

Abstract
The ever-increasing number of sequenced genomes and subsequent sequence-based analysis has provided tremendous insight into cellular processes; however, the ability to experimentally manipulate this genomic information in the laboratory requires the development of new high-throughput methods. To translate this genomic information into information on protein function, molecular and cell biological techniques are required. One strategy to gain insight into protein function is to observe where each specific protein is subcellularly localized. We have developed a pipeline of methods that allows rapid, efficient, and scalable gene cloning, imaging, and image analysis. This work focuses on a high-throughput screen of the Caulobacter crescentus proteome to identify proteins with unique subcellular localization patterns. The cloning, imaging, and image analysis techniques described here are applicable to any organism of interest.
1. Introduction
It has become clear that subcellular protein localization is as essential for bacterial cells as it is for their eukaryotic counterparts (Gitai, 2005; Thanbichler and Shapiro, 2008). Since bacteria do not contain membrane-bound organelles to sequester specific enzymatic functions from one another, it is essential that they tightly coordinate the structural, enzymatic, and regulatory activities required for viability. Recent studies have established that the function of many proteins depends on their spatial and temporal localization: division proteins go to the division plane, polar development proteins go the pole, DNA replication proteins go to the replisome, etc. Thus, we are now in a position to use protein localization as a systematic method for characterizing protein function. With this understanding and the emergence of new molecular and cell biological methods, protein localization studies can be performed on a proteomic scale.
The development of advanced methods for observing specific subcellular protein localizations by high-resolution imaging of fluorescent protein fusions has coincided with an explosion of genomic resources and techniques. While protein localization studies have traditionally been performed on small subsets of individual proteins, genome-scale analysis of protein localization has the potential to identify functions for uncharacterized proteins and enhance understanding of previously studied proteins, identify potential protein interactions, and propose localization mechanisms. Several groups have performed large-scale efforts that have cataloged the localizations of most of the proteins of Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Escherichia coli (Huh et al., 2003; Kitagawa et al., 2005; Matsuyama et al., 2006); however, the laborious nature of generating, imaging, and scoring these libraries has limited their reanalysis under different conditions. In this work, we developed a pipeline of high-throughput cloning and imaging methods that allowed rapid, efficient, and scalable analysis of protein localization.
The pipeline of methods we describe here can be applied to a wide range of species for a variety of experimental applications. Here, we will focus on our initial application of the pipeline to a high-throughput protein localization screen in the gram-negative aquatic bacterium Caulobacter crescentus (Werner et al., 2009). Caulobacter cells represent excellent models for studying bacterial cell biology due to their asymmetric polarity that can be readily visualized by the presence of a stalk that protrudes from one cell pole. This polarity provides cellular landmarks for specifically defining protein localization. Also, Caulobacter has well-studied cell shape, cell division, and cell-cycle regulated proteins known to have specific subcellular localization patterns that serve as positive controls for our protein localization screen (Ausmees et al., 2003; Collier and Shapiro, 2007; Gitai et al., 2004; Thanbichler and Shapiro, 2006). These characteristics along with the relative ease of manipulating Caulobacter made this an attractive subject for this study.
2. Pipeline Overview
The effort to generate a library of fluorescent protein fusions in C. crescentus involved five steps that we will detail in the sections below: (1) construction of a Caulobacter ORFeome library, (2) creating expression vectors by mobilization of the open reading frames (ORFs) into Caulobacter mCherry fusion destination vectors, (3) transferring of the expression vectors into Caulobacter, (4) imaging the resulting fusion strains, and (5) analysis of the imaging data.
For ease and modularity we adopted the Gateway (Invitrogen) system of recombinational cloning (Walhout et al., 2000) in order to construct the expression clones. In the Gateway system, each ORF of interest is first cloned into a “donor” vector to create an “entry” vector (BP reaction). This library of entry vectors, also known as an ORFeome (Brasch et al., 2004), is then mobilized into any “destination” vector to create an “expression” vector (LR reaction) for specific applications. For this study, our destination vectors created an inducible fluorescent fusion protein for each Caulobacter ORF. The Gateway system greatly reduces the time and effort required for traditional cut and paste cloning methods, and we were able to implement an “in vivo LR” method that significantly reduced the labor and expense of mobilizing the ORFs into destination vectors with similar cloning efficiency to traditional LR reactions. Once the expression vectors were generated in E. coli via in vivo LRs, we developed a high-throughput conjugation strategy to transfer them into Caulobacter, thereby generating the final desired fusion strains.
Upon completion of strain construction, thousands of Caulobacter strains containing fluorescent protein fusions needed to be imaged. This quantity of imaging required its own strategy as there were no previously developed methods for high-throughput imaging at the resolution necessary to visualize protein localization in bacteria. Traditional methods of imaging only a few samples on a slide would not be an efficient use of time for the number of strains that needed to be imaged, and available high-throughput imaging methods could not provide the resolution necessary for this study. Therefore, we designed a “pedestal slide” system that enabled the imaging of 48 samples on a single slide. Coupled with a robotic stage and semiautomated image acquisition script, we were able to decrease the time and effort to a minimum for this large-scale project.
Besides strain construction and imaging, an undertaking of this magnitude required a means to organize, store, validate, and analyze thousands of strains and images. A web site was created that organized all of the information regarding strain and primer locations. Scoring of each image was performed by individuals with the assistance of specially designed image viewing and analysis programs. Upon scoring of these images, the final steps to our complete library were validating the localization patterns by reimaging and verifying gene identity.
In this work, we developed several new methods and adapted others to create a novel integrated cloning and imaging pipeline. This pipeline of methods is applicable to many projects and may be adapted to any organism. Here we describe the generation of a library of localized proteins along with resources that have already been useful in other studies. The following sections will provide a more detailed description of the methods used in creating this localization library and describe the results we obtained.
3. Construction of a Caulobacter ORFeome
3.1. Rationale of entry vector cloning
The goal of this work was to create a library of proteins with specific subcellular localizations. In the process of attaining this goal we built a resource with which future high-throughput screens can be performed by creating a Caulobacter ORFeome. This ORFeome contains entry vectors that each possesses a single Caulobacter ORF. The modular nature of the Gateway system allows these ORFs to be transferred to the destination vector created for this study and any destination vector created in the future. Because of the relative ease of transfer from the entry vector to the destination vector, the initial step of entry vector construction was one of the most critical steps in the cloning process.
Cloning efficiency using the Gateway system is greatly enhanced by selection features in the “Gateway cassette” contained in the donor and destination vectors (Walhout et al., 2000). The Gateway cassette is approximately 1.7 kb and is flanked by the phage lambda-derived attP and attR sequences in the donor and destination vectors, respectively. Within the attP and attR borders are the cytotoxic ccdB gene and a chloramphenicol resistance (CamR) marker (Walhout et al., 2000). When a successful BP or LR reaction occurs, the ccdB gene and CamR marker are recombined out of the vector. If the recombination reaction is unsuccessful the CcdB protein will kill the CcdB-sensitive recipient cell, ensuring that only reactions resulting in inserted DNA into the donor or destination vectors will be transformed into the recipient strain. This can be verified by checking for chloramphenicol sensitivity. Recipient strains will be chloramphenicol sensitive if the resistance marker has been removed by a successful recombination reaction. This selection method greatly enhances the likelihood of transformed colonies containing the desired entry vector or expression vector and provides a simple method for validation.
3.2. Procedure for entry vector cloning
To create a Caulobacter ORFeome we started by PCR amplifying every ORF in the genome. A total of 3763 primers (Operon Biotechnologies) were designed to PCR amplify every protein encoding ORF using chromosomal DNA from the wild-type Caulobacter strain CB15 (Nierman et al., 2001) as the template. Each primer was designed with a specific sequence added to the 5′ end of the “forward” (5′ end of the ORF) and “reverse” (3′ end of the ORF) primer that allowed specific recombination into the donor vector pDONR223 (Rual et al., 2004). An ATG start codon was also included in the Forward primer that replaced the endogenous start codon. The 3′ end of each primer contained 15–20 bases of homology to the ORF of interest. The donor vector-specific sequence for each primer is shown below (Rual et al., 2004):
Forward—5′ GGGGACAACTTTGTACAAAAAAGTTGGCATG 3′
Reverse—5′ GGGGACAACTTTGTACAAGAAAGTTGGG 3′
These sequences contain the attB sites that recombine with attP sites (BP reaction) on the donor vector. All PCR reactions were performed in a 96-well format with KOD polymerase (Novagen) using the manufacturer’s instructions. Initial verification of a successful PCR reaction was done by examining the length of the PCR product on E-gels (Invitrogen).
After confirming the size of each PCR product, BP reactions were performed using the Gateway protocols and reagents provided by Invitrogen. The BP reaction, consisting of the recombination of the attB sites on the PCR product with the attP sites on pDONR223, was performed using BP clonase II and its specific protocol (Invitrogen). Upon completion of the BP reaction, samples were transformed into competent DH5α E. coli cells. After this validation method, sequencing (Agencourt) of the 5′ and 3′ end of each ORF in its entry vector was performed using M13 primers. Quality trimmed sequences were analyzed using BLAST analysis. This analysis confirmed that the correct ORF was present in its full length with no detectable point mutations.
3.3. Results
All cloning and imaging results and analysis, along with comprehensive tables and figures, are present in our earlier publication (Werner et al., 2009). In order to create the Caulobacter ORFeome, we attempted PCR amplification of all 3763 ORFs for which we had PCR primers designed. Several rounds of PCR with slightly altered conditions were performed in order to meet the appropriate PCR conditions for as many ORFs as possible. Combining the results from all of the PCR attempts, we recovered PCR products for 3744 ORFs (99.5% of all ORFs); however, only 3184 PCR products were the correct length when observed on a gel (85.0% of all ORFs). BP reactions were performed with these PCR products and sequencing of the entry vector insert was performed to verify the identity, size, and sequence integrity of the ORF. Upon analysis of the sequencing results, we had obtained 2786 entry vectors containing a unique correct-size ORF. This represented 74.0% of the ORFs for which we designed primers. This set of clones represents version 1.0 of the Caulobacter ORFeome. Future work may attempt to increase this number of successfully cloned entry vectors for even better coverage of the Caulobacter genome.
4. Construction of the Fluorescently Tagged Protein Library
4.1. Overview
While the construction of the Caulobacter ORFeome generally followed manufacturers procedures, many of the subsequent methods were developed or adapted for this project (but applicable to many others). In the process of constructing inducible fluorescent fusion Caulobacter strains, we designed and created unique destination vectors, implemented new high-throughput methods for performing LR reactions (in vivo LR), and utilized new high-throughput conjugation methods for moving vectors from E. coli to Caulobacter cells. To build our library of fluorescently tagged proteins each ORF needed to be moved into a vector that would create a fluorescent protein fusion. We created our desired expression vectors by moving ORFs from entry vectors to destination vectors containing the mCherry fluorescent protein (Shaner et al., 2004) via in vivo LR reactions. The resulting constructs also allowed us to control the expression of the fluorescent fusions by using the xylose-inducible xylX promoter (Meisenzahl et al., 1997). The use of an inducible promoter allowed us to minimize potential toxic effects of protein overexpression and enables future studies on how protein localization responds to a range of protein concentrations. The expression vectors were constructed in E. coli and then transferred into Caulobacter for examination of protein localization.
4.2. Designing xylose-inducible mCherry fusion destination vectors
The first step in creating the fluorescent fusion library was to design and construct destination vectors. Our goal was to create fluorescent fusions to both the N- and C-terminus of each protein. Initially, we started by constructing the destination vector, gXRC (Gateway, Xylose-inducible, Red fluorescent protein, C-terminal fusion), that would create an mCherry fusion to the C-terminus of each protein. This was followed by the construction of the N-terminal fusion destination vector gXRN. We chose the mCherry fluorescent protein because of its ability to fold and fluoresce in all cellular compartments (cytoplasm, periplasm, inner membrane, and outer membrane). We also included the xylX promoter so that the resulting fusion proteins would be under the control of a xylose-inducible promoter. In the absence of xylose no fluorescence is visible in cells; however, in the presence of xylose there is enough expression to visualize the localization of a fluorescent protein in Caulobacter.
The gXRC destination vector was created from the Kanamycin resistant (KanR) pXGFP4 (gift of M.R. Alley) which has an origin of replication (oriV) that allows replication in E. coli but not in Caulobacter. For Caulobacter to survive on media containing Kan the vector must integrate into the chromosome. pXGFP4 contains approximately 2.3 kilobases of the Caulobacter chromosome upstream and containing the xylX promoter which greatly increases the probability that the expression vector will integrate into this specific region when moved into Caulobacter. We generated gXRC by digesting pXGFP4 with NdeI and Asp718 and ligating with a similarly digested Gateway cassette PCR product amplified from pTGW (Drosophila Genomics Resource Center). The green fluorescent protein (GFP) in pXGFP4 was removed using NotI and Asp718 and replaced with the similarly digested mCherry PCR product amplified from pmCherry (Clontech). Thus, this cloning effort creates a vector with the Gateway cassette flanked on its 5′ end with the xylose promoter and its 3′ end with mCherry which creates the xylose-inducible C-terminal fusion.
Once gXRC was constructed and validated, gXRN was generated. The vector pXGFP4-C1 (gift of M. R. Alley), which creates N-terminal GFP fusions, was used to create this destination vector. pXGFP4-C1 was digested with BglII and Asp718 and ligated with a similarly digested PCR product amplified from pTGW. The GFP was removed from pXGFP4-C1 by digesting with BglII and NdeI and replaced with the appropriately digested mCherry PCR product amplified from pmCherry. This creates a vector with mCherry flanked by the xylose promoter and the Gateway cassette creating an N-terminal fusion vector. Successful construction of gXRC and gXRN allowed us to proceed to the next step of performing LR reactions with the Caulobacter ORFeome.
4.3. Procedure for the in vivo LR reaction
With the completion of the construction of gXRC and gXRN, expression vectors were made by performing LR reactions. The LR reaction recombines the attL sequences created on the entry vector, as a result of the BP reaction, with attR sequences in the destination vectors. As with the BP reaction, Invitrogen offers a commercial enzyme to facilitate this recombination step. However, this purified enzyme, while cost effective on a small scale, is a large expense when used at the scale of cloning the Caulobacter genome. Also, performing the traditional LR reaction in vitro required purifying destination vectors and transforming the LR reaction product into competent cells. These steps would be very labor intensive to perform at this scale. Therefore, we implemented a method to perform this LR reaction in vivo without the need for purified recombinase enzymes. We have named this approach the “in vivo LR” reaction.
The in vivo LR method builds upon work studying the xis and int genes that mediate the LR recombination reaction (Platt et al., 2000), as well as earlier low-throughput methods to perform Gateway reactions in living cells (Schroeder et al., 2005). The in vivo LR requires three features besides the entry clone and destination vectors needed for a traditional LR reaction. First, an E. coli strain containing the pXINT129 plasmid (Platt et al., 2000) that expresses the xis and int genes under the control of an isopropyl β-D-thiogalactopyranoside (IPTG)-inducible promoter was necessary to provide the recombinase enzymes for the LR reaction. Second, destination vectors must contain an origin of transfer (oriT) that allows the vector to be transferred from one organism to another via conjugation. Third, a conjugation helper strain (LS980) was used to facilitate conjugation between multiple strains, allowing the oriT-containing destination vector to be transferred from one strain to another.
The in vivo LR reaction is a series of recombination and conjugation events that transfers an ORF from the entry vector to the destination vector. The first step in the process was to transform isolated Spectinomycin/Streptomycin resistant (SpecR/StrepR) entry vectors into a chemically competent E. coli strain containing pXINT129 using a traditional heat shock transformation protocol. Four microliters of isolated entry vectors were added to each well of a 96-well plate containing 50 μl of chemically competent pXINT129 E. coli cells in each well. Each plate was heat shocked in a 42 °C water bath for 60 s. Immediately following the heat shock, plates were placed on ice and 100 μl LB broth (Silhavy et al., 1984) was added to each well. Plates were then incubated at 37 °C for 1 h. Fifty microliters of each transformation was added to 750 μl LB broth containing Spectinomycin (Calbiochem), Streptomycin (Fisher Bioreagents), and Kanamycin (Agribio, Inc.) to isolate E. coli strains containing the pXINT129 (KanR) plasmid and the entry vector and grown overnight.
To perform the in vivo LR, the entry vector/pXINT129 containing strains were combined with an E. coli strain containing gXRC (KanR), a conjugation helper E. coli strain (Chloramphenicol resistant; CamR), and a ccdB-sensitive, Rifampin resistant (CcdBS Rif R) recipient DH5α E. coli strain at a 1:1:1:1 ratio (50 μl each). This mixture of cultures was spotted with a 48-pin pinning tool (frogger, DanKar Corporation) on LB agar plates containing 1 mM IPTG (Ambion, Inc.) in order to express the recombinase enzymes. After overnight growth, the spots containing the E. coli strain mixtures were transferred with a frogger to LB broth containing Rifampin (Fisher Bioreagents) and Kanamycin to select for expression vectors that have been transferred to the recipient strain.
While we have not studied the specific course of events that occurs in the in vivo LR step, we can describe the likely path gXRC follows to become an expression vector. gXRC contains an oriT so it can be transferred from one strain to another via conjugation. It is likely gXRC is first transferred to the strain containing the entry vector and pXINT129. The Xis and Int proteins expressed from pXINT129 mediate the recombination of the ORF from the entry vector into gXRC. The recombined gXRC that now contains the gene of interest in place of the ccdB gene is then transferred again via conjugation from this strain to the recipient Rif R DH5α strain. This desired strain is purified from the other E. coli strains by growing in LB broth containing Rifampin and Kanamycin. All E. coli strains but the recipient strain containing the expression vector are killed by one or both of these antibiotics or the CcdB protein resulting from the unrecombined gXRC. It is probable that this is an inefficient process at the cellular level but we only need a small fraction to work for the procedure to be successful.
4.4. Protocol for fast and efficient transfer of expression vectors from E. coli to Caulobacter
The final step in constructing the strains to perform the large-scale localization screen was to transfer the expression vectors from E. coli to Caulobacter. Again, we performed this in a high-throughput manner using conjugation. In 96-well plates, we combined cultures of the E. coli strains containing expression vectors, the conjugation helper strain, and the wild-type Caulobacter strain CB15N at a 1:1:1 ratio (50 μl each). This mixture was spotted on PYE plates (Ely, 1991) with a 48-pin frogger to allow conjugation to occur between the E. coli and Caulobacter strains. The spots were incubated at 30 °C for 48 h and then streaked on PYE plates containing Nalidixic acid (Nal, Fisher Bioreagents) and Kanamycin. Kanamycin selected for the expression vector while the Nalidixic acid selected against E. coli. Only after completing this step did we discover that DH5α cells have resistance to Nalidixic acid which explained much of the difficulty we had obtaining pure cultures of Caulobacter without E. coli contamination. For future studies we will use an E. coli recipient strain that is sensitive to Nalidixic acid or use a different selection to isolate Caulobacter from E. coli. To verify that the KanR and NalR Caulobacter strain we obtained by purification actually possessed an expression vector containing an insert, we patched two colonies from each strain on PYE plates containing either Kanamycin or Kanamycin and Chloramphenicol (Fisher Bioreagents). Strains that were KanR but CamS were deemed to have a successfully cloned expression vector due to the removal of the CamR Gateway cassette.
The generation of multiple sets of strains along the path to creating the final Caulobacter strains required careful handling and storage. At each step along the cloning and conjugation pipeline E. coli and Caulobacter strains were stored at −80 °C. For future use, we stored the E. coli strains containing entry vectors, E. coli strains containing pXINT129 and the entry vectors, E. coli strains containing expression vectors, and the Caulobacter strains containing expression vectors. All strains were stored in 96-well plates; E. coli were stored in LB broth containing 20% glycerol and Caulobacter were stored in PYE broth containing 20% glycerol.
4.5. Results
For this step of the pipeline we created a system that allowed us to perform LR reactions without purified recombinase enzymes or isolated entry vectors and destination vectors. This system involved mixing four cultures together and letting the bacteria perform the work. We also developed a high-throughput way of transferring expression vectors from E. coli to Caulobacter without the need to isolate vectors and perform transformations. Once these methods were optimized, the entire process (from the transformation of entry vectors into the pXINT129-containing E. coli cells, to the final step of patching to confirm the isolation of the desired Caulobacter strains) could be performed in less than 2 weeks.
5. Imaging the Caulobacter Protein Localization Library
5.1. Overview
Once strain construction was complete we were ready to image the Caulobacter protein localization library. To visualize the Caulobacter cell borders and the fluorescent protein it was necessary to use phase contrast and fluorescent imaging. Traditional methods of examining protein localization in bacterial strains were not sufficient for the throughput necessary to image this volume of strains. Air immersion objectives could be used for high-throughput work but did not provide the resolution necessary to observe protein localization. Therefore, we developed a method utilizing a robotic stage and custom-made pedestal slides that allowed us to image strains in high-throughput at maximal diffraction-limited resolution using oil immersion objectives.
5.2. Protocol for high-throughput imaging of fluorescent fusion proteins in Caulobacter
To prepare this volume of strains for imaging we needed to optimize cell growth and induction conditions. All strains were grown overnight in a 96-well format containing PYE broth with Kanamycin. Four hours prior to imaging, cultures were diluted 1:15 in PYE broth with Kanamycin to allow the strains to recover from stationary phase. Two hours prior to imaging, cultures were induced with 0.03% xylose. This level of xylose was sufficient to observe protein localization in control strains but low enough to reduce toxicity effects caused by overexpression.
High-throughput high-resolution imaging was performed by developing a 48-pad “pedestal slide” and by writing a script so that the microscope automatically performs many of the repetitive imaging tasks. The 48-pad pedestal slide was developed by designing a stainless steel mold as shown in Fig. 11.1A and using custom-made coverglass slips of the same length and width dimensions as the mold. To create the pedestal slide (Fig. 11.1B), one coverglass was placed on a lab bench with two molds placed on top flush with each other and the bottom coverglass. Molten 1% agarose was poured over the molds and coverglass filling all of the holes. While the agarose was still in liquid form, a second coverglass was placed on top of the molds expelling the excess agarose from the top of the molds. The agarose was then allowed to solidify in the mold for approximately 5 min. Once the agarose solidified, the sandwich of coverglasses and molds was flipped over so the bottom coverglass was on top and this coverglass was removed by carefully sliding it off the sandwich. The top mold was then pried off, exposing 48 agarose pads 1 mm in height. These pads were allowed to dry for 10–15 min prior to the application of culture samples.
Figure 11.1.
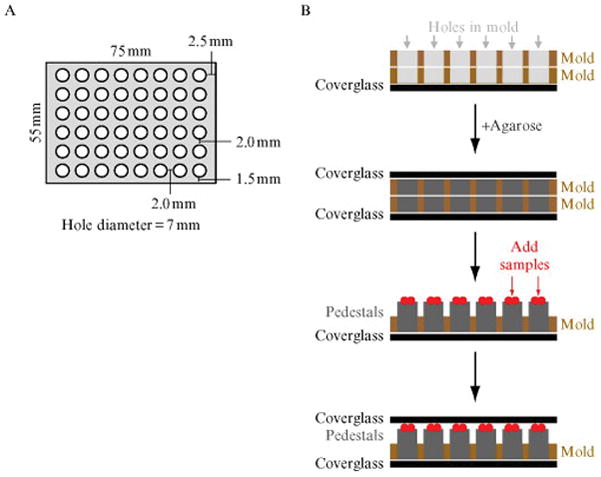
Schematic of the 48-well pedestal slide mold and pedestal slide assembly. (A) Measurements of mold are 75 mm long by 55 mm wide by 1 mm thick. The diameter of each hole is 7 mm. The width of the long border, short border, and width between holes is 1.5, 2.5, and 2.0 mm, respectively. (B) Flow chart of pedestal slide assembly as described in the text. A side view of a slice through the assembly sandwich is shown with the stainless steel molds in brown, empty holes in the mold in light gray, coverglass in black, agarose pedestals in dark gray, and culture samples in red.
Imaging of the fluorescent fusion library in Caulobacter was ready to be performed after the preparation of the pedestal slides. Culture growth and induction was timed to coincide with the preparation of the pedestal slides. Culture samples (3 μl) were pipetted on the 48-pedestal slides using a multichannel pipettor and fields of approximately 50–200 cells were chosen for imaging. All imaging was performed with a Nikon90i epifluorescent microscope equipped with a 100× 1.4 NA objective (Nikon), Rolera XR cooled CCD camera (QImaging), and NIS Elements software (Nikon). A script automated the tasks of taking phase and fluorescent images, naming and saving each image, and moving the objective to the next pedestal on the slide. After optimization, a single 48-pedestal slide could be imaged in as little as 15–20 min.
5.3. Results
Without the development of these high-throughput methods for imaging, this project would not have been feasible. Over 6400 strains (gXRC and gXRN expression vectors) were imaged at least once and most were imaged multiple times. It would have taken months to image that volume of strains using traditional imaging methods and would have been prohibitory to any future efforts to reimage this library. With these methods, three or four 96-well plates were comfortably imaged per day, which allowed the completion of this effort in merely a few weeks. We currently estimate that the entire library of C- and N-terminal fusions could be reimaged by a single person in less than 1 month.
6. Image Scoring and Analysis
6.1. Overview
The next step, after imaging all of the C- and N-terminal fusion proteins, was to examine the images to identify proteins that localized to discrete nonuniform patterns in Caulobacter. While image analysis software is constantly improving and certain aspects of localization analysis are immensely aided by quantitation, we found that the best initial analysis method was to simply examine each image by eye. Since the images were collected using a script that automated standardized file naming, we used a MATLAB (Matworks) script to automate opening the image files, aid in adjusting contrast levels, provide a standardized menu of scoring options, and save the scoring results in an easily accessible format. The use of these scripts allowed us to efficiently view each image, assign a localization pattern, and easily access and manipulate the scoring information. This scoring data allowed us to readily pare down our set of fluorescent protein fusions to a library of proteins with specific localization patterns.
In order to collect meaningful data from thousands of images of bacterial cells we needed to define what constituted a “localized” protein. For our study, we defined a localized protein as having a specific subcellular non-diffuse fluorescence pattern. The most abundant and easily recognized pattern was diffuse fluorescence defined as a confluent localization across the entire cell body. It is possible that we missed some cells with a subtle localization pattern that we were unable to detect by eye, but as a first pass we were satisfied to categorize these proteins as diffuse. Another set of proteins localized to the periphery of the cell creating a fluorescent cell outline suggesting that these proteins may reside in an extracytoplasmic compartment; however, these proteins did not show any other specific localization pattern. We chose to categorize both the diffuse and peripherally located proteins as “not localized” because both patterns appeared diffuse even if confined to a specific compartment of the cell.
While cells containing diffuse and peripherally localized proteins were the most common and easiest to identify, there were also strains that exhibited obvious localization patterns that were not difficult to discern. Many proteins localized as a tight focus in the cell. Interestingly, there could be one or multiple foci localized in several locations in the cell. A single focus could be localized at a pole or at a central location in the cell. One of the benefits of using Caulobacter as a model organism is the ability to differentiate the stalked and swarmer poles. Although we did not utilize this feature of Caulobacter for this study, future work will define polar localization more specifically. Some cells displayed more than one focus in the cell. Cells containing foci at both poles were termed “bipolar” while cells with foci at a pole and in the central region of the cell were called “Middle and Pole.” Other cells displayed a single focus at different locations in the cell. Cells that consistently showed a single focus at either a pole or a central location were defined as “Middle or Pole” while cells that displayed a focus at an inconsistent location throughout the cell were called “Variable Focus.”
Some fusions also localized in patterns other than a focus. Several proteins localized as a band across the middle of the cell while other proteins localized as a line of varying lengths along the long axis of the cell. Also, a previously unidentified localization category was proteins that were localized exclusively in the stalk. A final category was created that encompassed other obviously nonuniform yet less precise localization patterns. The “patchy/spotty” category encompassed proteins that may localize as helices or other structures. Patchy referred to light and dark patches in the cell that were not as defined as foci or bands while spotty referred to more than two foci in the cell often at less reproducible locations. The patchy/spotty category was the largest of the localized proteins but the most difficult to define.
6.2. Protocol for image scoring and validation
In order to score each image for the localization patterns described above we needed a program to assist collecting and organizing the scoring data. Besides localization patterns we could also describe problems with the image that may have prevented choosing a localization pattern such as too little fluorescence, a blurry picture, or E. coli contamination. All of this information was saved to a Microsoft Excel spreadsheet file that allowed us to easily summarize and manipulate the localization scoring data.
In order to eliminate some of the scoring bias that would be present by having a single individual score all of the pictures, several lab members were involved in the scoring process. Initially, two individuals scored all of the images and created a library of “possibly localized” proteins. Once this more manageable set of images was created, seven members of the lab independently scored each image to decide if each protein was “localized” or “not localized.” Finally, if five of the seven individuals deemed a protein to be localized, a specific pattern, as described above, was assigned to its localization. The “Localization Scorer” program is freely available upon request.
Once the set of localized proteins was collected it was necessary to validate the identities of the fusion proteins in each Caulobacter strain. Sequencing of PCR products amplified with primers from inside the xylX promoter and mCherry gene for gXRC and from within the mCherry gene to a region downstream from the Gateway cassette in gXRN verified the expected ORF was present in the expression vector. Only the Caulobacter strains in the localized set were sequence verified. Proteins that were both deemed localized by the consensus and were sequence verified constituted our final library of localized Caulobacter proteins.
The creation of a large set of localized proteins allowed us to perform statistical and quantitative analyses to examine general localization trends and to analyze the accuracy and distributions of localization patterns. For example, we statistically compared our set of localized proteins with their gene ontology (GO) functional classifications (Harris et al., 2004) to identify cellular activities that are more or less likely to function in precise cellular locations. To quantitate the localization patterns themselves, we used a software suite termed PSICIC (Guberman et al., 2008). PSICIC allowed us to perform a precise examination of protein localization and fluorescence intensity using interpolated contours to achieve subpixel resolution and generate an internal coordinate system for each cell. This allowed us to directly compare localization data between multiple cells regardless of cell geometries.
6.3. Results
After scoring and validating the localization library we were able to analyze the abundance of information we collected. Of the potential 5572 fusions in gXRC and gXRN, we identified a total of 352 localized fusion proteins (6.3% of all fusions) of which 187 localized as C-terminal fusions and 165 localized as N-terminal fusion proteins. Of these 352 fusions, 63 proteins localized with both a C- or N-terminal fusion, which gave a total of 289 unique proteins with localization patterns (Werner et al., 2009). These results testify to the value of the high-throughput methods we developed. Without the ability to create both C- and N-terminal fusion libraries we would have missed roughly one-third of the localized proteins.
After identifying whether the fusion proteins were localized, they were categorized into the specific classes described above. Of the 63 proteins that localized as both a C- or N-terminal fusion, 58 produced the same localization pattern giving us a “high-confidence” set of localized proteins. Another means to validate the localized proteins was to compare our set to proteins that have previously been reported as localized as fluorescent fusions. We identified 29 proteins whose genes were present in our ORFeome that localized as fluorescent protein fusions in previous studies. The localization patterns of 24 of the 29 proteins (83%) were recapitulated in our study indicating a relatively low rate of false-negative localizations. It is possible that the five localizations that did not match could be due to expression levels or the fluorescent protein used.
The statistical analysis correlating GO classifications to localized proteins provided insight into the cellular location of specific processes. This analysis found that proteins involved in small molecule metabolism were underrepresented in our localized protein library. This suggests that rapid diffusion of small molecules throughout the cell may not require precisely localized biosynthetic machinery. As expected, proteins involved in cellular processes such as cell division and chromosome partitioning, motility, and DNA replication/recombination/repair were overrepresented in the library of localized proteins along with less obvious processes such as signal transduction, intracellular trafficking and secretion, and membrane and cell wall biogenesis (Werner et al., 2009). These overenriched GO classes suggest that processes involved in spatial and temporal regulation inside the cell require localized proteins. This is consistent with the finding that genes whose transcription is cell-cycle regulated (Laub et al., 2000) are overrepresented in our set of localized proteins.
The use of PSICIC to precisely analyze protein localization provided intriguing information on general localization trends in Caulobacter. For analysis using PSICIC we excluded proteins classified as having a “patchy/spotty” localization pattern to reduce complexity. By examining the distribution of the mean position of each fluorescent fusion we found that there was an enrichment of localized proteins at the cell pole and near the middle of the cell. The standard deviation of the peak intensity position of these proteins was low suggesting that these localization patterns are highly reproducible. Interestingly, there were relatively few proteins that localized to the region between the pole and midcell. Proteins that did localize to this region had higher standard deviations of the peak intensity position, suggesting their localization patterns were more variable. These data suggest that from the cell pole to quartercell protein, localization becomes less accurate, while accuracy increases again between quarterand midcell (Werner et al., 2009). PSICIC also allowed us to examine how tightly a protein is localized in a single cell by examining the distance between the peak fluorescence intensity and its half-maximal value. These data found that polar proteins localized more tightly than proteins localized to the midcell (Werner et al., 2009). The combination of the findings that proteins are more likely to localize to the poles or midcell but proteins localized to the pole are more tightly localized suggests that there may be fundamentally different mechanisms involved in localizing these proteins.
An organized and accessible database was created to allow other individuals to access all of the information produced in this study. We designed a web site that allows public access to the list of proteins represented in our library and images of Caulobacter strains with localized proteins. Access to the list and images is found at the Gitai lab web site: www.molbio1.princeton.edu/labs/gitai/.
7. Conclusion
Methods and resources created in this study proved valuable for providing interesting data for this large-scale localization project but also provided the means to perform future high-throughput experimentation. In this study, we created a Caulobacter ORFeome that can be used to easily clone any ORF into any destination vector. To do this in a high-throughput manner, a protocol for performing the Gateway LR reaction was developed that reduces the effort and expense of traditional methods. Also, a technique to perform high-throughput high-resolution microscopy was developed that allows the reimaging of the current library or imaging of other libraries quickly and in a highly organized way. As useful as our library of localized proteins is for studying protein function and finding protein localization mechanisms, the techniques and resources we generated are just as valuable.
Generating our library of localized Caulobacter proteins identified new aspects of protein localization. Prior to this study there were no proteins known to specifically localize to the stalk. Also, there was only one protein, crescentin (Ausmees et al., 2003), shown to localize as a line while our study has identified two others. Computational analysis using PSICIC identified regions of the cell that lack as many localized proteins as other regions when examining only the proteins that localized in patterns other than patchy/spotty. Correlating protein localization to GO classifications discovered trends that were previously suspected, such as that motility and cell division proteins are more likely to be localized, but also found that other groups such as secretion and cell wall/membrane biogenesis proteins are also more likely to be localized. The creation of this library has provided a resource for future studies to be performed on the scale of an individual protein or at a proteomic scale of looking for localization trends or mechanisms.
Looking ahead only at the future projects that can be performed with the localization library provides just a small glimpse of the usefulness of this resource. While the feats of performing large-scale localization projects in yeast and E. coli were tremendous achievements, the difficulty associated with reimaging these libraries under various conditions without huge resource expenditures limited their subsequent utility. While the construction and imaging of our initial library took months to complete, subsequent imaging of the localized set of proteins can be performed in a single day. Future studies may include screening the localized set of proteins under various nutrient conditions or examining this library in the presence or absence of toxic compounds or antibiotics.
Creation of the Caulobacter ORFeome coupled with the in vivo LR reaction and high-throughput microscopy has provided a resource for future studies. Further examination of these proteins is only limited by the destination vector that can be created. Vectors are commercially available that produce tags for protein purification or pull-down assays and we are in the process of generating other destination vectors for a range of functional genomic studies. While we plan to increase the comprehensiveness of this ORFeome in the future, it has already provided a resource to perform many high-throughput studies.
This study has provided a template for manipulating genomic data at an experimental level. The ability to clone ORFs for an entire genome into a modular system such as the Gateway system has enabled researchers to perform new screens or comprehensive studies of protein function at a proteomic scale. We were able to couple the Gateway cloning system with new microscopy methods to perform high-throughput high-resolution microscopy-based screens for the first time. These methods are not confined to Caulobacter studies. Nearly all of the methods described in this work can be adapted to work in any bacteria or higher organism. These microscopy methods can be used to study any organism that fits under a microscope. This work has thus created a resource for studying protein localization, a more efficient and cost-effective method for high-throughput cloning, and a template for performing microscopy-based screens at the genomic level.
Acknowledgments
The authors thank Eric Chen for his assistance in creating the Caulobacter ORFeome and imaging, Jonathan Guberman for writing the image acquisition and image scoring programs, Angela Zippilli and Joe Irgon for gene sequencing and validation, and the entire Gitai lab for assistance with image scoring. We also thank Greg Phillips (Iowa State University, Ames, IA), Michael Kahn (Washington State University, Pullman, WA), Martin Thanbichler (Max Planck Institute for Terrestrial Microbiology, Marburg, Germany), and M. R. Alley (Anacor Pharmaceuticals, Palo Alto, CA) for materials, and Denis Dupuy and the rest of Marc Vidal’s lab for help in constructing the Caulobacter ORFeome entry library. John N. Werner is supported by a postdoctoral fellowship, Grant 1F32AI073043-01A1, from the National Institute of Allergy and Infectious Diseases and Zemer Gitai is supported by funding from Grant DE-FG02-05ER64136 from the U.S. Department of Energy Office of Science (Biological and Environmental Research) and a National Institute of Health New Innovator Award Number 1DP2OD004389-01.
References
- Ausmees N, Kuhn JR, Jacobs-Wagner C. The bacterial cytoskeleton: An intermediate filament-like function in cell shape. Cell. 2003;115:705–713. doi: 10.1016/s0092-8674(03)00935-8. [DOI] [PubMed] [Google Scholar]
- Brasch MA, Hartley JL, Vidal M. ORFeome cloning and systems biology: Standardized mass production of the parts from the parts-list. Genome Res. 2004;14:2001–2009. doi: 10.1101/gr.2769804. [DOI] [PubMed] [Google Scholar]
- Collier J, Shapiro L. Spatial complexity and control of a bacterial cell cycle. Curr Opin Biotechnol. 2007;18:333–340. doi: 10.1016/j.copbio.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991;204:372–384. doi: 10.1016/0076-6879(91)04019-k. [DOI] [PubMed] [Google Scholar]
- Gitai Z. The new bacterial cell biology: Moving parts and subcellular architecture. Cell. 2005;120:577–586. doi: 10.1016/j.cell.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Gitai Z, Dye N, Shapiro L. An actin-like gene can determine cell polarity in bacteria. Proc Natl Acad Sci USA. 2004;101:8643–8648. doi: 10.1073/pnas.0402638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guberman JM, Fay A, Dworkin J, Wingreen NS, Gitai Z. PSICIC: Noise and asymmetry in bacterial division revealed by computational image analysis at sub-pixel resolution. PLoS Comput Biol. 2008;4:e1000233. doi: 10.1371/journal.pcbi.1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, et al. The gene ontology (GO) database and informatics resource. Nucleic Acid Res. 2004;32:258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:671–672. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Laub MT, McAdams HH, Feldblyum T, Fraser CM, Shapiro L. Global analysis of the genetic network controlling a bacterial cell cycle. Science. 2000;290:2144–2148. doi: 10.1126/science.290.5499.2144. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, Horinouchi S, Yoshida M. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–847. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- Meisenzahl AC, Shapiro L, Jenal U. Isolation and characterization of a xylose-dependent promoter from Caulobacter crescentus. J Bacteriol. 1997;179:592–600. doi: 10.1128/jb.179.3.592-600.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen JA, Heidelberg JF, Alley MR, Ohta N, Maddock JR, Potocka I, Nelson WC, et al. Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci USA. 2001;98:6533. doi: 10.1073/pnas.061029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt R, Drescher C, Park SK, Phillips GJ. Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid. 2000;43:12–23. doi: 10.1006/plas.1999.1433. [DOI] [PubMed] [Google Scholar]
- Rual JF, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A, Li N, Rosenberg J, Lamesch P, Vidalain PO, Clingingsmith TR, Hartley JL, et al. Human ORFeome version 1.1: A platform for reverse proteomics. Genome Res. 2004;14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BK, House BL, Mortimer MW, Yurgel SN, Maloney SC, Ward KL, Kahn ML. Development of a functional genomics platform for Sinorhizobium meliloti: Construction of an ORFeome. Appl Environ Microbiol. 2005;71:5858–5864. doi: 10.1128/AEM.71.10.5858-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Silhavy TJ, Berman ML, Enquist LW. Experiments with Gene Fusions. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1984. [Google Scholar]
- Thanbichler M, Shapiro L. MipZ, a spatial regulator coordinating chromosome segregation with cell division in Caulobacter. Cell. 2006;126:147–162. doi: 10.1016/j.cell.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Thanbichler M, Shapiro L. Getting organized—How bacterial cells move proteins and DNA. Nat Rev Microbiol. 2008;6:28–40. doi: 10.1038/nrmicro1795. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: Application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc Natl Acad Sci USA. 2009;106:7858–7863. doi: 10.1073/pnas.0901781106. [DOI] [PMC free article] [PubMed] [Google Scholar]