalyzed allylic alkylation. Chem. Commun. 2009, 3868–3870 10.1039/b907722g. [DOI] [PubMed] [Google Scholar]
- Langlois J.-B.; Alexakis A. Copper-catalyzed asymmetric allylic alkylation of racemic cyclic substrates: Application of dynamic kinetic asymmetric transformation (DYKAT). Adv. Synth. Catal. 2010, 352, 447–457 10.1002/adsc.200900790. [DOI] [Google Scholar]
- Langlois J.-B.; Emery D.; Mareda J.; Alexakis A. Mechanistic identification and improvement of a direct enantioconvergent transformation in copper-catalyzed asymmetric allylic alkylation. Chem. Sci. 2012, 3, 1062–1069 10.1039/C2SC00868H. [DOI] [Google Scholar]
- Langlois J.-B.; Alexakis A. Identification of a valuable kinetic process in copper-catalyzed asymmetric allylic alkylation. Angew. Chem., Int. Ed. 2011, 50, 1877–1881 10.1002/anie.201005373. [DOI] [PubMed] [Google Scholar]
- Fiaud J.-C.; Aribi-Zouioueche L. Palladium-catalyzed asymmetric coupling reactions between allylic acetates and organozinc reagents. Mechanistic implications. J. Organomet. Chem. 1985, 295, 383–387 10.1016/0022-328X(85)80323-5. [DOI] [Google Scholar]
- Fiaud J.-C.; Legros J.-Y. Preparation of optically pure 1,2,5-triphenylphospholane. Use as ligand for enantioselective transition-metal catalysis. Tetrahedron Lett. 1991, 32, 5089–5092 10.1016/S0040-4039(00)93435-X. [DOI] [Google Scholar]
- Dübner F.; Knochel P. Copper(I)-catalyzed enantioselective substitution of allyl chlorides with diorganozinc compounds. Angew. Chem., Int. Ed. 1999, 38, 379–381. [DOI] [PubMed] [Google Scholar]
- Son S.; Fu G. C. Nickel-catalyzed asymmetric Negishi cross-couplings of secondary allylic chlorides with alkylzincs. J. Am. Chem. Soc. 2008, 130, 2756–2757 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- Tamaru Y.; Tanaka A.; Yasui K.; Goto S.; Tanaka S. Highly stereoselective allylation of benzaldehyde: Generation of a stereochemically defined allylzinc species from a π-allylpalladium intermediate and diethylzinc. Angew. Chem., Int. Ed. Engl. 1995, 34, 787–789 10.1002/anie.199507871. [DOI] [Google Scholar]
- Howell G. P.; Minnaard A. J.; Feringa B. L. Asymmetric allylation of aryl aldehydes: Studies on the scope and mechanism of the palladium catalysed diethylzinc mediated umpolung using phosphoramidite ligands. Org. Biomol. Chem. 2006, 4, 1278–1283 10.1039/b518165h. [DOI] [Google Scholar]
- Misale A.; Niyomchon S.; Luparia M.; Maulide N. Asymmetric palladium-catalyzed allylic alkylation using dialkylzinc reagents: A remarkable ligand effect. Angew. Chem., Int. Ed. 2014, 53, 7068–7073 10.1002/anie.201309074. [DOI] [PubMed] [Google Scholar]
- Alexakis A.; Hajjaji S. E.; Polet D.; Rathgeb X. Iridium-catalyzed asymmetric allylic substitution with aryl zinc reagents. Org. Lett. 2007, 9, 3393–3395 10.1021/ol0713842. [DOI] [PubMed] [Google Scholar]
- Polet D.; Rathgeb X.; Falciola C. A.; Langlois J.-B.; El Hajjaji S.; Alexakis A. Enantioselective iridium-catalyzed allylic arylation. Chem. - Eur. J. 2009, 15, 1205–1216 10.1002/chem.200801879. [DOI] [PubMed] [Google Scholar]
- Tosatti P.; Nelson A.; Marsden S. P. Recent advances and applications of iridium-catalysed asymmetric allylic substitution. Org. Biomol. Chem. 2012, 10, 3147–3163 10.1039/c2ob07086c. [DOI] [PubMed] [Google Scholar]
- Helmchen G.; Dahnz A.; Dübon P.; Schelwies M.; Weihofen R. Iridium-catalysed asymmetric allylic substitutions. Chem. Commun. 2007, 675–691 10.1039/B614169B. [DOI] [PubMed] [Google Scholar]
- Yoshikai N.; Miura K.; Nakamura E. Enantioselective copper-catalyzed allylic substitution reaction with aminohydroxyphosphine ligand. Adv. Synth. Catal. 2009, 351, 1014–1018 10.1002/adsc.200900054. [DOI] [Google Scholar]
- Jennequin T.; Wencel-Delord J.; Rix D.; Daubignard J.; Crévisy C.; Mauduit M. Chelating hydroxyalkyl NHC as efficient chiral ligands for room-temperature copper-catalyzed asymmetric allylic alkylation. Synlett 2010, 2010, 1661–1665 10.1055/s-0029-1220127. [DOI] [Google Scholar]
- Börner C.; Goldsmith P. J.; Woodward S.; Gimeno J.; Gladiali S.; Ramazzotti D. Asymmetric chemo- and regiospecific addition of organozinc reagents to Baylis–Hillman derived allylic electrophiles. Chem. Commun. 2000, 2433–2434 10.1039/b006943o. [DOI] [Google Scholar]
- Lee Y.; Akiyama K.; Gillingham D. G.; Brown M. K.; Hoveyda A. H. Highly site- and enantioselective Cu-catalyzed allylic alkylation reactions with easily accessible vinylaluminum reagents. J. Am. Chem. Soc. 2008, 130, 446–447 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]
- Akiyama K.; Gao F.; Hoveyda A. H. Stereoisomerically pure trisubstituted vinylaluminum reagents and their utility in copper-catalyzed enantioselective synthesis of 1,4-dienes containing Z or E alkenes. Angew. Chem., Int. Ed. 2010, 49, 419–423 10.1002/anie.200905223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; McGrath K. P.; Lee Y.; Hoveyda A. H. Synthesis of quaternary carbon stereogenic centers through enantioselective Cu-catalyzed allylic substitutions with vinylaluminum reagents. J. Am. Chem. Soc. 2010, 132, 14315–14320 10.1021/ja106829k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; Lee Y.; Mandai K.; Hoveyda A. H. Quaternary carbon stereogenic centers through copper-catalyzed enantioselective allylic substitutions with readily accessible aryl- or heteroaryllithium reagents and aluminum chlorides. Angew. Chem., Int. Ed. 2010, 49, 8370–8374 10.1002/anie.201005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumagin N. A.; Ponomaryov A. B.; Beletskaya I. P. Ketone synthesis via palladium-catalyzed carbonylation of organoaluminium compounds. Tetrahedron Lett. 1985, 26, 4819–4822 10.1016/S0040-4039(00)94960-8. [DOI] [Google Scholar]
- Lu B. Z.; Jin F.; Zhang Y.; Wu X.; Wald S. A.; Senanayake C. H. New general sulfinylating process for asymmetric synthesis of enantiopure sulfinates and sulfoxides. Org. Lett. 2005, 7, 1465–1468 10.1021/ol0501020. [DOI] [PubMed] [Google Scholar]
- Gao H.; Knochel P. New preparation and reactions of arylaluminum reagents using Barbier conditions. Synlett 2009, 2009, 1321–1325 10.1055/s-0028-1088127. [DOI] [Google Scholar]
- Merino E.; Melo R. P.; Ortega-Guerra M.; Ribagorda M.; Carreno M. C. Stereocontrolled approach to phenyl cyclitols from (SR)-[(p-tolylsulfinyl)methyl]-p-quinol. J. Org. Chem. 2009, 74, 2824–2831 10.1021/jo900109p. [DOI] [PubMed] [Google Scholar]
- Dabrowski J. A.; Gao F.; Hoveyda A. H. Enantioselective synthesis of alkyne-substituted quaternary carbon stereogenic centers through NHC-Cu-catalyzed allylic substitution reactions with (i-bu)2(alkynyl)aluminum reagents. J. Am. Chem. Soc. 2011, 133, 4778–4781 10.1021/ja2010829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski J. A.; Haeffner F.; Hoveyda A. H. Combining NHC-Cu and Bronsted base catalysis: Enantioselective allylic substitution/conjugate additions with alkynylaluminum reagents and stereospecific isomerization of the products to trisubstituted allenes. Angew. Chem., Int. Ed. 2013, 52, 7694–7699 10.1002/anie.201303501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham D. G.; Hoveyda A. H. Chiral N-heterocyclic carbenes in natural product synthesis: Application of Ru-catalyzed asymmetric ring-opening/cross-metathesis and Cu-catalyzed allylic alkylation to total synthesis of baconipyrone C. Angew. Chem., Int. Ed. 2007, 46, 3860–3864 10.1002/anie.200700501. [DOI] [PubMed] [Google Scholar]
- Novak A.; Fryatt R.; Woodward S. Nickel-catalysed asymmetric SN2′ substitution chemistry of Baylis-Hillman derived allylic electrophiles. C. R. Chim. 2007, 10, 206–212 10.1016/j.crci.2006.10.008. [DOI] [Google Scholar]
- Novak A.; Calhorda M. J.; Costa P. J.; Woodward S. Mapping the mechanism of nickel-Ferrophite catalysed methylation of Baylis-Hillman-derived SN2′ electrophiles. Eur. J. Org. Chem. 2009, 2009, 898–903 10.1002/ejoc.200801029. [DOI] [Google Scholar]
- Chung K.-G.; Miyake Y.; Uemura S. Nickel(0)-catalyzed asymmetric cross-coupling reactions of allylic compounds with arylboronic acids. J. Chem. Soc., Perkin Trans. 1 2000, 15–18 10.1039/a908076g. [DOI] [Google Scholar]
- Hayashi T.; Senda T.; Ogasawara M. Rhodium-catalyzed asymmetric conjugate addition of organoboronic acids to nitroalkenes. J. Am. Chem. Soc. 2000, 122, 10716–10717 10.1021/ja002805c. [DOI] [Google Scholar]
- Dong L.; Xu Y.-J.; Cun L.-F.; Cui X.; Mi A.-Q.; Jiang Y.-Z.; Gong L.-Z. Asymmetric nitroallylation of arylboronic acids with nitroallyl acetates catalyzed by chiral rhodium complexes and its application in a concise total synthesis of optically pure (+)-γ-lycorane. Org. Lett. 2005, 7, 4285–4288 10.1021/ol051795n. [DOI] [PubMed] [Google Scholar]
- Dong L.; Xu Y.-J.; Yuan W.-C.; Cui X.; Cun L.-F.; Gong L.-Z. Rhodium-catalyzed asymmetric nitroallylation of arylmetallics with cyclic nitroallyl acetates and applications in organic synthesis. Eur. J. Org. Chem. 2006, 2006, 4093–4105 10.1002/ejoc.200600275. [DOI] [Google Scholar]
- Miura T.; Takahashi Y.; Murakami M. Rhodium-catalysed substitutive arylation of cis-allylic diols with arylboroxines. Chem. Commun. 2007, 595–597 10.1039/B612710J. [DOI] [PubMed] [Google Scholar]
- Kiuchi H.; Takahashi D.; Funaki K.; Sato T.; Oi S. Rhodium-catalyzed asymmetric coupling reaction of allylic ethers with arylboronic acids. Org. Lett. 2012, 14, 4502–4505 10.1021/ol3019902. [DOI] [PubMed] [Google Scholar]
- Yu B.; Menard F.; Isono N.; Lautens M. Synthesis of homoallylic alcohols via Lewis acid assisted enantioselective desymmetrization. Synthesis 2009, 2009, 853–859 10.1055/s-0028-1083368. [DOI] [Google Scholar]
- Menard F.; Chapman T. M.; Dockendorff C.; Lautens M. Rhodium-catalyzed asymmetric allylic substitution with boronic acid nucleophiles. Org. Lett. 2006, 8, 4569–4572 10.1021/ol061777l. [DOI] [PubMed] [Google Scholar]
- Menard F.; Perez D.; Sustac Roman D.; Chapman T. M.; Lautens M. Ligand-controlled selectivity in the desymmetrization of meso cyclopenten-1,4-diols via rhodium(I)-catalyzed addition of arylboronic acids. J. Org. Chem. 2010, 75, 4056–4068 10.1021/jo100391e. [DOI] [PubMed] [Google Scholar]
- Shintani R.; Takatsu K.; Takeda M.; Hayashi T. Copper-catalyzed asymmetric allylic substitution of allyl phosphates with aryl- and alkenylboronates. Angew. Chem., Int. Ed. 2011, 50, 8656–8659 10.1002/anie.201103581. [DOI] [PubMed] [Google Scholar]
- Takeda M.; Takatsu K.; Shintani R.; Hayashi T. Synthesis of quaternary carbon stereocenters by copper-catalyzed asymmetric allylic substitution of allyl phosphates with arylboronates. J. Org. Chem. 2014, 79, 2354–2367 10.1021/jo500068p. [DOI] [PubMed] [Google Scholar]
- Gao F.; Carr J. L.; Hoveyda A. H. Copper-catalyzed enantioselective allylic substitution with readily accessible carbonyl- and acetal-containing vinylboron reagents. Angew. Chem., Int. Ed. 2012, 51, 6613–6617 10.1002/anie.201202856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; Carr J. L.; Hoveyda A. H. A broadly applicable NHC-Cu-catalyzed approach for efficient, site-, and enantioselective coupling of readily accessible (pinacolato)alkenylboron compounds to allylic phosphates and applications to natural product synthesis. J. Am. Chem. Soc. 2014, 136, 2149–2161 10.1021/ja4126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B.; Hoveyda A. H. Site- and enantioselective formation of allene-bearing tertiary or quaternary carbon stereogenic centers through NHC-Cu-catalyzed allylic substitution. J. Am. Chem. Soc. 2012, 134, 1490–1493 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J. Y.; Sarlah D.; Carreira E. M. Iridium-catalyzed enantioselective allylic vinylation. J. Am. Chem. Soc. 2013, 135, 994–997 10.1021/ja311422z. [DOI] [PubMed] [Google Scholar]
- Hamilton J. Y.; Sarlah D.; Carreira E. M. Iridium-catalyzed enantioselective allylic alkynylation. Angew. Chem., Int. Ed. 2013, 52, 7532–7535 10.1002/anie.201302731. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Brozek L. A.; Morken J. P. Pd-catalyzed enantioselective allyl-allyl cross-coupling. J. Am. Chem. Soc. 2010, 132, 10686–10688 10.1021/ja105161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozek L. A.; Ardolino M. J.; Morken J. P. Diastereocontrol in asymmetric allyl-allyl cross-coupling: Stereocontrolled reaction of prochiral allylboronates with prochiral allyl chlorides. J. Am. Chem. Soc. 2011, 133, 16778–16781 10.1021/ja2075967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Le H.; Kyne R. E.; Morken J. P. Enantioselective construction of all-carbon quaternary centers by branch-selective Pd-catalyzed allyl allyl cross-coupling. J. Am. Chem. Soc. 2011, 133, 9716–9719 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le H.; Kyne R. E.; Brozek L. A.; Morken J. P. Catalytic enantioselective allyl-allyl cross-coupling with a borylated allylboronate. Org. Lett. 2013, 15, 1432–1435 10.1021/ol400088g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster C. H.; Coombs J. R.; Kasun Z. A.; Morken J. P. Enantioselective carbocycle formation through intramolecular Pd-catalyzed allyl-aryl cross-coupling. Org. Lett. 2014, 16, 4420–4423 10.1021/ol5019163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmiya H.; Yokobori U.; Makida Y.; Sawamura M. Copper-catalyzed γ-selective allyl-alkyl coupling between allylic phosphates and alkylboranes. J. Am. Chem. Soc. 2010, 132, 2895–2897 10.1021/ja9109105. [DOI] [PubMed] [Google Scholar]
- Shido Y.; Yoshida M.; Tanabe M.; Ohmiya H.; Sawamura M. Copper-catalyzed enantioselective allylic substitution with alkylboranes. J. Am. Chem. Soc. 2012, 134, 18573–18576 10.1021/ja3093955. [DOI] [PubMed] [Google Scholar]
- Hojoh K.; Shido Y.; Ohmiya H.; Sawamura M. Construction of quaternary stereogenic carbon centers through copper-catalyzed enantioselective allylic cross-coupling with alkylboranes. Angew. Chem., Int. Ed. 2014, 53, 4954–4958 10.1002/anie.201402386. [DOI] [PubMed] [Google Scholar]
- Pérez M.; Fañanás-Mastral M.; Bos P. H.; Rudolph A.; Harutyunyan S. R.; Feringa B. L. Catalytic asymmetric carbon-carbon bond formation via allylic alkylations with organolithium compounds. Nat. Chem. 2011, 3, 377–381 10.1038/nchem.1009. [DOI] [PubMed] [Google Scholar]
- Guduguntla S.; Fañanás-Mastral M.; Feringa B. L. Synthesis of optically active β- or γ-alkyl-substituted alcohols through copper-catalyzed asymmetric allylic alkylation with organolithium reagents. J. Org. Chem. 2013, 78, 8274–8280 10.1021/jo401536u. [DOI] [PubMed] [Google Scholar]
- Fañanás-Mastral M.; Pérez M.; Bos P. H.; Rudolph A.; Harutyunyan S. R.; Feringa B. L. Enantioselective synthesis of tertiary and quaternary stereogenic centers: Copper/phosphoramidite-catalyzed allylic alkylation with organolithium reagents. Angew. Chem., Int. Ed. 2012, 51, 1922–1925 10.1002/anie.201107840. [DOI] [PubMed] [Google Scholar]
- Pérez M.; Fañanás-Mastral M.; Hornillos V.; Rudolph A.; Bos P. H.; Harutyunyan S. R.; Feringa B. L. Asymmetric allylic alkylation of acyclic allylic ethers with organolithium reagents. Chem. - Eur. J. 2012, 18, 11880–11883 10.1002/chem.201202251. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Yamamoto A.; Hagihara T. Stereo- and regiochemistry in palladium-catalyzed nucleophilic substitution of optically active (E)- and (Z)-allyl acetates. J. Org. Chem. 1986, 51, 723–727 10.1021/jo00355a025. [DOI] [Google Scholar]
- Evans P. A.; Uraguchi D. Regio- and enantiospecific rhodium-catalyzed arylation of unsymmetrical fluorinated acyclic allylic carbonates: Inversion of absolute configuration. J. Am. Chem. Soc. 2003, 125, 7158–7159 10.1021/ja035216q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit B.; Demel P. Copper-mediated and -catalyzed o-DPPB-directed allylic substitution. Adv. Synth. Catal. 2001, 343, 429–432. [DOI] [Google Scholar]
- Breit B.; Demel P.; Studte C. Stereospecific and stereodivergent construction of quaternary carbon centers through switchable directed/nondirected allylic substitution. Angew. Chem., Int. Ed. 2004, 43, 3786–3789 10.1002/anie.200453991. [DOI] [PubMed] [Google Scholar]
- Breit B.; Demel P.; Grauer D.; Studte C. o-DPPB-directed copper-mediated allylic substitution: Part 3. Stereospecific and stereodivergent construction of tertiary and quaternary carbon centers through switchable directed/nondirected allylic substitution. Chem. - Asian J. 2006, 1, 586–597 10.1002/asia.200600100. [DOI] [PubMed] [Google Scholar]
- Demel P.; Keller M.; Breit B. o-DPPB-directed copper-mediated and -catalyzed allylic substitution with Grignard reagents. Chem. - Eur. J. 2006, 12, 6669–6683 10.1002/chem.200600225. [DOI] [PubMed] [Google Scholar]
- Herber C.; Breit B. Iterative deoxypropionate synthesis based on a copper-mediated directed allylic substitution: Formal total synthesis of borrelidin (C3-C11 fragment). Chem. - Eur. J. 2006, 12, 6684–6691 10.1002/chem.200600343. [DOI] [PubMed] [Google Scholar]
- Rein C.; Demel P.; Outten R. A.; Netscher T.; Breit B. Reagent directing group controlled organic synthesis: Total synthesis of (R,R,R)-α-tocopherol. Angew. Chem., Int. Ed. 2007, 46, 8670–8673 10.1002/anie.200703268. [DOI] [PubMed] [Google Scholar]
- Reiss T.; Breit B. Total synthesis of (+)-bourgeanic acid utilizing o-DPPB-directed allylic substitution. Org. Lett. 2009, 11, 3286–3289 10.1021/ol9011635. [DOI] [PubMed] [Google Scholar]
- Reiss T.; Breit B. A unified strategy for the stereospecific construction of propionates and acetate-propionates relying on a directed allylic substitution. Chem. - Eur. J. 2009, 15, 6345–6348 10.1002/chem.200901064. [DOI] [PubMed] [Google Scholar]
- Norinder J.; Bäckvall J.-E. Dynamic processes in the copper-catalyzed substitution of chiral allylic acetates leading to loss of chiral information. Chem. - Eur. J. 2007, 13, 4094–4102 10.1002/chem.200601684. [DOI] [PubMed] [Google Scholar]
- Norinder J.; Bogár K.; Kanupp L.; Bäckvall J.-E. An enantioselective route to α-methyl carboxylic acids via metal and enzyme catalysis. Org. Lett. 2007, 9, 5095–5098 10.1021/ol702261t. [DOI] [PubMed] [Google Scholar]
- Thalén L. K.; Sumic A.; Bogár K.; Norinder J.; Persson A. K. Å.; Bäckvall J.-E. Enantioselective synthesis of α-methyl carboxylic acids from readily available starting materials via chemoenzymatic dynamic kinetic resolution. J. Org. Chem. 2010, 75, 6842–6847 10.1021/jo1011653. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Tang X.-L.; Tian S.-K. Cross-coupling of Grignard reagents with sulfonyl-activated sp3 carbon-nitrogen bonds. Adv. Synth. Catal. 2011, 353, 1980–1984 10.1002/adsc.201100285. [DOI] [Google Scholar]
- Lauer A. M.; Mahmud F.; Wu J. Cu(I)-catalyzed, α-selective, allylic alkylation reactions between phosphorothioate esters and organomagnesium reagents. J. Am. Chem. Soc. 2011, 133, 9119–9123 10.1021/ja202954b. [DOI] [PubMed] [Google Scholar]
- Ohmiya H.; Makida Y.; Tanaka T.; Sawamura M. Palladium-catalyzed γ-selective and stereospecific allyl-aryl coupling between allylic acetates and arylboronic acids. J. Am. Chem. Soc. 2008, 130, 17276–17277 10.1021/ja808673n. [DOI] [PubMed] [Google Scholar]
- Li D.; Tanaka T.; Ohmiya H.; Sawamura M. Synthesis of α-arylated allylsilanes through palladium-catalyzed γ-selective allyl-aryl coupling. Org. Lett. 2010, 12, 3344–3347 10.1021/ol101114r. [DOI] [PubMed] [Google Scholar]
- Ohmiya H.; Makida Y.; Li D.; Tanabe M.; Sawamura M. Palladium-catalyzed γ-selective and stereospecific allyl-aryl coupling between acyclic allylic esters and arylboronic acids. J. Am. Chem. Soc. 2010, 132, 879–889 10.1021/ja9092264. [DOI] [PubMed] [Google Scholar]
- Makida Y.; Ohmiya H.; Sawamura M. Sulfonamidoquinoline/palladium(II)-dimer complex as a catalyst precursor for palladium-catalyzed γ-selective and stereospecific allyl-aryl coupling reaction between allylic acetates and arylboronic acids. Chem. - Asian J. 2011, 6, 410–414 10.1002/asia.201000721. [DOI] [PubMed] [Google Scholar]
- Li C.; Xing J.; Zhao J.; Huynh P.; Zhang W.; Jiang P.; Zhang Y. J. Pd-catalyzed regioselective and stereospecific Suzuki-Miyaura coupling of allylic carbonates with arylboronic acids. Org. Lett. 2012, 14, 390–393 10.1021/ol203154j. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Ye J.; Zhang Y. J. Stereospecific allyl-aryl coupling catalyzed by in situ generated palladium nanoparticles in water under ambient conditions. Adv. Synth. Catal. 2013, 355, 491–498 10.1002/adsc.201200704. [DOI] [Google Scholar]
- Srinivas H. D.; Zhou Q.; Watson M. P. Enantiospecific, nickel-catalyzed cross-couplings of allylic pivalates and arylboroxines. Org. Lett. 2014, 16, 3596–3599 10.1021/ol5016724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmiya H.; Yokokawa N.; Sawamura M. Copper-catalyzed γ-selective and stereospecific allyl-aryl coupling between (Z)-acyclic and cyclic allylic phosphates and arylboronates. Org. Lett. 2010, 12, 2438–2440 10.1021/ol100841y. [DOI] [PubMed] [Google Scholar]
- Ye J.; Zhao J.; Xu J.; Mao Y.; Zhang Y. J. Pd-catalyzed stereospecific allyl-aryl coupling of allylic alcohols with arylboronic acids. Chem. Commun. 2013, 49, 9761–9763 10.1039/c3cc45053h. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Wang Y.; Tian S.-K. Regioselective and stereospecific cross-coupling of primary allylic amines with boronic acids and boronates through palladium-catalyzed C-N bond cleavage. Angew. Chem., Int. Ed. 2012, 51, 2968–2971 10.1002/anie.201109171. [DOI] [PubMed] [Google Scholar]
- Wu H.-B.; Ma X.-T.; Tian S.-K. Palladium-catalyzed stereospecific cross-coupling of enantioenriched allylic alcohols with boronic acids. Chem. Commun. 2014, 50, 219–221 10.1039/C3CC45772A. [DOI] [PubMed] [Google Scholar]
- Stokes B. J.; Opra S. M.; Sigman M. S. Palladium-catalyzed allylic cross-coupling reactions of primary and secondary homoallylic electrophiles. J. Am. Chem. Soc. 2012, 134, 11408–11411 10.1021/ja305403s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao K.; Yokobori U.; Makida Y.; Ohmiya H.; Sawamura M. Reversible 1,3-anti/syn-stereochemical courses in copper-catalyzed γ-selective allyl-alkyl coupling between chiral allylic phosphates and alkylboranes. J. Am. Chem. Soc. 2012, 134, 8982–8987 10.1021/ja302520h. [DOI] [PubMed] [Google Scholar]
- Ardolino M. J.; Morken J. P. Construction of 1,5-enynes by stereospecific Pd-catalyzed allyl-propargyl cross-couplings. J. Am. Chem. Soc. 2012, 134, 8770–8773 10.1021/ja302329f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardolino M. J.; Eno M. S.; Morken J. P. Stereocontrol in palladium-catalyzed propargylic substitutions: Kinetic resolution to give enantioenriched 1,5-enynes and propargyl acetates. Adv. Synth. Catal. 2013, 355, 3413–3419 10.1002/adsc.201300720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le H.; Batten A.; Morken J. P. Catalytic stereospecific allyl-allyl cross-coupling of internal allyl electrophiles with allylB(pin). Org. Lett. 2014, 16, 2096–2099 10.1021/ol500456s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem., Int. Ed. 2005, 44, 5384–5427 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- Colter A. K.; Clemens L. M. Solvent effects in the racemization of 1,1′-binaphthyl. A note on the influence of internal pressure on reaction rates. J. Phys. Chem. 1964, 68, 651–654 10.1021/j100785a503. [DOI] [Google Scholar]
- Bott G.; Field L. D.; Sternhell S. Steric effects. A study of a rationally designed system. J. Am. Chem. Soc. 1980, 102, 5618–5626 10.1021/ja00537a036. [DOI] [Google Scholar]
- Tamao K.; Minato A.; Miyake N.; Matsuda T.; Kiso Y.; Kumada M. Nickel-phosphine complex catalyzed Grignard synthesis of sterically hindered, unsymmetrical biaryls: Approach to asymmetric synthesis of biaryl atropisomers. Chem. Lett. 1975, 133–136 10.1246/cl.1975.133. [DOI] [Google Scholar]
- Frejd T.; Klingstedt T. 2,2′-Dimethyl-6,6′-bis(diphenylphosphino)biphenyl (BIPHEMP) as a chiral ligand for transition metal catalyzed asymmetric synthesis of binaphthyls and for asymmetric hydrogenation. A comparison with BINAP. Acta Chem. Scand. 1989, 43, 670–675 10.3891/acta.chem.scand.43-0670. [DOI] [Google Scholar]
- Hayashi T.; Hayashizaki K.; Kiyoi T.; Ito Y. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition-metal complexes. 6. Practical asymmetric synthesis of 1,1′-binaphthyls via asymmetric cross-coupling with a chiral [(alkoxyalkyl)ferrocenyl]monophosphine/nickel catalyst. J. Am. Chem. Soc. 1988, 110, 8153–8156 10.1021/ja00232a030. [DOI] [Google Scholar]
- Hayashi T.; Hayashizaki K.; Ito Y. Asymmetric synthesis of axially chiral 1,1′-5′,1″- and 1,1′-4′,1″-ternaphthalenes by asymmetric cross-coupling with a chiral ferrocenylphosphine-nickel catalyst. Tetrahedron Lett. 1989, 30, 215–218 10.1016/S0040-4039(00)95163-3. [DOI] [Google Scholar]
- Hayashi T.; Niizuma S.; Kamikawa T.; Suzuki N.; Uozumi Y. Catalytic asymmetric synthesis of axially chiral biaryls by palladium-catalyzed enantioposition-selective cross-coupling. J. Am. Chem. Soc. 1995, 117, 9101–9102 10.1021/ja00140a041. [DOI] [Google Scholar]
- Kamikawa T.; Hayashi T. Enantioposition-selective arylation of biaryl ditriflates by palladium-catalyzed asymmetric Grignard cross-coupling. Tetrahedron 1999, 55, 3455–3466 10.1016/S0040-4020(98)01154-5. [DOI] [Google Scholar]
- Kamikawa T.; Uozumi Y.; Hayashi T. Enantioposition-selective alkynylation of biaryl ditriflates by palladium-catalyzed asymmetric cross-coupling. Tetrahedron Lett. 1996, 37, 3161–3164 10.1016/0040-4039(96)00517-5. [DOI] [Google Scholar]
- Shimada T.; Cho Y.-H.; Hayashi T. Nickel-catalyzed asymmetric Grignard cross-coupling of dinaphthothiophene giving axially chiral 1,1′-binaphthyls. J. Am. Chem. Soc. 2002, 124, 13396–13397 10.1021/ja0282588. [DOI] [PubMed] [Google Scholar]
- Cho Y.-H.; Kina A.; Shimada T.; Hayashi T. Asymmetric synthesis of axially chiral biaryls by nickel-catalyzed Grignard cross-coupling of dibenzothiophenes. J. Org. Chem. 2004, 69, 3811–3823 10.1021/jo035880p. [DOI] [PubMed] [Google Scholar]
- Genov M.; Fuentes B.; Espinet P.; Pelaz B. Asymmetric Negishi reaction for sterically hindered couplings: Synthesis of chiral binaphthalenes. Tetrahedron: Asymmetry 2006, 17, 2593–2595 10.1016/j.tetasy.2006.10.008. [DOI] [Google Scholar]
- Genov M.; Almorín A.; Espinet P. Efficient synthesis of chiral 1,1 ′-binaphthalenes by the asymmetric Suzuki-Miyaura reaction: Dramatic synthetic improvement by simple purification of naphthylboronic acids. Chem. - Eur. J. 2006, 12, 9346–9352 10.1002/chem.200600616. [DOI] [PubMed] [Google Scholar]
- Genov M.; Almorín A.; Espinet P. Microwave assisted asymmetric Suzuki-Miyaura and Negishi cross-coupling reactions: Synthesis of chiral binaphthalenes. Tetrahedron: Asymmetry 2007, 18, 625–627 10.1016/j.tetasy.2007.03.001. [DOI] [Google Scholar]
- Wolfe J. P.; Singer R. A.; Yang B. H.; Buchwald S. L. Highly active palladium catalysts for Suzuki coupling reactions. J. Am. Chem. Soc. 1999, 121, 9550–9561 10.1021/ja992130h. [DOI] [Google Scholar]
- Littke A. F.; Dai C.; Fu G. C. Versatile catalysts for the Suzuki cross-coupling of arylboronic acids with aryl and vinyl halides and triflates under mild conditions. J. Am. Chem. Soc. 2000, 122, 4020–4028 10.1021/ja0002058. [DOI] [Google Scholar]
- Cammidge A. N.; Crépy K. V. L The first asymmetric Suzuki cross-coupling reaction. Chem. Commun. 2000, 1723–1724 10.1039/b004513f. [DOI] [Google Scholar]
- Nicolaou K. C.; Li H.; Boddy C. N. C.; Ramanjulu J. M.; Yue T.-Y.; Natarajan S.; Chu X.-J.; Bräse S.; Rübsam F. Total synthesis of vancomycin - Part 1: Design and development of methodology. Chem. - Eur. J. 1999, 5, 2584–2601. [DOI] [Google Scholar]
- Cammidge A. N.; Crépy K. V. L Synthesis of chiral binaphthalenes using the asymmetric Suzuki reaction. Tetrahedron 2004, 60, 4377–4386 10.1016/j.tet.2003.11.095. [DOI] [Google Scholar]
- Bringmann G.; Hamm A.; Schraut M. Atroposelective biaryl coupling with chiral catalysts: Total synthesis of the antileishmanial naphthylisoquinoline alkaloids ancistrotanzanine B and ancistroealaine A. Org. Lett. 2003, 5, 2805–2808 10.1021/ol0347693. [DOI] [PubMed] [Google Scholar]
- Yin J.; Buchwald S. L. A catalytic asymmetric Suzuki coupling for the synthesis of axially chiral biaryl compounds. J. Am. Chem. Soc. 2000, 122, 12051–12052 10.1021/ja005622z. [DOI] [Google Scholar]
- Herrbach A.; Marinetti A.; Baudoin O.; Guénard D.; Guéritte F. Asymmetric synthesis of an axially chiral antimitotic biaryl via an atropo-enantioselective Suzuki cross-coupling. J. Org. Chem. 2003, 68, 4897–4905 10.1021/jo034298y. [DOI] [PubMed] [Google Scholar]
- Shen X.; Jones G. O.; Watson D. A.; Bhayana B.; Buchwald S. L. Enantioselective synthesis of axially chiral biaryls by the Pd-catalyzed Suzuki-Miyaura reaction: Substrate scope and quantum mechanical investigations. J. Am. Chem. Soc. 2010, 132, 11278–11287 10.1021/ja104297g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Wang S.; Zhou Y.; He Y.; Zhuang Y.; Li L.; Wan P.; Wang L.; Zhou Z.; Qiu L. Highly diastereoselective synthesis of atropisomeric bridged P,N-ligands and their applications in asymmetric Suzuki-Miyaura coupling reaction. Adv. Synth. Catal. 2012, 354, 2395–2402 10.1002/adsc.201200095. [DOI] [Google Scholar]
- Wang S.; Li J.; Miao T.; Wu W.; Li Q.; Zhuang Y.; Zhou Z.; Qiu L. Highly efficient synthesis of a class of novel chiral-bridged atropisomeric monophosphine ligands via simple desymmetrization and their applications in asymmetric Suzuki-Miyaura coupling reaction. Org. Lett. 2012, 14, 1966–1969 10.1021/ol300721p. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Akai Y.; Nagata Y.; Suginome M. Highly enantioselective synthesis of axially chiral biarylphosphonates: Asymmetric Suzuki-Miyaura coupling using high-molecular-weight, helically chiral polyquinoxaline-based phosphines. Angew. Chem., Int. Ed. 2011, 50, 8844–8847 10.1002/anie.201103792. [DOI] [PubMed] [Google Scholar]
- Suginome M.; Yamamoto T.; Nagata Y.; Yamada T.; Akai Y. Catalytic asymmetric synthesis using chirality-switchable helical polymer as a chiral ligand. Pure Appl. Chem. 2012, 84, 1759–1769 10.1351/PAC-CON-11-08-23. [DOI] [Google Scholar]
- Nagata Y.; Kuroda T.; Takagi K.; Suginome M. Ether solvent-induced chirality inversion of helical poly(quinoxaline-2,3-diyl)s containing L-lactic acid derived side chains. Chem. Sci. 2014, 5, 4953–4956 10.1039/C4SC01920B. [DOI] [Google Scholar]
- Zhou Y.; Zhang X.; Liang H.; Cao Z.; Zhao X.; He Y.; Wang S.; Pang J.; Zhou Z.; Ke Z.; Qiu L. Enantioselective synthesis of axially chiral biaryl monophosphine oxides via direct asymmetric Suzuki coupling and DFT investigations of the enantioselectivity. ACS Catal. 2014, 4, 1390–1397 10.1021/cs500208n. [DOI] [Google Scholar]
- Wu Z.; Wang C.; Zakharov L. N.; Blakemore P. R. Enantioselective synthesis of biaryl compounds via Suzuki-Miyaura cross-coupling using a palladium complex of 7′-butoxy-7-(diphenylphosphino)-8,8′-biquinolyl: Investigation of a new chiral ligand architecture. Synthesis 2014, 46, 678–685 10.1055/s-0033-1340519. [DOI] [Google Scholar]
- Kamei T.; Sato A. H.; Iwasawa T. Asymmetric Suzuki-Miyaura cross-coupling of aryl chlorides with enhancement of reaction time and catalyst turnover. Tetrahedron Lett. 2011, 52, 2638–2641 10.1016/j.tetlet.2011.03.051. [DOI] [Google Scholar]
- Schaarschmidt D.; Grumbt M.; Hildebrandt A.; Lang H. A planar-chiral phosphino(alkenyl)ferrocene for Suzuki-Miyaura C-C coupling reactions. Eur. J. Org. Chem. 2014, 2014, 6676–6685 10.1002/ejoc.201402861. [DOI] [Google Scholar]
- Zhou Y.; Wang S.; Wu W.; Li Q.; He Y.; Zhuang Y.; Li L.; Pang J.; Zhou Z.; Qiu L. Enantioselective synthesis of axially chiral multifunctionalized biaryls via asymmetric Suzuki-Miyaura coupling. Org. Lett. 2013, 15, 5508–5511 10.1021/ol402666p. [DOI] [PubMed] [Google Scholar]
- Tang W.; Patel N. D.; Xu G.; Xu X.; Savoie J.; Ma S.; Hao M.-H.; Keshipeddy S.; Capacci A. G.; Wei X.; Zhang Y.; Gao J. J.; Li W.; Rodriguez S.; Lu B. Z.; Yee N. K.; Senanayake C. H. Efficient chiral monophosphorus ligands for asymmetric Suzuki-Miyaura coupling reactions. Org. Lett. 2012, 14, 2258–2261 10.1021/ol300659d. [DOI] [PubMed] [Google Scholar]
- Xu G.; Fu W.; Liu G. D.; Senanayake C. H.; Tang W. Efficient syntheses of korupensamines A, B and michellamine B by asymmetric Suzuki-Miyaura coupling reactions. J. Am. Chem. Soc. 2014, 136, 570–573 10.1021/ja409669r. [DOI] [PubMed] [Google Scholar]
- Uozumi Y.; Matsuura Y.; Arakawa T.; Yamada Y. M. A. Asymmetric Suzuki-Miyaura coupling in water with a chiral palladium catalyst supported on an amphiphilic resin. Angew. Chem., Int. Ed. 2009, 48, 2708–2710 10.1002/anie.200900469. [DOI] [PubMed] [Google Scholar]
- Jensen J. F.; Johannsen M. New air-stable planar chiral ferrocenyl monophosphine ligands: Suzuki cross-coupling of aryl chlorides and bromides. Org. Lett. 2003, 5, 3025–3028 10.1021/ol034943n. [DOI] [PubMed] [Google Scholar]
- Bronger R. P. J.; Guiry P. J. Aminophosphine-oxazoline and phosphoramidite-oxazoline ligands and their application in asymmetric catalysis. Tetrahedron: Asymmetry 2007, 18, 1094–1102 10.1016/j.tetasy.2007.04.020. [DOI] [Google Scholar]
- Mešková M.; Putala M. Highly sterically hindered binaphthalene-based monophosphane ligands: Synthesis and application in stereoselective Suzuki-Miyaura reactions. Tetrahedron: Asymmetry 2013, 24, 894–902 10.1016/j.tetasy.2013.06.007. [DOI] [Google Scholar]
- Zhang S.-S.; Wang Z.-Q.; Xu M.-H.; Lin G.-Q. Chiral diene as the ligand for the synthesis of axially chiral compounds via palladium-catalyzed Suzuki-Miyaura coupling reaction. Org. Lett. 2010, 12, 5546–5549 10.1021/ol102521q. [DOI] [PubMed] [Google Scholar]
- Benhamou L.; Besnard C.; Kündig E. P. Chiral PEPPSI complexes: Synthesis, characterization, and application in asymmetric Suzuki-Miyaura coupling reactions. Organometallics 2014, 33, 260–266 10.1021/om4009982. [DOI] [Google Scholar]
- Takemoto T.; Iwasa S.; Hamada H.; Shibatomi K.; Kameyama M.; Motoyama Y.; Nishiyama H. Highly efficient Suzuki-Miyaura coupling reactions catalyzed by bis(oxazolinyl)phenyl-pd(II) complex. Tetrahedron Lett. 2007, 48, 3397–3401 10.1016/j.tetlet.2007.03.063. [DOI] [Google Scholar]
- Debono N.; Labande A.; Manoury E.; Daran J.-C.; Poli R. Palladium complexes of planar chiral ferrocenyl phosphine-NHC ligands: New catalysts for the asymmetric Suzuki-Miyaura reaction. Organometallics 2010, 29, 1879–1882 10.1021/om100125k. [DOI] [Google Scholar]
- Loxq P.; Debono N.; Gülcemal S.; Daran J.-C.; Manoury E.; Poli R.; Çetinkaya B.; Labande A. Palladium(II) complexes with planar chiral ferrocenyl phosphane-(benz)imidazol-2-ylidene ligands. New J. Chem. 2014, 38, 338–347 10.1039/C3NJ00863K. [DOI] [Google Scholar]
- Castanet A.-S.; Colobert F.; Broutin P.-E.; Obringer M. Asymmetric Suzuki cross-coupling reaction: Chirality reversal depending on the palladium-chiral phosphine ratio. Tetrahedron: Asymmetry 2002, 13, 659–665 10.1016/S0957-4166(02)00169-6. [DOI] [Google Scholar]
- Mikami K.; Miyamoto T.; Hatano M. A highly efficient asymmetric Suzuki-Miyaura coupling reaction catalyzed by cationic chiral palladium(II) complexes. Chem. Commun. 2004, 2082–2083 10.1039/b407250b. [DOI] [PubMed] [Google Scholar]
- Sawai K.; Tatumi R.; Nakahodo T.; Fujihara H. Asymmetric Suzuki-Miyaura coupling reactions catalyzed by chiral palladium nanoparticles at room temperature. Angew. Chem., Int. Ed. 2008, 47, 6917–6919 10.1002/anie.200802174. [DOI] [PubMed] [Google Scholar]
- Bermejo A.; Ros A.; Fernández R.; Lassaletta J. M. C2-symmetric bis-hydrazones as ligands in the asymmetric Suzuki-Miyaura cross-coupling. J. Am. Chem. Soc. 2008, 130, 15798–15799 10.1021/ja8074693. [DOI] [PubMed] [Google Scholar]
- Ros A.; Estepa B.; Bermejo A.; Álvarez E.; Fernández R.; Lassaletta J. M. Phosphino hydrazones as suitable ligands in the asymmetric Suzuki-Miyaura cross-coupling. J. Org. Chem. 2012, 77, 4740–4750 10.1021/jo300548z. [DOI] [PubMed] [Google Scholar]
- Vinci D.; Martins N.; Saidi O.; Bacsa J.; Brigas A.; Xiao J. Ferrocenyl phosphine-oxazaphospholidine oxide ligands for the Suzuki-Miyaura coupling of hindered aryl bromides and chlorides. Can. J. Chem. 2009, 87, 171–175 10.1139/v08-113. [DOI] [Google Scholar]
- Grach G.; Pieters G.; Dinut A.; Terrasson V.; Medimagh R.; Bridoux A.; Razafimahaleo V.; Gaucher A.; Marque S.; Marrot J.; Prim D.; Gil R.; Planas J. G.; Viñas C.; Thomas I.; Roblin J.-P.; Troin Y. N-heterocyclic pyridylmethylamines: Synthesis, complexation, molecular structure, and application to asymmetric Suzuki-Miyaura and oxidative coupling reactions. Organometallics 2011, 30, 4074–4086 10.1021/om200375s. [DOI] [Google Scholar]
- Wu L.; Salvador A.; Ou A.; Shi M. W.; Skelton B. W.; Dorta R. Monodentate chiral N-heterocyclic carbene-palladium-catalyzed asymmetric Suzuki-Miyaura and Kumada coupling. Synlett 2013, 24, 1215–1220 10.1055/s-0033-1338864. [DOI] [Google Scholar]
- Shigeng G.; Tang J.; Zhang D.; Wang Q.; Chen Z.; Weng L. Synthesis, structure, and catalytic activity of palladium complexes with new chiral cyclohexane-1,2-based di-NHC-ligands. J. Organomet. Chem. 2012, 700, 223–229 10.1016/j.jorganchem.2011.12.008. [DOI] [Google Scholar]
- Li Y.; Tang J.; Gu J.; Wang Q.; Sun P.; Zhang D. Chiral 1,2-cyclohexane-bridged bis-NHC palladium catalysts for asymmetric Suzuki-Miyaura coupling: Synthesis, characterization, and steric effects on enantiocontrol. Organometallics 2014, 33, 876–884 10.1021/om400825e. [DOI] [Google Scholar]
- Jumde V. R.; Iuliano A. Deoxycholic acid derived monophosphites as chiral ligands in the asymmetric Suzuki-Miyaura cross-coupling. Tetrahedron: Asymmetry 2011, 22, 2151–2155 10.1016/j.tetasy.2011.12.006. [DOI] [Google Scholar]
- Castillo A. B.; Perandones B. F.; Zangrando E.; Gladiali S.; Godard C.; Claver C. Pd-catalysed asymmetric Suzuki-Miyaura reactions using chiral mono- and bidentate phosphorus ligands. J. Organomet. Chem. 2013, 743, 31–36 10.1016/j.jorganchem.2013.06.022. [DOI] [Google Scholar]
- Wang T.; Hao X.-Q.; Huang J.-J.; Wang K.; Gong J.-F.; Song M.-P. Chiral CNN pincer palladium(II) complexes with 2-aryl-6-(oxazolinyl)pyridine ligands: Synthesis, characterization, and application to enantioselective allylation of isatins and Suzuki-Miyaura coupling reaction. Organometallics 2014, 33, 194–205 10.1021/om400945d. [DOI] [Google Scholar]
- Yamaguchi K.; Yamaguchi J.; Studer A.; Itami K. Hindered biaryls by C-H coupling: Bisoxazoline-Pd catalysis leading to enantioselective C-H coupling. Chem. Sci. 2012, 3, 2165–2169 10.1039/c2sc20277h. [DOI] [Google Scholar]
- Yamaguchi K.; Kondo H.; Yamaguchi J.; Itami K. Aromatic C-H coupling with hindered arylboronic acids by Pd/Fe dual catalysts. Chem. Sci. 2013, 4, 3753–3757 10.1039/c3sc51206a. [DOI] [Google Scholar]
- Ros A.; Estepa B.; Ramírez-López P.; Álvarez E.; Fernández R.; Lassaletta J. M. Dynamic kinetic cross-coupling strategy for the asymmetric synthesis of axially chiral heterobiaryls. J. Am. Chem. Soc. 2013, 135, 15730–15733 10.1021/ja4087819. [DOI] [PubMed] [Google Scholar]
- Bhat V.; Wang S.; Stoltz B. M.; Virgil S. C. Asymmetric synthesis of QUINAP via dynamic kinetic resolution. J. Am. Chem. Soc. 2013, 135, 16829–16832 10.1021/ja409383f. [DOI] [PubMed] [Google Scholar]
- Mosquera A.; Pena M. A.; Pérez Sestelo J.; Sarandeses L. A. Synthesis of axially chiral 1,1′-binaphthalenes by palladium-catalysed cross-coupling reactions of triorganoindium reagents. Eur. J. Org. Chem. 2013, 2013, 2555–2562 10.1002/ejoc.201300042. [DOI] [Google Scholar]
- Ye J.; Ma S. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 2014, 1, 1210–1224 10.1039/C4QO00208C. [DOI] [Google Scholar]
- de Graaf W.; Boersma J.; van Koten G.; Elsevier C. J. Chiral induction in the synthesis of 4,4-dimethyl-1-phenylpenta-1,2-diene (1-Ph-3-t-bu-allene) catalyzed by chiral phosphine complexes of palladium. J. Organomet. Chem. 1989, 378, 115–124 10.1016/0022-328X(89)85013-2. [DOI] [Google Scholar]
- Li H. L.; Müller D.; Guénée L.; Alexakis A. Copper-catalyzed enantioselective synthesis of axially chiral allenes. Org. Lett. 2012, 14, 5880–5883 10.1021/ol302790e. [DOI] [PubMed] [Google Scholar]
- Elsevier C. J.; Mooiweer H. H.; Kleijn H.; Vermeer P. Stereochemistry of the Pd(PPh3)4-catalyzed conversion of 1-bromoallenes into phenyl substituted allenes. Tetrahedron Lett. 1984, 25, 5571–5572 10.1016/S0040-4039(01)81629-4. [DOI] [Google Scholar]
- Elsevier C. J.; Vermeer P. Stereochemistry of the palladium(0)-catalyzed phenylation of 1-halogenoallenes. J. Org. Chem. 1985, 50, 3042–3045 10.1021/jo00217a004. [DOI] [Google Scholar]
- Woerly E. M.; Cherney A. H.; Davis E. K.; Burke M. D. Stereoretentive Suzuki-Miyaura coupling of haloallenes enables fully stereocontrolled access to (−)-peridinin. J. Am. Chem. Soc. 2010, 132, 6941–6943 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B.; Pereira R.; Pérez M.; Álvarez R.; de Lera A. R. Stereoselective Stille coupling of enantiopure haloallenes and alkenylstannanes for the synthesis of allenyl carotenoids. Experimental and computational studies. J. Org. Chem. 2008, 73, 6534–6541 10.1021/jo800756b. [DOI] [PubMed] [Google Scholar]
- Elsevier C. J.; Stehouwer P. M.; Westmijze H.; Vermeer P. Anti stereoselectivity in the palladium(0)-catalyzed conversion of propargylic esters into allenes by phenylzinc chloride. J. Org. Chem. 1983, 48, 1103–1105 10.1021/jo00155a036. [DOI] [Google Scholar]
- Elsevier C. J.; Kleijn H.; Boersma J.; Vermeer P. Synthesis, structure, and reactivity of some (σ-allenyl)palladium(II) and (σ-prop-2-ynyl)palladium(II) complexes. Organometallics 1986, 5, 716–720 10.1021/om00135a015. [DOI] [Google Scholar]
- Konno T.; Tanikawa M.; Ishihara T.; Yamanaka H. Palladium-catalyzed coupling reaction of fluoroalkylated propargyl mesylates with organozinc reagents: Novel synthesis of optically active fluorine-containing trisubstituted allenes. Chem. Lett. 2000, 1360–1361 10.1246/cl.2000.1360. [DOI] [Google Scholar]
- Dixneuf P. H.; Guyot T.; Ness M. D.; Roberts S. M. Synthesis of optically active allenes using tandem enzyme and palladium-catalysed reactions. Chem. Commun. 1997, 2083–2084 10.1039/a704374k. [DOI] [Google Scholar]
- Yoshida M.; Gotou T.; Ihara M. Palladium-catalyzed direct coupling reaction of propargylic alcohols with arylboronic acids. Tetrahedron Lett. 2004, 45, 5573–5575 10.1016/j.tetlet.2004.05.147. [DOI] [Google Scholar]
- Yoshida M.; Okada T.; Shishido K. Enantiospecific synthesis of 1,3-disubstituted allenes by palladium-catalyzed coupling of propargylic compounds with arylboronic acids. Tetrahedron 2007, 63, 6996–7002 10.1016/j.tet.2007.05.035. [DOI] [Google Scholar]
- Yoshida M.; Ueda H.; Ihara M. Palladium-catalyzed coupling reaction of propargylic oxiranes with arylboronic acids in aqueous media. Tetrahedron Lett. 2005, 46, 6705–6708 10.1016/j.tetlet.2005.07.134. [DOI] [Google Scholar]
- Ohmiya H.; Yokobori U.; Makida Y.; Sawamura M. General approach to allenes through copper-catalyzed γ-selective and stereospecific coupling between propargylic phosphates and alkylboranes. Org. Lett. 2011, 13, 6312–6315 10.1021/ol202866h. [DOI] [PubMed] [Google Scholar]
- Yokobori U.; Ohmiya H.; Sawamura M. Synthesis of allenylsilanes through copper-catalyzed γ-selective coupling between γ-silylated propargylic phosphates and alkylboranes. Organometallics 2012, 31, 7909–7913 10.1021/om300552f. [DOI] [Google Scholar]
- Uehling M. R.; Marionni S. T.; Lalic G. Asymmetric synthesis of trisubstituted allenes: Copper-catalyzed alkylation and arylation of propargylic phosphates. Org. Lett. 2012, 14, 362–365 10.1021/ol2031119. [DOI] [PubMed] [Google Scholar]
- Yang M.; Yokokawa N.; Ohmiya H.; Sawamura M. Synthesis of conjugated allenes through copper-catalyzed γ-selective and stereospecific coupling between propargylic phosphates and aryl- or alkenylboronates. Org. Lett. 2012, 14, 816–819 10.1021/ol2033465. [DOI] [PubMed] [Google Scholar]
- Kamikawa K.; Uemura M. Stereoselective synthesis of axially chiral biaryls utilizing planar chiral (arene)chromium complexes. Synlett 2000, 938–949. [Google Scholar]
- Uemura M.; Nishimura H.; Hayashi T. Catalytic asymmetric induction of planar chirality by palladium-catalyzed asymmetric cross-coupling of a meso (arene)chromium complex. Tetrahedron Lett. 1993, 34, 107–110 10.1016/S0040-4039(00)60069-2. [DOI] [Google Scholar]
- Uemura M.; Nishimura H.; Hayashi T. Catalytic asymmetric induction of planar chirality: Palladium-catalyzed asymmetric cross-coupling of meso tricarbonyl(arene)chromium complexes with alkenylboronic and arylboronic acids. J. Organomet. Chem. 1994, 473, 129–137 10.1016/0022-328X(94)80113-4. [DOI] [Google Scholar]
- Urbaneja X.; Mercier A.; Besnard C.; Kündig E. P. Highly efficient desymmetrisation of a tricarbonylchromium 1,4-dibromonaphthalene complex by asymmetric Suzuki-Miyaura coupling. Chem. Commun. 2011, 47, 3739–3741 10.1039/c1cc10347d. [DOI] [PubMed] [Google Scholar]
- Bergin E.; Hughes D. L.; Richards C. J. Application of the Suzuki reaction to the asymmetric desymmetrisation of 1,2-and 1,3-disubstituted bulky cobalt metallocenes. Tetrahedron: Asymmetry 2010, 21, 1619–1623 10.1016/j.tetasy.2010.05.012. [DOI] [Google Scholar]
- Gao D.-W.; Shi Y.-C.; Gu Q.; Zhao Z.-L.; You S.-L. Enantioselective synthesis of planar chiral ferrocenes via palladium-catalyzed direct coupling with arylboronic acids. J. Am. Chem. Soc. 2013, 135, 86–89 10.1021/ja311082u. [DOI] [PubMed] [Google Scholar]