

Abstract
Sphingosine-1-phosphate (S1P), a lipid second messenger formed upon phosphorylation of sphingosine by sphingosine kinase (SK), plays a crucial role in natural killer (NK) cell proliferation, migration, and cytotoxicity. Dysregulation of the S1P pathway has been linked to a number of immune system disorders and therapeutic manipulation of the pathway has been proposed as a method of disease intervention. However, peripheral blood NK cells, as identified by surface markers (CD56+CD45+CD3−CD16) consist of a highly diverse population with distinct phenotypes and functions and it is unknown whether the S1P pathway is similarly diverse across peripheral blood NK cells. In this work, we measured the phosphorylation of sphingosine–fluorescein (SF) and subsequent metabolism of S1P fluorescein (S1PF) to form hexadecanoic acid fluorescein (HAF) in 111 single NK cells obtained from the peripheral blood of four healthy human subjects. The percentage of SF converted to S1PF or HAF was highly variable amongst the cells ranging from 0% to 100% (S1PF) and 0% to 97% (HAF). Subpopulations of cells with varying levels of S1PF formation and metabolism were readily identified. Across all subjects, the average percentage of SF converted to S1PF or HAF was 37 ± 36% and 12 ± 19%, respectively. NK cell metabolism of SF by the different subjects was also distinct with hierarchical clustering suggesting two possible phenotypes: low (<20%) or high (>50%) producers of S1PF. The heterogeneity of SK and downstream enzyme activity in NK cells may enable NK cells to respond effectively to a diverse array of pathogens as well as incipient tumor cells. NK cells from two subjects were also loaded with S1PF to assess the activity of S1P phosphatase (S1PP), which converts S1P to sphingosine. No NK cells (n = 41) formed sphingosine, suggesting that S1PP was minimally active in peripheral blood NK cells. In contrast to the SK activity, S1PP activity was homogeneous across the peripheral blood NK cells, suggesting a bias in the SK pathway towards proliferation and migration, activities supported by S1P.
Introduction
Natural killer (NK) cells are effector lymphocytes that play a vital role in the immune response. NK cells can rapidly attack tumor cells and pathogen-infected cells in the absence of antigen-specific cell surface receptors.1 To accomplish this task, NK cells express major histocompatibility complex (MHC) class 1-specific inhibitory receptors, which enable them to recognize “self” markers. Upon interaction with foreign or stressed cells missing “self” markers, NK cells lose these inhibitory signals and become activated, releasing cytotoxic granules to lyse or initiate apoptosis in target cells. NK cells also express activating receptors, such as natural killer group 2, member D (NKG2D), which recognize ligands overexpressed in distressed or damaged cells.2 Additionally, NK cells are major producers of proinflammatory and immunosuppressive cytokines, such as tumor necrosis factor α (TNF-α) and interleukein-10 (IL-10), respectively.3 NK cells are typically grouped into two major subsets, CD56bright and CD56dim cells.4 CD56bright cells represent up to 10% of NK cells in peripheral blood, and are generally weakly cytotoxic but highly active cytokine producers.5 The remaining 90% of peripheral blood NK cells are CD56dim and are more efficient at lysing target cells but poor producers of cytokines. A recent study employing mass cytometry to characterize 36 cell surface proteins in NK cells, revealed extraordinary phenotypic heterogeneity amongst primary NK cells.6 The level of inhibitory receptor expression was primarily determined by genetics, while the expression level of activating receptors was heavily influenced by the environment. Based on these results, NK cells were proposed to consist of 6000 to 30 000 distinct subsets in the peripheral blood of a single person.7 This extreme level of heterogeneity suggests that each NK cell is likely unique and that technologies capable of single-cell measurements are vital to characterizing the physiology and function of NK cells.
The sphingosine-1-phosphate (S1P) pathway is a key regulator of lymphocyte migration, differentiation, and cytokine production.8-10 Sphingosine and S1P are interconverted by the actions of sphingosine kinase (SK) and S1P phosphatase (S1PP).11 Sphingosine can also be acetylated by ceramide synthase (CerS) to form ceramide, while S1P is also degraded by S1P lyase (S1PL) to form hexadecenal and ethanolamine phosphate. The relative amounts of these sphingolipids have been shown to determine cell fate i.e. death or survival.12 S1P drives lymphocyte proliferation, differentiation, egress through blood-vessels, and production of pro-inflammatory cytokines. Increases in sphingosine and ceramide typically act in opposition to S1P and promote inflammation, apoptosis, and autophagy.13 Dysregulation of the S1P pathway has been linked to a number of diseases of the immune system, including asthma, rheumatoid arthritis, and inflammatory bowel disease. The S1P pathway is particularly important for regulating NK cell migration and cytotoxicity. Gradients of S1P drive NK cell chemotaxis, and S1PR5, a member of the S1P receptor (S1PR) family, is essential for the mobilization of NK cells to inflamed organs.14 Despite promoting increased NK cell mobility, S1P also inhibits NK cell cytotoxicity.15 Conversely, glycosylated ceramides enhance NK cell cytotoxicity.16 The intracellular balance of these sphingolipids within single NK cells has not yet been measured, leading to significant gaps in understanding how the competing roles of these lipids are managed within a single cell and across a population of cells.
Although single-cell measurements are important for characterizing cellular diversity in NK cells, developing technologies capable of directly measuring lipid signaling in single primary cells has proven extremely challenging. These cells are fragile with high death rates in response to extensive handling and difficult to load with exogenous reagents. Their very small (6–7 μm diameter) size also requires analytical methods with very low limits of detection (LODs). Antibodies directed against lipids, including S1P, have been used to label cells from primary samples; however, these antibodies are plagued by nonspecific binding.17 The poor immunogenicity of most lipids also limits the affinity of the antibodies for their targets.18
Further, most antibody-based assays are indirect, qualitative measures of signaling processes since they detect only substrate or only product and do not quantify the number of moles of these species present in a cell. Characterization of multiple elements in a signaling pathway e.g. substrate, product and metabolites, is often required to achieve an accurate view of pathway signaling. Fluorescently and radiometrically-labeled sphingosines have been developed for monitoring SK activity in assays using lysates or purified enzyme but rarely used for measurements on single cells.19-21 To distinguish sphingosine from S1P, these assays incorporate an environment-sensitive fluorophore such as nitrobenzoxadiazole or utilize [32P]ATP. In these assays, an additional separation step thin-layer chromatography (TLC)19 or high performance liquid chromatography (HPLC)22 is required to remove unmetabolized ATP or to separate sphingosine from S1P. This increases assay complexity and nonspecific loss of the lipids. Analysis of sphingosine and S1P is also frequently accomplished using HPLC followed by mass spectrometry (MS) which permits detection of other sphingolipids but at a cost of poor sensitivity and therefore large sample sizes.23 None of the above technologies meet the requirements necessary for single-cell assays of sphingosine and its metabolites, i.e. ultra-high sensitivity, quantification of substrate and product simultaneously, and the ability to detect additional metabolites.
Capillary electrophoresis (CE) is well suited for separating the components of very small samples, routinely achieving subattomole limits of detection and yielding highly efficient separations of metabolites, often up to 1 million theoretical plates. With inclusion of internal standards, CE is capable of highly accurate quantification of molecules, including that obtained from a single-cell. The analysis of single cells by CE or chemical cytometry has been applied to measure a wide range of biological attributes including enzymatic activities such as kinases, lipases, and phosphatases.24-26 CE has been utilized to measure sphingosine kinase activity in cell lysates by separating fluorescently-labeled sphingosine and sphingosine-1-phosphate.27 Previously, an automated CE system was employed to rapidly quantify the conversion of a fluorescent sphingosine reporter (SF) to fluorescent S1P (S1PF) in single cells.28 The technology also detected the presence of additional metabolites of SF and S1PF, although these were not identified. The automated single-cell CE system increased throughput 100-fold compared to that attained by non-automated systems, permitting the analysis of sufficiently large sample sizes to identify statistically significant differences of S1P pathway signaling.
This work describes the use of automated single-cell CE system to characterize the S1P pathway in single primary human NK cells from peripheral blood. The conversion of SF to S1PF was measured in NK cells from four healthy subjects. The metabolism of S1PF to hexadecanoic acid fluorescein (HAF) was also characterized. The relative activities of SK and S1PP were assayed in the NK cells of two subjects by comparing cells loaded with SF to cells loaded with S1PF. The heterogeneity in S1P pathway activity was quantified amongst cells from a single subject as well as between different subjects. Measuring the single-cell sphingolipid rheostat revealed novel information about the regulation of the S1P pathway in human NK cells.
Experimental methods
Reagents and materials
Sphingosine 5/6-fluorescein (SF) and sphingosine-1-phosphate 5/6-fluorescein (S1PF) were procured from Echelon Biosciences Inc. (Salt Lake City, UT). Monosodium phosphate, 1-propanol, isopropanol, methanol, and pre-cleaned glass slides (50 mm × 45 mm × 1.5 mm) were acquired from Fisher Scientific (Pittsburgh, PA). Sodium chloride, sodium azide, potassium chloride, paraformaldehyde, magnesium chloride, calcium chloride, piperazine-1-ethanesulfonic acid (HEPES), formic acid, and glucose were purchased from Sigma-Aldrich Inc. (St Louis, MO). Ethanol was procured from Decon Labs (King of Prussia, PA). Roswell Park Memorial Institute Media (RPMI) with l-glutamine, fetal bovine serum (FBS), Dulbecco’s phosphate buffered saline, and penicillin/streptomycin were purchased from Invitrogen (Grand Island, NY). EPON resin 1002F (phenol, 4,4′-(1-methylethylidene)bis-, polymer with 2,2′-[(1-methylethylidene)-bis(4,1-phenyleneoxymethylene)]bis-[oxirane]) was acquired from Miller–Stephenson (Sylmar, CA, USA). SU-8 developer (1-methoxy-2-propyl acetate) was purchased from MicroChem (Newton, MA, USA). Poly(dimethylsiloxane) (PDMS) (Sylgard 184 Silicone Elastomer) was procured from Dow Corning (Midland, MI). Acetonitrile was purchased from VWR (Radnor, PA).
Purification of natural killer cells from PBMCs
Blood was drawn from study participants using a protocol approved by the University of North Carolina Institutional Review Board for Biomedical Research. Written consent was provided by each study participant. PBMCs were isolated using a Lymphoprep gradient (Axis Shield, Scotland, UK), similar to that used in previous studies.29-31 NK cells were isolated from PBMCs by negative selection using Dynabeads NK isolation kit (Invitrogen, Carlsbad, CA), which depletes T cells, B cells, monocytes, platelets, dendritic cells, granulocytes, and erythrocytes using monoclonal antibodies directed against CD3, CD14, CD36, HLC Class II, CD123, and CD235a. After isolation, the NK cells were suspended in media (RMPI 1640 with l-glutamine, 10% heatinactivated FBS, and 1% penicillin/streptomycin) for single-cell analysis or flow buffer (1% heat-inactivated FBS and 0.09% sodium azide in Dulbecco’s phosphate buffered saline without magnesium or calcium) for flow cytometry analysis.
Flow cytometry characterization of NK cell purity was conducted by incubating NK cells with antibodies directed against CD45-allophycocyanin cyanine 7 (APCCy7) (BD Biosciences, San Jose, CA), CD56-phycoerythrin (PE) (BD Biosciences), CD3-allophycocyanin (APC) (BD Biosciences), and CD16-Pacific Blue (BD Biosciences), as in previous studies.29-31 The cells were fixed in 0.5% paraformaldehyde, and analyzed within 24 hours of fixation by flow cytometer (BD LSRII, BD Biosciences). Data were analyzed with FLOWJO (TreeStar, Ashland, OR). Dead cells were excluded using a LIVE/DEAD® fixable far-red viability dye (Molecular Probes, Grand Island, NY). The purity of the NK cell preparation was quantified by flow cytometric measurement of CD45 and CD56 protein expression (CD45+CD56+). The population was comprised of 92 ± 4% CD45+CD56+ cells which define NK cells (Fig. S1, ESI†). This was comparable to previously published NK cell purifications using this method.29-31 The absence of cells stained with anti-CD3-APC antibodies indicated that there were no contaminating T cells.
Fabrication of microwells
The arrays of 10 × 10 microwell cell traps were fabricated by patterning holes into a photoresist layer on a coverslip using standard photolithography as described previously.28 Briefly, coverslips were spin-coated with 1002F photoresist, and baked at 95 °C for 30 min.32 An iron oxide photomask was then placed above the 1002F-coated coverslip and the assembly illuminated with UV light (600 mJ). The photoresist-coated coverslip was subsequently developed in SU-8 developer, rinsed with isopropanol, and then baked at 120 °C for 30 min. The microwells or cell traps were 20 μm in depth and separated from adjacent wells by 100 μm.
Fabrication of poly(dimethylsiloxane) channel system
The PDMS channel system used to store the captured cells in a physiologic buffer solution as well as to contain the electrophoretic buffer for CE separations was similar to that described previously.28 A PDMS layer with cutouts for the physiologic and electrophoretic-buffer channels was bonded to the microwell array. The electrophoretic-buffer channel was parallel to the physiologic buffer-channel. These buffer channels were connected by a perpendicular channel filled with air. The electrophoretic and physiologic buffers remained in their respective channels due to surface tension. The cell trap array was placed in the physiologic buffer channel aligned with the center of the air-channel.
Reporter loading
For single-cell experiments, SF and S1PF were loaded into cells by incubating 5 × 105 cells in 100 μL NK cell media containing 80 μM SF or S1PF for 30 min at 37 °C in a 5% carbon dioxide atmosphere. For cell lysate experiments, cells were incubated with 10 μM SF for 1 h. After reporter loading, cells were pelleted and then washed 5 times with 200 μL physiologic buffer (135 mM sodium chloride, 5 mM potassium chloride, 1 mM magnesium chloride, 1 mM calcium chloride, 10 mM HEPES, and 10 mM glucose at pH 7.4). For single-cell experiments, cells were then resuspended in physiologic buffer at a concentration of 1 × 104 cells per μL and immediately loaded into cell traps. The sum of the 30 min incubation time with the reporter and the additional waiting time before cell lysis is termed the “reporter exposure time”. For cell lysate experiments, cells were resuspended in 10 μL of physiologic buffer and then chemically lysed by adding 10 μL of methanol. Cell lysates were immediately stored at −80 °C until analysis. Statistical differences in reporter loading with SF vs. S1PF were calculated using a Mann–Whitney U-test.33
Ensemble measurements of the sphingosine kinase pathway using cell lysates
Two types of cells were used to perform analysis of the S1P pathway in cell lysates: NK cells from peripheral blood and tissue-cultured K562 cells originally derived from a chronic myelogenous leukemia patient. Conversion of SF to S1PF was measured in both cell types by loading cells with reporter, as described above, lysing the cells, and separating the fluorescent compounds using CE. For experiments testing the effect of SK inhibition, K562 cells were incubated in media (RMPI 1640 with l-glutamine, 10% heat-inactivated FBS, and 1% penicillin/streptomycin) with varying concentrations of SK Inhibitor 2 (SKI 2) (Cayman Chemical Co., Ann Arbor, MI) for 30 min.34 SF reporter was then added to this media, as detailed above, and phosphorylation of the reporter was quantified. The cells were therefore exposed to SKI 2 for a total period of 1.5 h. In order to test the effects of ceramide synthase inhibition, K562 cells were incubated with 10 μM fumonisin B1 (Sigma-Aldrich), a ceramide synthase inhibitor, for 2 h.35 Cells were then loaded with the reporter, lysed, and analyzed using CE. Cells were exposed to fumonisin B1 for a total of 3 h. In order to generate lysates for HPLC-MS analysis, 10 million K562 cells were incubated with 80 μM SF for 1 h, washed as described above, resuspended in 50 μL of ECB, and lysed by the addition of 50 μL of methanol. The cell suspension was then filtered to remove debris prior to HPLC analysis.
Characterizing lysates using HPLC-MS
Analysis of SF and its metabolites were performed with an HPLC-MS system consisting of an Agilent 1200 HPLC and Agilent 6520 Accurate Mass Quadruple Time-Of-Flight (Q-TOF) mass spectrometer (Agilent Technologies, Milford, MA). Gradient separations were performed using a 2.1 mm × 50 mm Agilent Eclipse XDB-C18 column with 1.8 mm particles. Separations were conducted for a total of 12 minutes at a flow rate of 0.2 mL min−1 and a pressure of 400 bar. The mobile phase composition was 30% acetonitrile and 0.1% formic acid for 1.5 min followed by a linear increase to 90% acetonitrile and 0.1% formic acid over 7.5min, and then 90% acetonitrile and 0.1% formic acid for 3 min. Negative mode electrospray ionization (ESI) MS was conducted with a fragmentor voltage of 200 V, nebulizing gas pressure of 45 psi, and a capillary voltage of 3500 V. Sample (35 μL) was used for each separation. Fractions of HPLC eluent were collected manually for subsequent CE analysis. Data analysis was performed using Agilent Qualitative Analysis B.04.00 software.
Cell capture in microwells
SF or S1PF-loaded NK cells were captured in microwells by pipetting 10 μL of cells (1 × 106 cells per mL) onto the microwell array. The cells were incubated on the array for 20 min, and then excess cells were rinsed from the array with physiologic buffer. The cells trapped in the wells were then continually bathed in physiologic buffer at 35 °C (1 mm s−1 flow velocity). A student t-test was used to compare the capture efficiency of microwells with different diameters.
Automation of single-cell analysis
The PDMS channel system was mounted on an inverted microscope (Ti-E, Nikon, Melville, NY) with a computer-controlled motorized stage (Ti-SER, Nikon). The microscope stage was fitted with a temperature controlled stage insert. The insert was maintained at 37 °C for the duration of the experiment. Customized software (Python, Wolfeboro Falls, NH and Micro-Manager, Vale Lab, UCSF) identified the address of each cell trap and moved the motorized stage.28 Each cell trap that did not contain a single cell was manually deleted from the program’s list of well addresses to be interrogated, so that only wells with single-cells were analyzed. All other tasks were performed using LabVIEW (National Instruments, Austin, TX).
Measurement of sphingosine kinase activity in single cells
During automated single-cell analysis, the following events occurred sequentially under computer control. The microscope stage moved to the address of the first cell to be analyzed, placing the cell directly below the inlet of a capillary (30 μm i.d.). The capillary was aligned 50 μm above the plane of the microwells. A focused laser pulse lysed the cell by creating a cavitation bubble36 and the cell’s contents were then electrokinetically loaded into the capillary. Electrokinetic injections were performed by the application of 5 kV across the capillary for 1 s. The voltage was then set to zero as the stage moved through the air channel to the center of the electrophoretic buffer channel. A voltage of 18 kV (0.5 kV cm−1) was applied across the capillary for 55 ± 10 s to separate the cellular contents. The voltage was then set to zero for 1 s as the stage moved back to place the capillary at the address of the next cell to be assayed. The next cell was lysed, then injected, and this entire process was repeated until all of the cells in the array were analyzed.
The capillary was interrogated 4 cm from the inlet end by the focused beam of a fiber-coupled diode pumped solid state 473 nm laser and the fluorescence of the analytes detected with a photomultiplier tube as described previously.37 The analytes were identified by comparison of their migration times to that of standards. The peak areas of SF, S1PF, and unidentified compounds were calculated using a custom program written in MATLAB (Natick, MA).38 The peak areas of known amounts of SF and S1PF standards were used to calculate the molar amount of SF and S1PF contained in each cell. A brightfield image of each cell, taken immediately prior to the single-cell experiment, was used to measure cell diameter.
In order to characterize the activity of S1P pathway enzymes, the molar amounts of SF, S1PF, and HAF in each cell were calculated using a calibration curve. The total reporter loaded per cell was defined as the sum of the number of moles of SF, S1PF, and HAF (SFmol + S1PFmol + HAFmol). The percent of each compound was calculated by dividing the molar amount of the compound by the total moles of reporter loaded into cells. For instance, the percent of S1PF formed was calculated as S1PFmol/(SFmol + S1PFmol + HAFmol). The “reporter exposure time” for each cell was defined as the time between the start of the cell incubation with SF or S1PF and the time of cell lysis.
Statistical analysis
Linear regressions were performed to determine coefficients of determination (R2). A t test was used to determine whether correlations were statistically significant.39 Hierarchical clustering was performed using a cosine distance function to compare the total distribution of percent S1PF between the four subjects.40
Results and discussion
Optimization of the automated CE system for NK cell analysis
The S1P pathway in NK cells was characterized using an automated CE system comprised of a three-channel microdevice mounted on an automated microscope operated with customized software (Fig. 1a).28 Two parallel channels, one containing electrophoretic buffer and the other a physiologic buffer, were connected by a perpendicular air-filled channel. Surface tension forces at the air-buffer interfaces prevented the electrophoretic and physiologic buffers from mixing. This three-channel design enabled fast transfer of the capillary between physiologic and electrophoretic buffers with minimal carryover of the buffers. Individual cells were captured in the physiologic buffer channel within cell traps, or microwells, microfabricated into the lower surface of the channel (Fig. 1b and c). The capillary moved under software control to the address of an initial cell trap, after which the cell beneath the capillary was lysed using a laser pulse,36 and the cellular contents electrokinetically injected into the capillary. The capillary traversed through the air channel to the electrophoretic buffer and separation of the cell contents was initiated. After a defined time, the capillary moved back through the air gap to a position above the next cell to be analyzed and the entire process was repeated in a fully automated fashion.
Fig. 1.
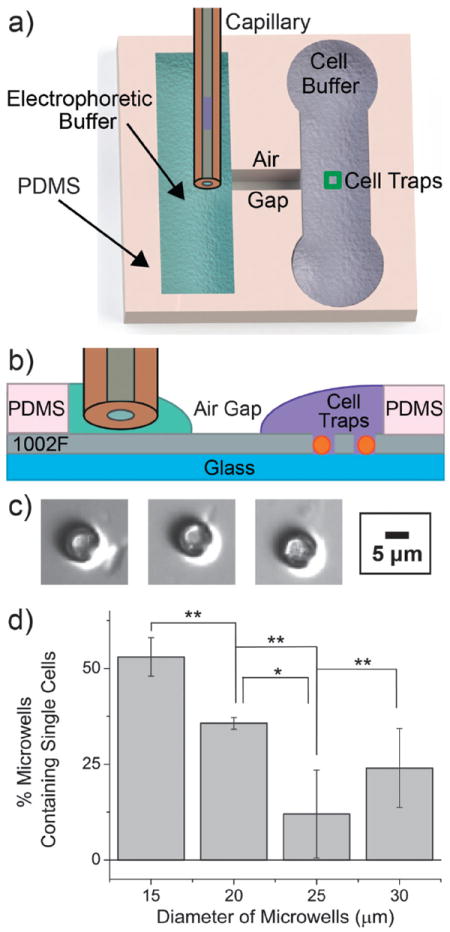
Optimization of the automated single-cell CE system for NK cells. (a) Schematic (top view) of the channel system used for automated singlecell CE analysis. (b) Schematic (cross section) through a section of the cell traps (not drawn to scale). Surface tension prevents the electrophoretic (green) and physiologic (purple) buffers from spilling into the air gap. The orange spheres represent cells within the traps. (c) Brightfield image of an NK cell trapped in each of three microwells. (d) The percentage of microwells entrapping a single cell is shown for microwells of varying diameter. Three replicates were obtained for each histogram (*p ≤ 0.05; **p ≤ 0.01).
In order to efficiently trap NK cells, which are small (6–7 μm diameter) relative to the size of most tissue-cultured cells (12–15 μm diameter), the cell-trap size was optimized for capture of single NK cells. Microwells (20 μm deep) with 4 different diameters (15, 20, 25, and 30 μm) were tested for their single-cell capture efficiency. NK cells were loaded onto the microwell array and permitted to settle by gravity. The array was then washed to remove the excess cells. The 15 μm microwells were significantly more effective at trapping single NK cells than the 20, 25, and 30 μm diameter wells (Fig. 1d). The percentages of microwells that captured more than one primary cell were 13 ± 13%, 22 ± 5%, 80 ± 20%, and 63 ± 6% for the 15, 20, 25, and 30 μm microwells, respectively. These results are consistent with previous work suggesting that microwells that are double the cell size aremost efficient at single-cell capture.37 Due to their higher efficiency at capturing single cells, cell traps with diameters of 15 μm were used to capture cells for all subsequent single-cell CE assays.
Assay of S1P signaling in single cells by CE
NK cells were incubated with the SF reporter and then loaded into microwell traps (Fig. S2a, ESI†). Traps with zero cells or >2 cells were not utilized in the assays. The diameter of each trapped, single cell was measured by brightfield microscopy and used to estimate cell volume. The cells were then sequentially analyzed using the automated, single-cell CE system(Fig. S2b, ESI†). Three major peaks were observed from the cells on the electropherograms (Fig. 2a and b). Two of the peaks were identified as SF and S1PF based on their co-migration with standards. A third peak was observed in a subpopulation of cells. This peak, which had an unknown identity, was named Unknown 50 (U50) since it migrated at 50 s. U50 has previously been reported to be an unidentified fluorescein-labeled metabolite of the S1P pathway (Fig. 2c).
Fig. 2.
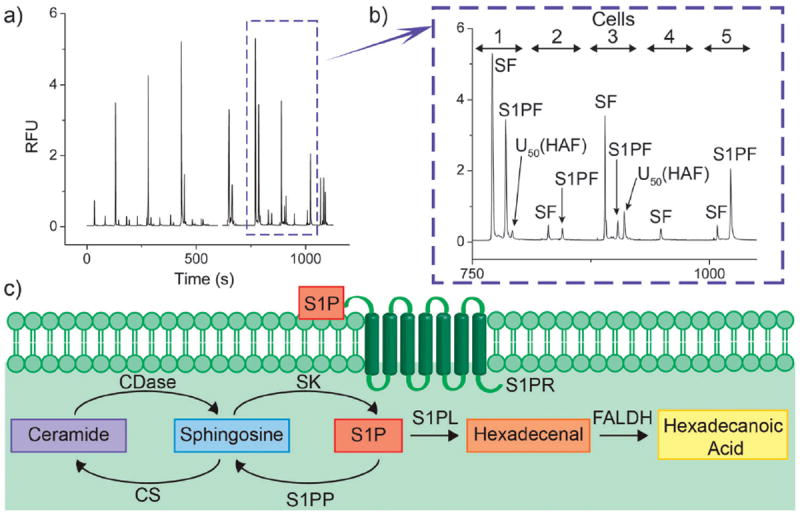
Single-cell analysis of peripheral blood NK cells. (a) Representative electropherogram demonstrating separation of SF, S1PF, and U50 (HAF) from 19 cells sequentially analyzed. (b) An expanded region of the electropherogram shown in panel (b) demonstrating the contents of 5 cells. (c) The S1P pathway in NK cells. Abbreviations: ceramidase (CDase) and ceramide synthase (CS).
To determine the identity of U50, K562 cells, which have previously been shown to form relatively large amounts of U50, were treated with inhibitors of S1P pathway enzymes.25 Inhibition of ceramide synthase by fumonisin B1 did not affect the production of U50 in cells (Fig. S3, ESI†). However, inhibition of SK by sphingosine kinase inhibitor 2 (SKI 2) was found to decrease formation of U50 as well as the production of S1PF (Fig. S4a, ESI†). In addition, increasing concentrations of SKI 2 decreased U50 formation in a dose-dependent manner (Fig. S4b, ESI†). These results suggested that U50 might be a metabolite downstream of S1PF. To identify U50, K562 cells were loaded with SF, incubated, lysed and the lysate separated using HPLC (Fig. S5a, ESI†). The mass of all fluorescent species was measured using mass spectrometry. Three fluorescent sphingolipids were identified: SF, S1PF, and an analyte with a mass (627.29) matching that of hexadecenoic acid fluorescein (HAF). Although no hexadecenal–fluorescein was observed, hexadecenal is rapidly converted to hexadecanoic acid by fatty aldehyde dehydrogenase (FALDH) in cells.41 These data suggested that hexadecenal–fluorescein was produced in the cells but rapidly converted to HAF by FALDH. To confirm the identity of the species with molecular weight 627.29 as HAF, MS–MS was performed. Species with molecular weights consistent with HAF with CO2 and/or water loss were identified confirming the presence of HAF in the cell lysate (Fig. S5b, ESI†). To determine whether U50 was indeed HAF, a fraction of the HPLC eluent containing HAF was collected and then electrophoresed. HAF possessed a migration time of 50 s and co-migrated with U50 when co-injected (Fig. S5c, ESI†). Thus, U50 is HAF, a downstream metabolite of S1PF.
Characterization of reporter exposure time and cell size on S1PF formation
NK cells from 4 healthy subjects were loaded with SF, placed in cell traps, and analyzed SF metabolites measured. In total, 133 cells (49, 32, 31, and 21 cells from subjects 1, 2, 3, and 4, respectively) were examined. The electropherograms from 14 ± 13% of the cells (32%, 7%, 35%, and 0% from subjects 1, 2, 3, and 4, respectively) did not contain fluorescent peaks above the limit of detection i.e. a peak height greater than or equal to three times the standard deviation of the background, suggesting that an insufficient amount of SF was loaded into these cells for detection (Fig. S6a and b, ESI†). Of the 111 cells with detectable peaks, the total amount of SF loaded per cell ranged from 0 to 196 amol, corresponding to 0–2.2 mM, assuming that the cells were spherical (6 μm diameter). Only the 111 cells with identifiable fluorescent peaks were further characterized. Within these 111 cells, there was no correlation between the total amount of reporter loaded per cell (R2 = 0.009), the cell diameter (R2 = 0.009, Fig. S7, ESI†) or the reporter exposure time (R2 = 0.02). The absence of a correlation between the total amount of loaded reporter in a cell and the cell diameter suggests that each cell possessed a different intracellular concentration of the reporter. The lack of correlation between the total amount of loaded reporter in a cell and the reporter exposure time suggested that significant amounts of reporter were not exported from the cells over time. There was additionally no correlation between the amount of moles of S1PF produced and the reporter exposure time (R2 = 0.01) for the combined cells from all subjects (Fig. S8a and b, ESI†). This lack of a correlation may be due to the formation of an equilibrium between the substrate, SF, and product, S1PF, within the cell. The number of moles of S1PF produced did increase with the total amount of reporter loaded into cells (R2 = 0.9, p ≤ 0.001, Fig. S9, ESI†). This relationship remained linear, even at very high amounts of loaded reporter, indicating that SK was not saturated in many of the cells. A significant fraction of cells (5% and 22%) from subjects 1 and 3, respectively, fully converted the SF reporter to S1PF even in cells with as much as 28 amol of total reporter. In some cells, over 98% of the SF reporter was converted despite extremely high quantities of loaded reporter 196 amol or approximately 2 mM demonstrating an exceptionally high capacity to produce S1P.
S1PF and HAF formation in single primary NK cells
The conversion of the reporter to S1PF and HAF was highly variable amongst individual NK cells (Fig. 3a). On average, the electropherograms of 70 ± 16% and 30 ± 10% of cells possessed identifiable S1PF or HAF, respectively. The average percent of reporter converted to S1PF or HAF was 37 ± 36% and 12 ± 19%, respectively, across all cells from all subjects. There was no correlation between % HAF formation and the total amount of reporter loaded into cells (R2 = 0.002), suggesting that the amount of HAF formed was not dependent on reporter uptake. In general, the percentage of SF converted to HAF was less than that converted to S1PF. Only 9 of 111 cells formed a greater amount of HAF than S1PF. This suggested that once formed, S1P was fairly stable in cells and that SK activity was proportionately higher than S1PL activity in the majority of NK cells supporting an accumulation of S1PF. The relatively high level of SK activity in most cells suggested that a major subpopulation of NK cells was geared towards increased proliferation, migration, and cytokine production. The small population of cells with large amounts of HAF suggested that in some cells, the high SK activity was overcome by metabolism of S1P to shut down S1P signaling. These peripheral blood NK cells formed a highly heterogeneous population with respect to S1P production and metabolism. Since S1P is implicated in the regulation of NK cell migration and cytotoxicity, traits that are known to vary substantially between NK cells, this cell-to-cell heterogeneity may be important for the wide range of immune functions in which NK cells must participate.
Fig. 3.
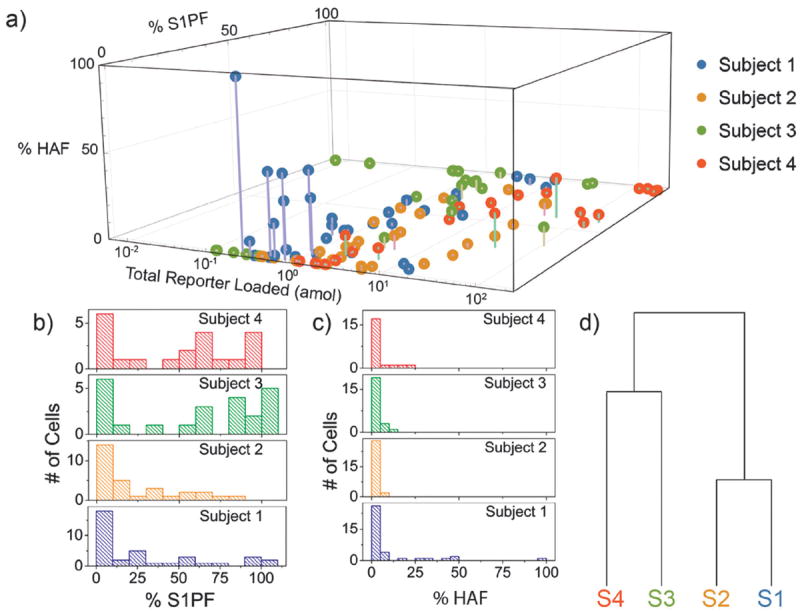
Metabolism of SF in single NK cells. (a) A 3D plot comparing the percentage of S1PF and HAF formed from the total SF loaded into a cell. The lower x axis displays the total concentration of reporter loaded into the cell (SFmol + S1PFmol + HAFmol). (b) Histograms expressing the number of cells with different percentages of S1PF for each subject. (c) Histograms comparing the percent of reporter converted to HAF for each subject. (d) The tree diagram showing hierarchical clustering comparing the overall distribution of percent S1PF formation between subjects 1–4.
Inter-subject comparison of the distribution of SF reporter conversion revealed similarities as well as subtle differences among all subjects. Overall there was a broad distribution of phosphorylation amongst the cells analyzed from subject 1; however, the majority of cells (51%) contained less than 20% S1PF with 45% of subject 1’s cells possessing less than 10% S1PF (Fig. 3b). The cells from subject 2 also possessed a wide range of levels of S1PF, and 43% of the cells produced less than 10% S1PF. Thus the SF-S1PF balance in the NK cells of subjects 1 and 2 suggested a relatively lower level of NK cell proliferation and mobility relative to the average of all subjects. The majority (65%) of subject 3’s cells possessed more than 50% S1PF with 30% of the cells displaying 490% S1PF suggesting that this individual’s NK cells might be biased towards proliferation and migration. Subject 4 also had a wide, fairly even, distribution of phosphorylation with the majority (57%) of cells possessing greater than 50% S1PF. The SF-S1PF balance in the NK cells of subjects 3 and 4 was biased towards higher S1P production, relative to the average of all subjects, suggesting that these cells might have higher levels of mobility and proliferation. These results highlight the intercellular heterogeneity of the S1P pathway in NK cells from a single individual as well as the S1P signaling variability between subjects. Hierarchical clustering was used to compare the overall distribution of S1PF formation between the subjects (Fig. 3d). Subjects 1 and 2 clustered together, since both possessed S1PF distributions skewed towards lower levels of S1PF relative to the distributions of subjects 3 and 4, which co-clustered (Fig. 3b).
The majority of cells from all four subjects displayed low percentages of reporter conversion to HAF (Fig. 3c). 73%, 93%, 83%, and 81% of cells from subjects 1–4 had less than 5% production of HAF, respectively. 16%, 0%, 0%, and 0% of the cells from subjects 1–4 had greater than 25% conversion of SF to HAF. This again suggests a relatively higher activity for SK compared to the downstream metabolic enzymes, which may permit rapid accumulation of S1P and cell activation during NK-cell encounters with infected cells. Of note is that although subjects 1 and 2 clustered together with respect to the percentage of S1P formed, the two individuals differed when producing HAF. Subject 1 formed much greater amounts of HAF than did subject 2, suggesting that subject 1 did produce S1PF, but also metabolized it quickly. While the NK cells of subjects 1 and 2 both possessed low amounts of S1PF, the cells may have used varying mechanisms to lower the percentage of S1PF. These data suggest that there may be multiple layers of the S1P concentration in NK cells and simultaneous measurement of multiple enzymatic products is required for an accurate understanding of S1P signaling.
The distinct patterns of S1PF and HAF formation observed in NK cells from different subjects may arise from a variety of sources other than the history of viral infections. NK cell activity varies with body mass index (BMI), gender, age, and race, among many other factors.42-44 Notably, subjects 1 and 2, whose cells were skewed towards lower levels of S1P, were both older than subjects 3 and 4 (Table S1, ESI†). However, the sample size of four subjects is too small to identify the sources of variations in the phosphorylation distribution. Further research will be needed to identify the sources of this heterogeneity and determine whether age, race, or BMI are contributors to the inter-subject variability.
Sphingosine-1-phosphate phosphatase activity in NK cells
To additionally characterize the flow of metabolites in the S1P pathway, S1PF was loaded into cells and its fluorescent metabolites measured. A total of 41 cells were assayed, with 11 from subject 5 and 31 from subject 6. On average, significantly less S1PF reporter (0.2 ± 0.2 amol) was loaded into cells compared to that for the SF reporter (13 ± 35 amol) (p = 2 × 10−6). S1PF is more polar than SF, likely creating an added barrier to traversing the membrane.27 17 ± 15% of cells did not load a detectable quantity of S1PF. The amount of S1PF reporter loaded did not correlate with the cell diameter for the combined cells from subjects 5 and 6 (R2 = 0.04) or for the cells for each subject individually (Fig. 4a). For both subjects, the amount of reporter loaded into the cells was linearly correlated with the time that the reporter was incubated in the solution surrounding the cell (R2 = 0.6). This correlation was significant for all cells (p < 0.001), as well as for the cells from subjects 5 (p < 0.05) and 6 (p < 0.001), respectively (Fig. 4b). This relationship may be due to the very slow cellular internalization of the S1PF reporter as a result of its increased polarity or to a different mechanism of import into the cell. The majority of cells (95 ± 8%) that did load with S1PF did not convert it to any other detectable compound (Fig. 4c). None of the cells dephosphorylated the reporter to form SF. This result suggested that the majority of NK cells analyzed had very low S1PP activity in comparison to SK activity. However, the low levels of S1PF conversion may also be due to differences in the location of S1PF in the cells. The fluorescence microscopy images of S1PFloaded cells suggested that the reporter was distributed predominantly in the cytoplasm of the cells and with minimal amounts present in the nucleus (Fig. S10, ESI†). 0% and 11% of S1PF-loaded cells from subject 5 and 6, respectively, were observed to convert the reporter to HAF, suggesting that the S1PF was accessible to the downstream metabolic enzymes. Again, these data suggested that in NK cells, the sphingosine metabolic pathways might be biased toward accumulation of S1P within the cells.
Fig. 4.
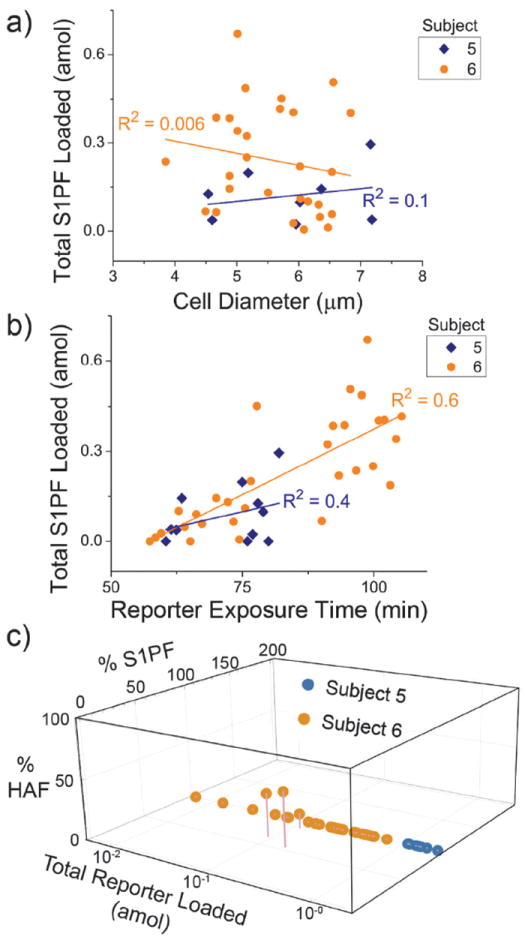
S1PF loading and metabolism in NK cells from two subjects (n = 36 cells). (a) The total amount of S1PF loaded into cells (S1PFmol + HAFmol) plotted against the diameter of the cells. (b) The total amount of S1PF loaded into cells (S1PFmol + HAFmol) was plotted against the reporter exposure time. (c) The percent of S1PF and HAF formed in a cell (relative to the total reporter loaded) was plotted against the total amount of S1PF reporter loaded into that cell.
Conclusion
Our findings suggest that activity within the S1P pathway is highly heterogeneous in NK cells obtained from an individual subject as well as between different subjects. These results are consistent with previous studies identifying a wide variety of NK cell types thought to be present in humans.4,14 Differing patterns of SF reporter processing were also observed amongst the subjects with the cells of some subjects displaying a bias towards lower S1PF levels per cell and other individuals exhibiting a very broad range of S1PF levels. Phosphorylation of the SF reporter was upregulated relative to S1PF breakdown in the majority of cells, but small subpopulations of cells, mostly from one subject, demonstrated high metabolism of the phosphorylated reporter. This indicated that NK cells may regulate S1P concentrations using different mechanisms. Based on the very low levels of back conversion of S1PF to SF, S1PP activity in human subjects appears to be significantly lower than that of SK activity. Additionally, no peaks were observed on the electropherograms that could have been attributable to fluorescent ceramide formation, suggesting that the sphingosine–ceramide pathway in healthy humans is skewed to rapidly metabolize sphingosine to sphingosine-1-phosphate. The rapid conversion into sphingosine-1-phosphate would then act to favor cell proliferation and cell egress out of blood vessels into tissue as well as production of pro-inflammatory cytokines rather than apoptosis. All of these attributes would contribute towards rapid activation of the immune system toward pathogens.
The automated single-cell CE technology enabled the rapid assay by electrophoretic separation of hundreds of single cells. Sphingosine metabolism was characterized in the primary NK cells derived from the peripheral blood of six different humans. The high separation capacity of CE enabled measurement of multiple sphingolipids, permitting characterization of the activity of four enzymes (SK, S1PP, S1PL/FALDH). Increasing the automated system’s throughput and synthesizing additional fluorescent sphingolipid reporters will enable simultaneous measurement of the activity of dozens of kinases and lipases in thousands of cells per human, providing a more comprehensive picture of each individual cell’s signaling pathways.
Supplementary Material
Insight, innovation, integration.
Quantification of lipid signaling in single primary human cells has been challenging due to the hydrophobicity of lipids, very small size of mammalian cells, and fragility of primary cells. An automated single-cell, capillary electrophoresis system was used to characterize the sphingosine pathway in primary, human NK cells. The technology enabled simultaneous measurement of fluorescent sphingosine-1-phosphate (S1P) and hexadecanoic acid (HA) formed from a fluorescein-labeled sphingosine reporter. Substantial heterogeneity in S1P production and metabolism across cells within and between subjects was readily apparent. NK-cell subpopulations may exist with respect to SK activity and individual humans may possess distinct phenotypes. A deeper understanding of lipid signaling at the single-cell level will be critical to understand NK cell biology and disease.
Acknowledgments
This research was supported by the NIH (CA171631 to A.J.D. and CA177993 to N.L.A., and ES013611 and ES01366-S2 to I.J.). We also thank the UNC Biomarker Mass Spectrometry Facility, which is supported by NIEHS grant P30-ES10126.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00007f.
References
- 1.Cerwenka A, Lanier LL. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 3.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper MA, Fehniger TA, Caligiuri MA. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 5.Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. Immunology. 2009;126:458–465. doi: 10.1111/j.1365-2567.2008.03027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Dekker CL, Mackey S, Maecker H, Swan GE, Davis MM, Norman PJ, Guethlein LA, Desai M, Parham P, Blish CA. Sci Transl Med. 2013;5:11. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanier LL. Nat Biotechnol. 2014;32:140–142. doi: 10.1038/nbt.2810. [DOI] [PubMed] [Google Scholar]
- 8.Spiegel S, Milstien S. Nat Rev Immunol. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takabe K, Paugh SW, Milstien S, Spiegel S. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivera J, Proia RL, Olivera A. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuvillier O. Biochim Biophys Acta, Mol Cell Biol Lipids. 2002;1585:153–162. doi: 10.1016/s1388-1981(02)00336-0. [DOI] [PubMed] [Google Scholar]
- 12.Pyne NJ, Pyne S. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 13.Maceyka M, Spiegel S. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, Jacques Y, Baratin M, Tomasello E, Vivier E. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 15.Lagadari M, Lehmann K, Ziemer M, Truta-Feles K, Berod L, Idzko M, Barz D, Kamradt T, Maghazachi AA, Norgauer J. Int J Oncol. 2009;34:287–294. [PubMed] [Google Scholar]
- 16.Kobayashi E, Motoki K, Yamaguchi Y, Uchida T, Fukushima H, Koezuka Y. Bioorg Med Chem. 1996;4:615–619. doi: 10.1016/0968-0896(96)00049-1. [DOI] [PubMed] [Google Scholar]
- 17.Nincheri P, Bernacchioni C, Cencetti F, Donati C, Bruni P. Cell Signalling. 2010;22:1688–1699. doi: 10.1016/j.cellsig.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Gargir A, Ofek I, Meron-Sudai S, Tanamy MG, Kabouridis PS, Nissim A. Biochim Biophys Acta, Gen Subj. 2002;1569:167–173. doi: 10.1016/s0304-4165(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 19.Pitman MR, Pham DH, Pitson SM. Methods Mol Biol. 2012;874:21–31. doi: 10.1007/978-1-61779-800-9_2. [DOI] [PubMed] [Google Scholar]
- 20.Lima S, Milstien S, Spiegel S. J Lipid Res. 2014;55:1525–1530. doi: 10.1194/jlr.D048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Billich A, Ettmayer P. Anal Biochem. 2004;326:114–119. doi: 10.1016/j.ab.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Caligan TB, Peters K, Ou J, Wang E, Saba J, Merrill AH. Anal Biochem. 2000;281:36–44. doi: 10.1006/abio.2000.4555. [DOI] [PubMed] [Google Scholar]
- 23.Camera E, Picardo M, Presutti C, Catarcini P, Fanali S. J Sep Sci. 2004;27:971–976. doi: 10.1002/jssc.200301712. [DOI] [PubMed] [Google Scholar]
- 24.Proctor A, Herrera-Loeza SG, Wang QZ, Lawrence DS, Yeh JJ, Allbritton NL. Anal Chem. 2014;86:4573–4580. doi: 10.1021/ac500616q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickinson AJ, Hunsucker SA, Armistead PM, Allbritton NL. Anal Bioanal Chem. 2014;406:7027–7036. doi: 10.1007/s00216-014-7974-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Essaka DC, Prendergast J, Keithley RB, Palcic MM, Hindsgaul O, Schnaar RL, Dovichi NJ. Anal Chem. 2012;84:2799–2804. doi: 10.1021/ac2031892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K, Mwongela S, Kottegoda S, Borland L, Nelson A, Sims C, Allbritton N. Anal Chem. 2008;80:1620–1627. doi: 10.1021/ac702305q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickinson A, Armistead P, Allbritton N. Anal Chem. 2013;85:4797–4804. doi: 10.1021/ac4005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horvath KM, Herbst M, Zhou H, Zhang H, Noah TL, Jaspers I. Respir Res. 2011;12:102. doi: 10.1186/1465-9921-12-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller L, Brighton LE, Jaspers I. Am J Physiol : Lung Cell Mol Physiol. 2013;304:L332–L341. doi: 10.1152/ajplung.00256.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller L, Chehrazi CV, Henderson MW, Noah TL, Jaspers I. Part Fibre Toxicol. 2013;10:16. doi: 10.1186/1743-8977-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pai JH, Wang Y, Salazar GT, Sims CE, Bachman M, Li GP, Allbritton NL. Anal Chem. 2007;79:8774–8780. doi: 10.1021/ac071528q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glantz SA. Primer of Biostatistics. McGraw-Hill Companies, Inc.; New York, NY: 2012. [Google Scholar]
- 34.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 35.Wang E, Norred WP, Bacon CW, Riley RT, Merrill AH. J Biol Chem. 1991;266:14486–14490. [PubMed] [Google Scholar]
- 36.Quinto-Su P, Lai H, Yoon H, Sims C, Allbritton N, Venugopalan V. Lab Chip. 2008;8:408–414. doi: 10.1039/b715708h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang D, Sims C, Allbritton N. Electrophoresis. 2010;31:2558–2565. doi: 10.1002/elps.201000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovarik ML, Shah PK, Armistead PM, Allbritton NL. Anal Chem. 2013;85:4991–4997. doi: 10.1021/ac4002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schefler WC. Statistics for the Biological Sciences. Addison-Wesley Publishing Co.; Reading, MA: 1979. [Google Scholar]
- 40.Johnson SC. Psychometrika. 1967;32:241–254. doi: 10.1007/BF02289588. [DOI] [PubMed] [Google Scholar]
- 41.Nakahara K, Ohkuni A, Kitamura T, Abe K, Naganuma T, Ohno Y, Zoeller RA, Kihara A. Mol Cell. 2012;46:461–471. doi: 10.1016/j.molcel.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 42.Ogata K, An E, Shioi Y, Nakamura K, Luo S, Yokose N, Minami S, Dan K. Clin Exp Immunol. 2001;124:392–397. doi: 10.1046/j.1365-2249.2001.01571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Shea D, Cawood TJ, O’Farrelly C, Lynch L. PLoS One. 2010;5:8. doi: 10.1371/journal.pone.0008660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golden-Mason L, Stone AE, Bambha KM, Cheng LL, Rosen HR. Hepatology. 2012;56:1214–1222. doi: 10.1002/hep.25771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.