

Introduction
If you don't know where you're going, you might wind up someplace else—Yogi Berra
My professional journey, once I accepted the fact that I would not play first base for the Chicago Cubs, was not accomplished in a straight line. I entered medical school intending to become a family physician but soon became interested in research. I took an internal medicine residency at the University of California, San Francisco (UCSF), then a long research fellowship in the nascent field of molecular biology at the National Institutes of Health (NIH), and followed that with training in clinical endocrinology at the University of Wisconsin. I spent the next fifteen years dividing my time between the clinic and laboratory at the University of Iowa. Finally, I decided to concentrate on research and spent the next twenty-three years at Vanderbilt University. For the past eleven years, I have been at the University of Iowa, where I helped establish a new diabetes research center. My research journey also had several different directions. Upon reflection, I guess I ended up “someplace else” by choice, and it certainly has been an interesting trip. It is one I unreservedly recommend.
Formal Education, Initial Research Experiences, and My First Mentor
About half of the sixteen students in my high school class in a small Iowa town were destined to go to college. A rather amazing collection of teachers recognized this and gave us a de facto college preparatory education. I often regret that I did not ask the school superintendent whether this was accidental or deliberate. I certainly had plenty of opportunities to inquire about this, as he was my father. I suspect it was not accidental. My mother was also a schoolteacher, so spending a lifetime in various educational environments was probably predetermined.
Enrolled as an undergraduate student at the University of Iowa, I was a bit intimidated by the prospect of competing with students from schools with student bodies of one thousand or more. However, when I found that my freshman inorganic chemistry book was the same one used in our high school, I thought that college might not be so difficult after all. I relaxed a bit more when we got our first “unknown” inorganic compound to identify. Several years earlier, I had received a Gilbert chemistry set for Christmas, which was recently listed as one of the most dangerous “toys” ever sold. This was no surprise, as my favorite creations (first experiments?) were fireworks. I learned to identify several metals by the color of the flames they produced when heated, and so I found the “unknowns” to be old friends. The undergraduate years were fun, especially when a letter of admission to medical school at the University of Iowa arrived in 1957. My intention was to become a primary care physician.
Medical school was generally challenging, but Nicholas (Nick) S. Halmi was easily the most demanding and rigorous teacher in the first year. He nominally taught histology, but this was really a remedial course for all he thought that had been omitted or taught erroneously during the year, particularly in his area of special interest, endocrinology. Two classmates and I learned that he recruited two students from each class to help him do research, which he conducted only during the three months of summer “vacation.” I was selected to ask whether he would consider taking all three of us. To our delight, he agreed, and this led to a life-changing experience.
Nick studied thyroid iodide concentration and had perfected a technique of studying the transport of radioactive iodide from plasma across the thyroid membrane. He designed experiments that never seemed to fail. He initiated our education in the elements of experimental design, conduct, and interpretation. When he decided the story was complete, he wrote a draft in longhand in an evening, his secretary typed it, and he sent it to a journal, all in 24 h. In a few days, a note would come back saying the article was accepted for publication. I thought this was how it always worked.
Toward the end of that summer, Nick approached me with an unexpected opportunity. The United States Public Health Service had announced a two-year program that allowed second-year medical students to split half-time academic studies with half-time research/teaching in a discipline that led to a master's degree. (There were no formal M.D./Ph.D. programs at that time.) I was really intrigued by the summer research experience and had just gotten married, so I readily accepted this offer. I spent the next two years taking medical and graduate school classes, serving as a teaching assistant in the gross anatomy and histology laboratories, and doing research. By the time I went back to being a full-time medical student, I was seriously considering a career that involved research. Then I was offered a second surprising opportunity.
Nick decided to take a one-year sabbatical and suggested that I (a) teach the endocrinology part of his histology/endocrinology course to first-year medical students, (b) keep the laboratory functioning and productive, and (c) not overspend his grant. Lecturing is never easy, but it never has been as difficult as it was facing 120 medical students for the first time. The laboratory worked well. With the assistance of several classmates who served as able technicians and with occasional directions from Nick in the form of handwritten letters (no phone calls, E-mails, or computers), we completed three projects concerning how intracellular iodide influences the transport process (1–3). All of this was accomplished while I was a full-time third-year medical student, and it had the blessing of the department chairman. (As far as I know, the dean knew nothing about this!) This was my first experience with real mentoring. I was entrusted with a variety of responsibilities, given a great opportunity to achieve, and certainly learned a bit about multitasking.
After completing medical school, I was accepted for medical residency at UCSF, and I was there from July 1962 to July 1964. During the spring of 1964, one of my attending physicians, Mort Myers, an Iowa graduate and an internist in Berkeley, brought Gordon Tomkins (then at the NIH) to ward rounds one morning. This was my first exposure to Gordon's incredible intellect. Later that day, he gave a resident's conference during which he discussed the newly emerging field of molecular biology, with emphasis on the regulation of the Escherichia coli lac operon. I mentioned to him afterward that I was going to be joining the NIH as a research associate. He invited me to stop by his laboratory after I arrived.
The NIH Years and My Second Mentor
I arrived at the NIH in the summer of 1964, and a few months later, I visited Gordon, who was chief of the Laboratory of Molecular Biology. I mentioned being unhappy in my assigned laboratory and asked him for advice. He unexpectedly said he had space (a trait I later found was recurring), much to the surprise of Beverly Peterkofsky and Shin-ichi Hayashi, who became my lab mates. Both were exceptionally kind, generous, and patient as they initiated my training in biochemistry and molecular biology.
Gordon had visited Paris in the early 1960s and was completely familiar with the evolving story of the structure and regulation of the E. coli lac operon. He was convinced that similar work could be done using cultured mammalian cells. Brad Thompson, a fellow United States Public Health Service research associate, eagerly accepted this challenge. He began by using the ascites form of a rat hepatoma (7288c) to establish the hepatoma tissue culture (HTC) cell line. Brad then found that the addition of dexamethasone, a synthetic glucocorticoid, to HTC cells resulted in a 10-fold induction of tyrosine transaminase activity. The effect occurred after a lag period of about two hours and required ongoing protein and RNA synthesis (4). This landmark observation was the formal beginning of studies of gene regulation by hormones in cultured mammalian cells.
I arrived at the Tomkins laboratory just as Brad was completing the studies described above. Concomitantly, Robert Schimke and colleagues, then at the NIH, were establishing the conceptual framework for mechanistic studies of enzyme induction, a topic discussed in detail in a recent review (41). They postulated that an increased amount of an enzyme could result from an increased rate of synthesis, a decreased rate of degradation, or some combination of both (principles I was about to employ and would revisit years later when studying mRNA regulation). These investigators presented experimental evidence of both mechanisms in studies of the regulation of tryptophan oxygenase in rat liver (5). Shin-ichi, my new laboratory mate, was purifying tyrosine transaminase to make the specific antibody required for studies of its regulation in HTC cells. Shin-ichi knew he had to leave in a few months to accept a position in Japan, so we worked very hard to purify the enzyme, establish its kinetic properties, and prepare a highly specific antibody directed against the enzyme, which we renamed tyrosine aminotransferase (TAT) (6).
My first task upon developing the anti-TAT antibody was to validate its specificity with the intention of directly testing many possible features of enzyme induction that could not be addressed in animals. This was new experimental territory, so I performed tests that showed that the purified enzyme and the induced and basal enzymes from rat liver and HTC cells had identical antigenicity; thus, the antibody could be reliably used to study induction of the enzyme in the cultured cells (7). Antigenic and catalytic activities of TAT change in parallel in a variety of experimental conditions, so induction is not the result of the conversion of a cross-reacting material to an active enzyme. A double-isotope experiment showed that the increase in enzyme amount results from an enhanced rate of synthesis, not an inhibition of degradation (7). A subsequent study showed that a lag period of ∼60 min occurs before the rate of synthesis begins to increase, and this reaches its maximal level in 4–6 h (8). The induced steady-state synthesis of TAT is maintained as long as dexamethasone remains in the culture medium, but removal of the inducer results in a rapid decline to the basal level (8).
The enthusiasm that the members of the Tomkins laboratory had for this new way of studying mammalian gene regulation was not always met with excitement by those who had been using rats for this purpose. Some investigators refused to believe that anything useful could come from analyzing regulatory processes in tumor cells. Others thought that tissue culture was useful only for determining the minimal nutritional requirements for growth of a given cell line. Finally, some conceded the above points but felt the environment in culture bore little resemblance to that in an intact animal. We therefore had to go to great lengths to address as many of these concerns as possible, all while exploiting and defining the many advantages of tissue culture. The studies described above and subsequent studies by a number of the Tomkins lab members illustrated unique advantages of cell culture systems, including the ability to perform addition-removal-addition sequences to test for an essential mRNA step in the induction process (9); use synchronized cells to show that HTC cells are refractory to dexamethasone-mediated induction during certain phases of the cell cycle (10); precisely manipulate the hormonal environment (11); and select for variants, which was an early foray into mammalian cell genetics (12).
I was at the NIH from 1964 to 1968. The NIH in the 1960s was a very special place. The clinical and research associates were a bright, energetic, and happy-to-be-there group who scattered to leadership positions in academic medicine all around the country. The senior scientists in the Laboratory of Molecular Biology were an amazingly talented group, as were the postdoctoral fellows they attracted. Best of all was the experience of being around Gordon for four years. He had an infectious enthusiasm for science, a wonderful sense of humor, incredible imagination, and a legendary memory. He presented me with a wonderful opportunity, which included two extra years in his laboratory, and was a terrific mentor.
Early Mechanistic Studies of Enzyme Induction by Hormones
I planned to study hormonal regulation of gene expression when I established my own laboratory at the University of Iowa in 1970. Unfortunately, the techniques required to extend such studies beyond the level of protein synthesis were not available until the mid-1970s. In the meantime, I engaged in an interesting and fruitful collaboration with Roger Chalkley, also at the University of Iowa. Roger was studying histone phosphorylation in rapidly dividing cells using regenerating rat liver as a model system. I suggested the use of synchronized HTC cells as a means of studying this process in precisely defined segments of the cell replication cycle (13). We also studied the role of histone acetylation in the regulation of TAT gene expression a few years before this became a topic of broad interest (14).
When suitable techniques became available, hormonal regulation of abundant mRNAs, not surprisingly, was studied first. Ringold et al. (15) at UCSF used a very sensitive hybridization technique to demonstrate that dexamethasone, in a mechanism involving the newly discovered glucocorticoid receptor (GR), induces mammary tumor virus RNA. Chan et al. (16) at Baylor College of Medicine showed that estrogen and progesterone induce the mRNAs that encode avidin and ovalbumin, respectively, in chick oviduct. Subsequently, three groups, including ours, used different in vitro translation systems to demonstrate that glucocorticoid hormones cause proportionate increases in mRNATAT activity and enzyme-specific activity in rat liver (17–19), as does dibutyryl cAMP (Bt2cAMP) (20). (This compound was used because, unlike cAMP, it rapidly penetrates cells.) Marty Diesterhaft1 and Tamio Noguchi, who started these studies in my laboratory, showed that the kinetics and magnitude of the responses to the two inducers are quite different (21), which suggested that different mechanisms are involved, a topic that we and others studied in detail, as discussed below.
As our ability to isolate poly(A)+ RNA improved, we were able to do similar experiments using HTC cells. Dexamethasone increased mRNATAT activity from 0.04 to 0.4% of total poly(A)+ activity in the cells, an increase that matched the increase in catalytic activity (22). Proportionate changes in catalytic activity, the rate of synthesis of the protein, and mRNATAT activity were noted when various concentrations of dexamethasone were added to the culture medium. In all cases, this requires ongoing RNA synthesis (22). At this point, the hypothesis was that an increased amount of mRNA was responsible for these effects. However, this could be due to an enhanced rate of synthesis (transcription) or a decreased rate of degradation of the mRNA. The answer to this question required the cloning of a specific cDNATAT that could then be used as a hybridization probe to quantitate the amount of mRNATAT and isolate the gene.
If you come to a fork in the road, take it—Yogi Berra
Yogi Berra's quote does not suggest how many options there are for the road best taken. Indeed, in the late 1970s, I had several choices. Continuing on the route to the cloning of a cDNATAT was the obvious first choice. A second, rather unexpected option was to move in the heavily traveled direction of immunoglobulin gene regulation. A final intriguing possibility involved the exploration of whether insulin regulates specific gene expression. With the encouragement of a group of enthusiastic colleagues, I chose to move in all three directions at the same time.
Several Research Opportunities Are Explored
Path 1: TAT Gene Regulation
Jim Hargrove became interested in the synthesis, degradation, and post-translational modification of TAT and performed a number of studies on these topics (23) while another group in the lab attempted to isolate a cDNA specific for mRNATAT. Unfortunately, that effort was not successful. This is when I realized that selecting a good research project is of prime importance and knowing when to let go is equally important. In the words of Kenny Rogers, “You've got to know when to hold ‘em, know when to fold ‘em.” Because other projects were advancing, as described below, I regretfully said good-bye to TAT.
Path 2: Immunoglobulin Gene Regulation
Tris Parslow, an M.D./Ph.D. student, joined my lab to explore gene regulation but, seeing the challenges of working with low-abundance mRNAs, chose instead to work on immunoglobulin genes in B lymphocytes. Many groups were studying the structure and rearrangements of those genes, but few were examining how their expression is controlled. The results of his choice and hard work were remarkable. After cloning a cDNA for the κ light chain constant region, Tris showed that lipopolysaccharide treatment of the pre-B cell line 70Z/3 rapidly induced κ mRNA, accompanied by the appearance of a DNase-hypersensitive site within an intron of the gene that signified a change in chromatin structure (24, 25). This was surprising because it was assumed at the time that intron sequences were functionless and that gene regulation and inducible hypersensitive sites occurred only at promoters. In sequencing the κ promoter, Tris also discovered a sequence motif (ATGCAAAT), now called the octamer, situated 70 bp upstream from the transcription start site in all immunoglobulin light chain promoters and, in the opposite orientation (ATTTGCAT), in heavy chain promoters as well (26). These findings sparked tremendous interest and turned out to be vital clues to the mechanisms of B cell-specific gene regulation. Other labs soon showed that the site in the intron was a new type of eukaryotic regulatory sequence called an enhancer. It became hypersensitive when bound by a novel protein complex named the nuclear factor for kappa in B cells (or NF-κB), and the octamer is the binding site for a family of proteins called the Oct proteins, whose members include key regulators of B cell and stem cell genes. I chose not to continue along this path, but it was exciting to watch the stories unfold from our “hit-and-run” foray into immunology!
Path 3: Regulation of Gene Transcription by Insulin
By the late 1970s, the general features of steroid and peptide hormone action on specific gene expression were being elucidated; it was rapidly becoming a very crowded field. The one exception was insulin. The cell-surface insulin receptor had been described, but virtually nothing was known about its intracellular signal transduction pathway. Insulin modulates the extent of phosphorylation of a number of important cytoplasmic enzymes and thus influences several important metabolic pathways (27). But would insulin, which plays such an important role in glucose, protein, and lipid metabolism, do all this without an effect on specific genes? I doubted it, so the question was, “which gene to study?”
The rate of gluconeogenesis, a critical metabolic process, is increased by glucagon (cAMP) and glucocorticoids when plasma glucose levels decline. It is normally decreased by insulin when the plasma glucose exceeds the normal limit. The activity of cytosolic phosphoenolpyruvate carboxykinase (PEPCK),2 a key gluconeogenic enzyme, is regulated by the aforementioned hormones in accordance with their effects on gluconeogenesis in rat liver (28) and, most importantly for our intended purpose, in H4IIE cells (29, 30). Thus, the regulation of the PEPCK gene became the objective of our trek along path 3. We purified PEPCK, used it to produce a specific antibody, and began to study the effect of insulin and other hormones on PEPCK gene expression.
PEPCK and Its Regulation by Insulin as a Major Interest
Insulin Regulates the Amount of mRNAPEPCK
Patrick Iynedjian and Richard Hanson, then at Temple University, had shown that Bt2cAMP increases mRNAPEPCK activity in rat liver (31). Elmus Beale and others in my laboratory confirmed this observation by showing that this agent causes a 20-fold increase in mRNAPEPCK through a process that requires RNA synthesis (32). They also showed that the inducer does not affect the half-life of the mRNA and concluded that it must increase activity by promoting RNA synthesis or processing. Terry Andreone performed a set of experiments, using H4IIE cells treated with various combinations of Bt2cAMP and insulin, that helped set the stage for our future research. The cyclic nucleotide causes proportionate changes in the rate of synthesis of PEPCK and mRNAPEPCK activity (33). Physiologic concentrations of insulin prevent this induction through a receptor-mediated process and also inhibit basal mRNAPEPCK activity, an observation that provides evidence of the dominant role insulin plays in the regulation of this important enzyme. Elmus and Jim Hartley cloned cDNAPEPCK, proved its authenticity, and used it to show that the hormonally modulated changes in mRNAPEPCK activity are due to altered amounts of the mRNA (34). We later found an excellent correlation between the amount of mRNAPEPCK and glucose production from the gluconeogenic precursors lactate and pyruvate in H4IIE cells (35). Our next objective was to study how the multihormonal regulation of mRNAPEPCK is accomplished. This required isolation and characterization of the gene, which was not a trivial task at the time.
Isolation and Characterization of the Rat CytosolicPEPCK Gene (Pck1)
It was a landmark event for us when Elmus and colleagues successfully elucidated the structure of the PEPCK protein, mRNA, and gene. The 621-amino acid (69,289 Da) protein sequence was determined by mass spectrometry. The 2624-nucleotide mRNA has an open reading frame that corresponds exactly to the amino acid sequence. The 6-kb gene consists of 10 exons and 9 introns. The transcription start site was identified (important for numbering upstream regulatory elements), and a substantial portion of the 5′ upstream sequence was determined (36). A few differences were noted between this analysis and that reported by Richard Hanson and colleagues at Case Western Reserve University, as described in his Reflections article (37). We later sequenced the human PCK1 gene from position −1300 of the promoter through the first two exons. This region is similar in sequence and location of the regulatory elements (see below) to the complementary region of the rat gene (38).
Insulin Inhibits the Transcription of Pck1
The prevailing view held that insulin influenced gene expression indirectly through its activation of phosphodiesterase, which simply reduced the intracellular concentration of the inducer, cAMP. Kazuyuki Sasaki used 8-(4-chlorophenyl)thio-cAMP (8-CPT-cAMP), an analog that is not metabolized, to help dispel that notion. Using cDNAPEPCK and a transcription elongation assay, he showed that 8-CPT-cAMP causes a 10-fold increase in the de novo rate of synthesis of mRNAPEPCK in H4IIE cells. Dexamethasone causes a similarly rapid 6-fold increase. The effects of these two agents are additive. The addition of insulin rapidly inhibits the action of the individual inducers, and of the combination, without affecting total RNA synthesis. In addition, the insulin effect is dominant (39). In a subsequent study, probes directed against various regions of Pck1 were used to show that remarkably fewer nascent transcripts are located near the transcription initiation site in insulin-treated cells than in cells exposed to 8-CPT-cAMP (40). However, the reduced number of transcripts are distributed evenly across the gene; thus, insulin apparently does not also cause attenuation or premature release of the transcripts. Interestingly, insulin does reduce the rate of elongation from ∼2500 nucleotides/min estimated in 8-CPT-cAMP-treated cells to ∼1000/min (40). These studies showed that insulin primarily affects specific gene transcription, at transcription initiation in this case. The key question now became, “How does this happen?” But first, there was a necessary digression.
Hormone Response Elements Identified in the Pleiotropic Pck1 Promoter
Chimeric Reporter Genes Replicate the Hormonal Regulation of Pck1
By the mid-1980s, the general concept of steroid/thyroid hormone action had been established (41). Members of this class of hormones bind to a receptor that interacts with a specific segment of DNA, the hormone response element, which is usually located in the DNA sequence 5′ upstream from the transcription initiation site, and triggers the assembly of an active transcription complex. A number of glucocorticoid-responsive genes were studied, and a consensus sequence for the glucocorticoid response element (GRE) was established. The insertion of a consensus GRE into a reporter vector with a heterologous promoter was sufficient to confer a glucocorticoid response (41). Therefore, we expected to find such a GRE in the Pck1 promoter.
In 1984, I succeeded Rollo Park as the chair of the Department of Physiology (now Molecular Physiology and Biophysics) at Vanderbilt. I persuaded Mark Magnuson, who as a medical student had worked in my lab at Iowa, to rejoin my group as a postdoctoral fellow. Mark used the known sequence and restriction enzyme sites in the Pck1 promoter to produce a family of 3′, 5′, and internal deletions that span a large region upstream from the transcription initiation site (42). These were individually inserted into a plasmid vector that contained the chloramphenicol acetyltransferase (CAT) reporter gene, later switched to a luciferase reporter. H4IIE cells were first transfected with pPL9CAT, which contains the proximal 600 bp of the wild-type Pck1 promoter. Dexamethasone and cAMP both stimulated CAT expression of the chimeric PEPCK-CAT fusion gene, and their effects were additive. Insulin inhibited the individual or combined effects of these inducers, as it does with the endogenous gene in H4IIE cells. We concluded that this short segment of DNA contains the cis-acting elements responsible for the multihormonal regulation of Pck1 transcription (42).
Little attention had been given to inhibitory responses of hormones, so hormone response elements that mediate such a response had not been described. Although insulin inhibits basal transcription (39), the effect is relatively small. We elected to study the process in cells stimulated by either dexamethasone or 8-CPT-cAMP. To do this, we first had to identify the GRE and/or the cAMP response element (CRE). Short et al. (43) had identified a CRE in the Pck1 promoter; Pat Quinn confirmed this and showed that it overlaps with basal promoter elements (44), which we thought would complicate the search for an insulin response element. Therefore, we decided to digress from the insulin studies, concentrate on identifying the GRE, and use this information to try to figure out how insulin affects this gene.
The GRE Becomes a Glucocorticoid Response Unit (GRU)
We expected to find a consensus GRE that would confer glucocorticoid regulation upon a heterologous reporter (41). What we eventually found was something different, complex, and much more interesting. Enyu Imai did the critical experiment. He showed that the minimal DNA sequence required for a glucocorticoid response is located within −467 bp of the start site (45). The glucocorticoid response is lost when the DNA segment is truncated to position −402. No consensus GRE was found in this excluded region, but the purified GR binds, albeit with one-thirtieth the affinity of a consensus GRE, to two regions (GR1 and GR2) located between positions −395 and −349 (45). GR1 and GR2 resemble the consensus GRE at only 7/12 and 6/12 positions, respectively, which presumably explained the low binding affinity of the sites (consensus ≫ GR1 > GR2) (45, 46). Unlike other mammalian GREs described at the time, GR1 and GR2, singly or in combination, do not confer a glucocorticoid response on a heterologous reporter gene. Enyu showed that two different sequences located between positions −467 and −402, which bound factors present in a rat liver nuclear extract, are necessary for the glucocorticoid response. These regions, originally called accessory factor elements AF1 and AF2 (45), were later renamed gAF1 and gAF2. Neither gAF1 nor gAF2 supports the glucocorticoid response in the absence of GR1 or GR2; deletion of each reduces the dexamethasone response by 50%, as does deletion of either GR1 or GR2 (45). A third element, gAF3, was subsequently identified (47). The deletion of either gAF1/gAF3 (gAF1 > gAF3) or gAF2 reduces the glucocorticoid response by ∼50%; deletion of both abolishes the response.
A series of studies by numerous members of the laboratory revealed that gAF1 binds hepatocyte nuclear factor (HNF)-α and COUP-TF (48), gAF2 binds FoxA2/HNF-3β (49), and gAF3 binds COUP-TF (47). The domains required for assembly of the active transcription complex were defined by J.-C. Wang (50), and several co-regulators that bind to the GR and the gAF proteins were identified. These include CBP (CRE-binding protein-binding protein)/p300 (35, 51), Src-1 (52), and peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α (53). The CRE is required for a complete glucocorticoid response, apparently through the binding of CCAAT/enhancer-binding protein β (C/EBPβ), as demonstrated by Kazuya Yamada (54). This complex assembly is illustrated in Fig. 1 (upper panel).
FIGURE 1.
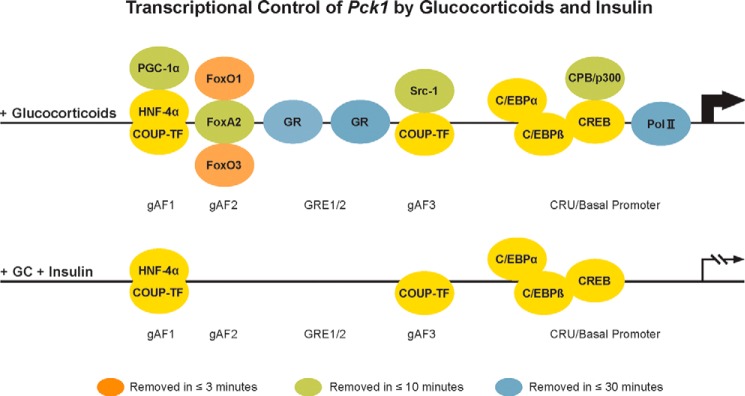
Transcriptional control of Pck1 by glucocorticoids and insulin. The ChIP assay was used to identify the DNA-binding proteins and transcription factors associated with the Pck1 promoter in response to glucocorticoids (upper panel) or glucocorticoids (GC) followed by insulin (lower panel). As described in the text, there are four classes of these proteins based on their rate of removal after insulin addition: rapid (<3 min; orange), intermediate (<10 min; green), delayed (<30 min; blue), and constitutive (yellow). Pol II, polymerase II.
It soon became apparent that there actually are no simple GREs. All require the participation of other DNA-binding proteins for maximal activity (41). Don Scott and Per-Erik Strömstedt had previously shown that the replacement of GR1 with a consensus GRE relieves the necessity of having gAF1 or gAF3 (46). This begged the question as to what these accessory protein-receptor interactions accomplish. John Stafford addressed this question using quantitative, equilibrium, and stopped-flow fluorescence anisotropy, which allowed him to measure cooperative protein-DNA interactions in real time using nuclear extracts and various fragments of the Pck1 promoter DNA. A DNA segment containing just the GR1-GR2 segment binds the GR poorly (30-fold less) compared with one containing the consensus GRE. Inclusion of either gAF1 or gAF2 adjacent to the GR1-GR2 segment restores the high-affinity binding of the GR, equivalent to that afforded by the consensus GRE. Although gAF1 and gAF2 function somewhat differently, both increase the affinity and stabilize the binding of the GR to the Pck1 GREs (55). Thus, the accessory factors serve a dual purpose. They enhance and stabilize binding of the GR to the GREs, and in combination with the GR, they help nucleate the formation of the complete transcription complex.
The in Vivo Composition of the GRU
Many of the studies described above were performed using a variety of biochemical techniques in cell-free systems, so it was reasonable to ask whether these results translate to the living H4IIE cell. The ChIP assay was used to answer this question. Certain of the gAF proteins, including HNF-4α, CREB (cAMP response element-binding protein), C/EBPα, C/EBPβ, and COUP-TF, are constitutively present on the Pck1 promoter (35, 56). Dexamethasone causes a 10-fold increase in GR occupancy, which promotes nucleation of a large complex that consists of, at least, polymerase II, Src-1, CBP, p300, FoxO1, FoxO3, and PGC-1α (35, 56). Thus, the results from the cell-free and intact cell studies are concordant. In an unpublished study, Rob Hall showed that the same process occurs on rat liver Pck1. This assembly of DNA elements and associated proteins, and the numerous co-regulators they bind, composes a large functional transcription complex that we termed the GRU (Fig. 1) (45).
Pck1 Is Also Regulated by Retinoic Acid
Peter Lucas, Don Scott, and Takashi Sugiyama performed a series of studies to show that gAF1 and gAF3 also bind heterodimers of the retinoic acid receptor (RAR) and retinoid X receptor (RXR). In the absence of dexamethasone, but in the presence of retinoic acid, gAF1 and gAF3 bound to RAR and RXR activate Pck1 transcription (57–59). Thus, in one context, gAF1 and gAF3 are GRU components, and in another context, a retinoic acid response unit (RARU). This makes gAF1 and gAF3 part of an integrated pleiotropic metabolic control domain consisting of the GRU, RARU, and cAMP response unit (CRU) (60, 61). As we were nearing completion of this investigation, an interesting paper was brought to our attention. In 1957, George Wolf and Connor Johnson at the University of Illinois and Daniel Lane (then at the University of Illinois) showed that in vitamin A-deficient rats, the citric acid cycle, glycolysis, glycogen metabolism, hepatic cholesterol, and fatty acid synthesis all function normally. However, gluconeogenesis was markedly impaired (62). Perhaps our results help explain this earlier observation.
We're lost, but we're making good time—Yogi Berra
Although some thought we were lost while traveling along path 3, this was really just a necessary detour that taught us many interesting things, and we did make good time. But now we were ready to address a major question.
How Does Insulin Inhibit Transcription of Pck1?
We initially posed the question, “Does insulin affect each of the multihormonal processes individually, or does it exert a more global effect on the entire promoter?” It was possible that some combination of both might be operative, although at the time, there was no way to address the latter possibility. Therefore, we took the only approach available, which was to search for a cis-acting element that mediated the effect of insulin. We hoped that this would then lead to the identification of a transcription factor that mediated the effect of insulin, which would be the final step in the signaling pathway from the receptor to Pck1. Because the insulin signaling pathway was unknown at the time, we further hoped that we would be able to work back to establish the steps between this transcription factor and the insulin receptor.
Using a combination of transient transfection and stably transfected H4IIE cells, Richard O'Brien and Claude Forest found that the insulin effect requires two insulin response sequences (IRSs) located between positions −468 and −271 (IRS-1) and positions −271 and +69 (IRS-2) (63, 64). IRS-1 was further localized to region −416 to −402, which corresponds to gAF2 in the GRU (45). The core sequence of IRS-1 (T(G/A)TTTT(G/T)) was later found in the glucose-6-phosphatase and insulin growth factor binding protein-1 genes, whose transcription is also inhibited by insulin (reviewed in Ref. 65). IRS-2 defied precise localization, but was within the general region of the multicomponent CRU and the basal promoter (43, 44). The colocation of these elements with components of other hormone response units suggested that insulin might act by interfering with the binding/function of the accessory protein(s) at IRS-1/gAF2 and/or IRS-2/CRU. However, an analysis of the candidate IRS-1/gAF2- and IRS-2/CRU-binding proteins showed that there was an uncoupling of insulin action and protein binding to these elements (49, 65). These experiments required a careful analysis, through point mutations, of the correlation between protein binding and insulin action, without which one would have mistakenly concluded that these candidate factors mediate the action of insulin (49, 65). Subsequent studies showed that the transcription factors FoxO1 and FoxO3 bind to the IRS-1/gAF2 region of the Pck1 promoter and may be involved in its regulation by glucocorticoids and/or insulin (reviewed in Ref. 65). A number of gene knock-out studies in animals, and overexpression studies in a variety of animal and cultured cell systems generally, although not universally, support this view (65, 66). Despite all these data, it is not clear exactly how the FoxO proteins work on the Pck1 promoter. Moreover, in contrast to the studies of the other response units, we were never able to find a combination of mutations of the promoter that completely abolished the insulin response, so another form of regulation seemed possible, at least as a complementary mechanism. However, from the work of Calum Sutherland, we did establish that the PI3K signal transduction pathway, and not the MAPK pathway, mediates the insulin response on Pck1 transcription (67).
Insulin Rapidly Disrupts the Pck1 Transcription Complex
The development of the ChIP assay allowed Rob Hall to assess what happens to the known accessory proteins and transcription factors on the proximal Pck1 promoter after insulin is added to H4IIE cells. He found that insulin very rapidly disrupts the structure of the transcription complex on Pck1 (56). The extent and rapidity of dissociation of the components differ. FoxO1 and FoxO3 are essentially gone within 3 min. An intermediate group consisting of CBP, FoxA2, p300, and Src-1 are significantly reduced by 3 min and gone by 10–30 min. The GR (binding stabilized by FoxA2) and polymerase II are markedly reduced by 30 min (Fig. 1); the occupancy of these proteins on the promoter correlates best with the rate of transcription of the gene (56). The occupancy of the constitutive DNA-binding accessory factors, including CREB, C/EBPα, C/EBPβ, HNF-4α, and COUP-TF, is unaffected by insulin. A similar disruption of the promoter complex on the glucose-6-phosphatase gene, the transcription of which is also inhibited by insulin, was also observed (56).
Rob assessed numerous chromatin modifications in an attempt to explain this remarkable effect. Rapid demethylation of Arg-17 on histone H3 correlates best with the results described above (56). How or if this epigenetic effect causes the repression of transcription of this key gluconeogenic gene remains to be established. How this signal might translate to and through the separate components of what might be called the insulin response unit is also unknown.
The Pck1 Promoter Is a Finely Tuned Machine
The hormone response domain in the promoter of Pck1 consists of a remarkable assemblage of DNA elements, DNA-binding transcription factors, and co-regulatory proteins. It receives and interprets signals from a number of hormones to provide additive, synergistic, positive, negative, and dominant control for the synthesis of this key gluconeogenic enzyme. This degree of regulation allows the gene to be finely tuned across a broad range of activities and underscores the importance of this enzyme. The complexity and redundancy built into the stimulatory side of this system preclude the possibility that the loss of one DNA element, one transcription factor, or one hormone could abolish the role of this gene in the preferred pathway of gluconeogenesis. The role of insulin in Pck1 is to restrain gluconeogenesis in the face of hyperglycemia, which it accomplishes by overriding the stimulatory loop.
Is the Pck1 Promoter a Nuclear Receptor Test Site?
I am struck by the number of members of the nuclear receptor family that operate through this relatively short segment of DNA. These include the GR, HNF-4α, COUP-TF II, RARα, RXRα, estrogen-related receptor-α, thyroid hormone receptor-α, peroxisome proliferator-activated receptor-α, liver X receptor-β, and farnesoid X receptor-β (41, 68). The 52 nuclear receptor genes are sorted into three major subfamilies and 20 groups. The nuclear receptor gene products that bind to the Pck1 promoter represent all three of the major subfamilies and at least nine of the 20 groups. Among other things, this promoter seems to have been chosen as a beta test site for this important class of transcription factors.
Structure and Function of Genes of the Hexokinase (HK) Family
The antiparallel processes of hepatic gluconeogenesis and glycolysis are highly regulated by a variety of agents, including insulin (69). We decided to isolate the glucokinase (GK) gene so that we could analyze how insulin stimulates the transcription of one gene (GK) while, at the same time and in the same cell, it inhibits the transcription of another (Pck1). We eventually characterized two genes in the mammalian HK family, which consists of three 100-kDa proteins (HK1–3) and a 50-kDa member (HK4), usually referred to as GK. The HKs catalyze the conversion of glucose to glucose 6-phosphate, the first committed step in glycolysis. The process is stimulated by insulin in target tissues such as skeletal muscle and adipose tissue, where HK2 is abundant, and in liver and pancreatic beta cells, where GK is abundant. Each protein is the product of a different gene. The 100-kDa forms are thought to have evolved from an ancestral 50-kDa form (the two yeast HKs are also 50-kDa proteins).
GK
Terry Andreone and colleagues purified GK to homogeneity and used oligonucleotide probes based on selected peptide sequences to screen a rat liver cDNA library (70). The cDNAs obtained were used to show that insulin causes a 30-fold induction of hepatic mRNAGK, the expected regulation, and to deduce most of the ∼2400-nucleotide mRNAGK sequence. The protein deduced from the latter consists of 465 amino acids with a molecular mass of 51,924 Da. If conservative amino acid replacements are considered, GK is very similar to the C-terminal half of rat brain HK1 (the only mammalian sequence available at the time) and to yeast HK at 75 and 63% of positions, respectively (70).
Mark Magnuson and colleagues screened a number of rat genomic libraries with the cDNA probes and succeeded in isolating the rat hepatic GK gene (Gck) (71). It spans 15.5 kb and has 10 exons (ranging in size from 96 to 977 bp) and nine introns. The transcription initiation site was identified along with 1.5 kb of promoter sequence. Insulin causes a rapid 20-fold increased rate of transcription of Gck while simultaneously inhibiting PEPCK transcription in rat liver (71, 72). Unfortunately, we never found a liver-based cell line that exhibited insulin-regulated endogenous Gck or Gck reporter gene expression, so we could not pursue the studies that started us on this quest. This study did lead to Mark's subsequent description of the closely related pancreatic beta cell Gck (73), which in turn helped define the partial deficiency of GK as a monogenic cause of diabetes (maturity onset diabetes of the young (MODY) type 2) (74).
HK2
The isolation of the HK2 gene (Hk2) was not straightforward. The structure of Hk1 was available; however, the Hk1–3 genes were presumed to be quite similar, and the HK1 protein is expressed in all of the tissues that express HK2. Richard Printz and colleagues devised a strategy that circumvented these obstacles. Reasoning that there might be some sequence similarities in this gene family, they constructed a probe based on a DNA sequence common to Gck and Hk1 and used this to screen a cDNA library made from adipose tissue mRNA that was expected to be enriched for mRNAHK2. They obtained two unique cDNAs that contained sequences not found in any of the GK or HK1 cDNAs. These overlapped and together composed an open reading frame that encodes a 103-kDa protein that, when expressed in E. coli, has the unique kinetic properties of HK2 (75). A number of genomic libraries were screened with cDNAHK2 to obtain fragments of the entire ∼41-kb Hk2 gene, which consists of 18 exons (75). In a memorable moment on a quiet Saturday afternoon, Richard and I compared the sequences of the 10 Gck exons with those of the 18 Hk2 exons. The results were astonishing. A different first exon is used in each gene, but exons 2 and 4–9 of Hk2 are identical in size to the corresponding ones in the Gck gene. Exon 10 in Hk2 is a tandem fusion corresponding to exons 10 and 2 of Gck, and then exons 12–18 of Hk2 match exons 4–10 of Gck. Only exon 3, which is two codons shorter in Hk2, differs in size (75). Hossein Ardehali showed that unlike HK1, both halves of HK2 have catalytic activity (76). Taken together, this evidence strongly suggests that Hk2 evolved from a Gck-like ancestral precursor, presumably by gene duplication with tandem ligation (75). Richard and Hossein also isolated and characterized the human HK2 gene. It has the same structure as the rat gene (18 exons of exactly the same size), and there is 94% identity of the 917 amino acids present in each enzyme (77).
The insulin-induced increase in HK2 activity in adipose tissue and skeletal muscle is the result of an increased rate of transcription of the gene (75). Haruhiko Osawa and Brooks Robey found that catecholamines, 8-CPT-cAMP, and insulin increase Hk2 transcription in L6 skeletal muscles cells, an effect accompanied by a corresponding increase in mRNAHK2, protein, and glucose phosphorylation and utilization. Similar changes were noted in adipose cell lines. They mapped the basal promoter elements and CRE and showed that the insulin effect is mediated through the PI3K and S6 protein kinase signal transduction pathways (78–80). Robbie O'Doherty and David Wasserman made the remarkable observation that a single bout of acute exercise increases rat skeletal muscle HK2 activity and mRNA (81) and transcription of the gene (82). They later continued this work in their own laboratories.
Epilogue
Preparing this article afforded me the opportunity to reflect on how my career in research happened (Fig. 2). The phrase that came to mind was “improbable outcome”; something must have aligned the stars. First, I had the good fortune to have two mentors who provided excellent training experiences. I deeply appreciate Nick Halmi and Gordon Tomkins for giving me the chance to explore my potential for biomedical research. Second, I found the physical and intellectual environments, first at Iowa and then at Vanderbilt, conducive for the research I wanted to pursue. Third, I learned from advice and experience to be prepared to change direction; to embrace new technology; and to expect, recognize, and exploit the unexpected. Fourth, I chose projects that had significant growth potential, although I did not realize the full extent of it at the time. Decades later, these projects still afford many interesting research opportunities.
FIGURE 2.

The author.
For example, early studies on the regulation of gene expression by glucocorticoids focused on two hepatic enzymes, TAT and tryptophan oxygenase. It is now apparent that >1000 genes in many different tissues are regulated by this class of hormones (41). Like Pck1, each of these genes probably has a complex constellation of transcription factors and DNA elements. Whether this complexity can be deciphered and translated into improved and safer therapy by these potent therapeutic agents remains to be seen. Likewise, the hormonal control of hepatic glucose production, or the lack thereof, remains a critical element in understanding the pathophysiology of type 2 diabetes, a scourge of our time. Because resistance to the action of insulin is a central feature of this genetic disease, it seems a reasonable possibility that the inability of insulin to properly regulate one or more of the genes involved in hepatic glucose production may contribute to this process. To date, to my regret, the analysis of some obvious candidate genes has failed to reveal useful information. Newer techniques, particularly if they can be used to detect subtle multiplicative changes in several genes along a metabolic pathway, might prove to be more informative.
Finally, the many postdoctoral fellows and students associated with me over the years formed the foundation of the intellectual environment alluded to above. I could not mention all of them in this paper or describe all their work, but I deeply appreciate the opportunity I had to participate in their early years of scientific exploration. I found to my delight that a career in science is constantly interesting, challenging, and rewarding. This, coupled with the opportunity to interact with similarly engaged colleagues from around the world, made it a fabulous trip. It would be great to be able to start over, but I trust future scientific generations to continue the quest with enthusiasm!
Acknowledgments
I thank Chris Adams, Richard O'Brien, Tris Parslow, and Mike Welsh for helpful comments and encouragement during the preparation of this manuscript and Stacie Vik for expert assistance. My family was more than patient with me over the years, especially when a major grant was due.
Footnotes
Unless indicated otherwise, the people named in this paper performed the research described while they were colleagues in my laboratory.
PEPCK exists in two forms, cytosolic and mitochondrial, which are encoded by different genes. My studies dealt with the cytosolic form, so PEPCK in this paper represents that protein.
References
- 1. Granner D. K., Curtis S. J., Scranton J. R., Halmi N. S. (1962) Differences in thyroid function between triiodothyronine-treated and hypophysectomized rats: blocked glands. Endocrinology 71, 816–821 [DOI] [PubMed] [Google Scholar]
- 2. Granner D. K., Curtis S. J., Scranton J. R., Halmi N. S. (1963) Differences in thyroid function between triiodothyronine-treated and hypophysectomized rats: binding glands. Endocrinology 72, 100–105 [DOI] [PubMed] [Google Scholar]
- 3. Granner D. K., Scranton J. R., Curtis S. J. (1963) Kinetic analysis of thyroidal iodine concentration in hypophysectomized rats fed high or low iodine diets. Endocrinology 72, 503–504 [DOI] [PubMed] [Google Scholar]
- 4. Thompson E. B., Tomkins G. M., Curran J. F. (1966) Induction of tyrosine α-ketoglutarate transaminase by steroid hormones in a newly established tissue culture cell line. Proc. Natl. Acad. Sci. U.S.A. 56, 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schimke R. T., Sweeney E. W., Berlin C. M. (1965) The roles of synthesis and degradation in the control of rat liver tryptophan pyrrolase. J. Biol. Chem. 240, 322–331 [PubMed] [Google Scholar]
- 6. Hayashi S., Granner D. K., Tomkins G. M. (1967) Tyrosine aminotransferase: purification and characterization. J. Biol. Chem. 242, 3998–4006 [PubMed] [Google Scholar]
- 7. Granner D. K., Hayashi S., Thompson E. B., Tomkins G. M. (1968) Stimulation of tyrosine aminotransferase synthesis by dexamethasone phosphate in cell culture. J. Mol. Biol. 35, 291–301 [DOI] [PubMed] [Google Scholar]
- 8. Granner D. K., Thompson E. B., Tomkins G. M. (1970) Dexamethasone phosphate induced synthesis of tyrosine aminotransferase in hepatoma tissue culture cells. Studies of the early phases of induction and of the steroid requirement for maintenance of the induced rate of synthesis. J. Biol. Chem. 245, 1472–1478 [PubMed] [Google Scholar]
- 9. Peterkofsky B., Tomkins G. M. (1968) Evidence for the steroid-induced accumulation of tyrosine-aminotransferase messenger RNA in the absence of protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 60, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin D. Jr., Tomkins G. M., Granner D. K. (1969) Synthesis and induction of tyrosine aminotransferase in synchronized hepatoma cells in culture. Proc. Natl. Acad. Sci. U.S.A. 62, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Samuels H. H., Tomkins G. M. (1970) Relation of steroid structure to enzyme induction in hepatoma tissue culture cells. J. Mol. Biol. 52, 57–74 [DOI] [PubMed] [Google Scholar]
- 12. Thompson E. B., Granner D. K., Gelehrter T. D., Simons S. S., Hager G. (1979) Unlinked control of multiple glucocorticoid-sensitive process in spontaneous HTC cell variants. in Hormones and Cell Culture: Sixth Cold Spring Harbor Conference on Cell Proliferation (Sato G. H., Ross R., eds) pp. 339–350, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 13. Balhorn R., Jackson V., Granner D. K., Chalkley R. (1975) Phosphorylation of the lysine-rich histone throughout the cell cycle. Biochemistry 14, 2504–2511 [DOI] [PubMed] [Google Scholar]
- 14. Plesko M. M., Hargrove J. L., Granner D. K., Chalkley R. (1983). Inhibition by sodium butyrate of enzyme induction by glucocorticoids and dibutyryl cAMP. A role for the rapid form of histone acetylation. J. Biol. Chem. 258, 13738–13744 [PubMed] [Google Scholar]
- 15. Ringold G. M., Yamamoto K. R., Tomkins G. M., Bishop M., Varmus H. E. (1975) Dexamethasone-mediated induction of mouse mammary tumor virus RNA: a system for studying glucocorticoid action. Cell 6, 299–305 [DOI] [PubMed] [Google Scholar]
- 16. Chan L., Means A. R., O'Malley B. W. (1973) Rates of induction of specific translatable messenger RNAs for ovalbumin and avidin by steroid hormones. Proc. Natl. Acad. Sci. U.S.A. 70, 1870–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nickol J. M., Lee K. L., Hollinger T. G., Kenney F. T. (1976) Translation of messenger RNA specific for tyrosine aminotransferase in oocytes of Xenopus laevis. Biochem. Biophys. Res. Commun. 72, 687–693 [DOI] [PubMed] [Google Scholar]
- 18. Roewekamp W. G., Hofer E., Sekeris C. E. (1976) Translation of mRNA from rat-liver polysomes into tyrosine aminotransferase and tryptophan oxygenase in a protein-synthesizing system from wheat germ. Effects of cortisol on the translatable levels of mRNA for these two enzymes. Eur. J. Biochem. 70, 259–268 [DOI] [PubMed] [Google Scholar]
- 19. Diesterhaft M., Noguchi T., Hargrove J., Thornton C., Granner D. K. (1977) Translation of tyrosine aminotransferase mRNA in a modified reticulocyte system. Biochem. Biophys. Res. Commun. 79, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 20. Noguchi T., Diesterhaft M., Granner D. K. (1978) Dibutyryl cyclic AMP increases the amount of functional messenger RNA coding for tyrosine aminotransferase in rat liver. J. Biol. Chem. 253, 1332–1335 [PubMed] [Google Scholar]
- 21. Diesterhaft M., Noguchi T., Granner D. K. (1980) Regulation of rat liver tyrosine aminotransferase mRNA by hydrocortisone and by dibutyryl cAMP. Eur. J. Biochem. 108, 357–365 [DOI] [PubMed] [Google Scholar]
- 22. Olson P. S., Thompson E. B., Granner D. K. (1980) Regulation of hepatoma tissue culture cell tyrosine aminotransferase mRNA by dexamethasone. Biochemistry 19, 1705–1711 [DOI] [PubMed] [Google Scholar]
- 23. Hargrove J. L., Granner D. K. (1983) Biosynthesis and intracellular processing of tyrosine aminotransferase. in Transaminases (Christian P., Metzler D. E., eds) pp. 511–532, John Wiley & Sons, New York [Google Scholar]
- 24. Parslow T. G., Granner D. K. (1982) Chromatin changes accompany immunoglobulin kappa gene activation: a potential control region inside the gene. Nature 299, 449–451 [DOI] [PubMed] [Google Scholar]
- 25. Parslow T. G., Granner D. K. (1983) Structure of a nuclease-sensitive region inside the immunoglobulin kappa gene: evidence for a role in gene regulation. Nucleic Acids Res. 11, 4775–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parslow T. G., Blair D. L., Murphy W. J., Granner D. K. (1984) Structure of the 5′ ends of immunoglobulin genes: a novel conserved sequence. Proc. Natl. Acad. Sci. U.S.A. 81, 2650–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larner J. (2013) Inositol, glycogen, insulin, and six Nobelists. J. Biol. Chem. 288, 12313–12324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shrago E., Lardy H. A., Nordlie R. C., Foster D. O. (1963) Metabolic and hormonal control of phosphoenolpyruvate carboxykinase and malic enzyme in rat liver. J. Biol. Chem. 238, 3188–3192 [PubMed] [Google Scholar]
- 29. Barnett C. A., Wicks W. D. (1971) Regulation of phosphoenolpyruvate carboxykinase and tyrosine transaminase in hepatoma cell cultures. J. Biol. Chem. 246, 7201–7206 [PubMed] [Google Scholar]
- 30. Gunn J. M., Tilghman S. M., Hanson R. W., Reshef L., Ballard F. J. (1975) Effects of cyclic adenosine monophosphate, dexamethasone and insulin on phosphoenolpyruvate carboxykinase synthesis in Reuber H-35 hepatoma cells. Biochemistry 14, 2350–2357 [DOI] [PubMed] [Google Scholar]
- 31. Iynedjian P. B., Hanson R. W. (1977) Increase in level of functional messenger RNA coding for phosphoenolpyruvate carboxykinase (GTP) during induction by cyclic adenosine 3′:5′-monophosphate. J. Biol. Chem. 252, 655–662 [PubMed] [Google Scholar]
- 32. Beale E. G., Katzen C. S., Granner D. K. (1981) Regulation of rat liver phosphoenolpyruvate carboxykinase (GTP) messenger RNA activity by N6,O2-dibutyryladenosine 3′,5′-phosphate. Biochemistry 20, 4878–4883 [DOI] [PubMed] [Google Scholar]
- 33. Andreone T. L., Beale E. G., Bar R. S., Granner D. K. (1982) Insulin decreases phosphoenolpyruvate carboxykinase (GTP) mRNA activity by a receptor-mediated process. J. Biol. Chem. 257, 35–38 [PubMed] [Google Scholar]
- 34. Beale E. G., Hartley J. L., Granner D. K. (1982) N6,O2-Dibutyryl cyclic AMP and glucose regulate the amount of messenger RNA coding for hepatic phosphoenolpyruvate carboxykinase (GTP). J. Biol. Chem. 257, 2022–2028 [PubMed] [Google Scholar]
- 35. Duong D. T., Waltner-Law M. E., Sears R., Sealy L., Granner D. K. (2002) Insulin inhibits hepatocellular glucose production by disrupting the association of CREB-binding protein and RNA polymerase II with the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 277, 32234–32242 [DOI] [PubMed] [Google Scholar]
- 36. Beale E. G., Chrapkiewicz N. B., Scoble H. A., Metz R. J., Quick D. P., Noble R. L., Donelson J. E., Biemann K., Granner D. K. (1985) Rat hepatic cytosolic phosphoenolpyruvate carboxykinase (GTP). Structures of the protein, messenger RNA, and gene. J. Biol. Chem. 260, 10748–10760 [PubMed] [Google Scholar]
- 37. Hanson R. W. (2005) Metabolism in the era of molecular biology. J. Biol. Chem. 280, 1705–1715 [DOI] [PubMed] [Google Scholar]
- 38. O'Brien R. M., Printz R. L., Halmi N., Tiesinga J. J., Granner D. K. (1995) Structural and functional analysis of the human phosphoenolpyruvate carboxykinase gene promoter. Biochem. Biophys. Acta 1264, 284–288 [DOI] [PubMed] [Google Scholar]
- 39. Sasaki K., Cripe T. P., Koch S. R., Andreone T. L., Petersen D. D., Beale E. G., Granner D. K. (1984) Multihormonal regulation of phosphoenolpyruvate carboxykinase gene transcription. The dominant role of insulin. J. Biol. Chem. 259, 15242–15251 [PubMed] [Google Scholar]
- 40. Sasaki K., Granner D. K. (1988) Regulation of phosphoenolpyruvate carboxykinase gene transcription by insulin and cAMP: reciprocal actions on initiation and elongation. Proc. Natl. Acad. Sci. U.S.A. 85, 2954–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Granner D. K., Wang J.-C., Yamamoto K. R. (2015) Regulatory actions of glucocorticoid hormones: from organisms to mechanisms. in Glucocorticoid Signaling: From Molecules to Mice to Man (Wang J.-C., Harris C., eds) pp. 3–31, Springer-Verlag, New York: [DOI] [PubMed] [Google Scholar]
- 42. Magnuson M. A., Quinn P. G., Granner D. K. (1987) Multihormonal regulation of phosphoenolpyruvate carboxykinase-chloramphenicol acetyltransferase fusion genes. Insulin's effects oppose those of cAMP and dexamethasone. J. Biol. Chem. 262, 14917–14920 [PubMed] [Google Scholar]
- 43. Short J. M., Wynshaw-Boris A., Short H. P., Hanson R. W. (1986) Characterization of the phosphoenolpyruvate carboxykinase (GTP) promoter-regulatory region. II. Identification of cAMP and glucocorticoid regulatory domains. J. Biol. Chem. 261, 9721–9726 [PubMed] [Google Scholar]
- 44. Quinn P. G., Wong T. W., Magnuson M. A., Shabb J. B., Granner D. K. (1988) Identification of basal and cyclic AMP regulatory elements in the promoter of the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 8, 3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imai E., Stromstedt P.-E., Quinn P. G., Carlstedt-Duke J., Gustafsson J.-A., Granner D. K. (1990) Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 10, 4712–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scott D. K., Stromstedt P.-E., Wang J.-C., Granner D. K. (1998) Further characterization of the glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol. Endocrinol. 12, 482–491 [DOI] [PubMed] [Google Scholar]
- 47. Scott D. K., Mitchell J. A., Granner D. K. (1996) The orphan receptor COUP-TF binds to a third glucocorticoid accessory factor element within the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 271, 31909–31914 [DOI] [PubMed] [Google Scholar]
- 48. Hall R. K., Sladek F. M., Granner D. K. (1995) The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of the phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc. Natl. Acad. Sci. U.S.A. 92, 412–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J.-C., Stromstedt P.-E., O'Brien R. M., Granner D. K. (1996) Hepatic nuclear factor 3 is an accessory factor required for the stimulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Mol. Endocrinol. 10, 794–800 [DOI] [PubMed] [Google Scholar]
- 50. Wang J.-C., Stromstedt P.-E., Sugiyama T., Granner D. K. (1999) The phosphoenolpyruvate carboxykinase gene glucocorticoid response unit. identification of the functional domains of accessory factors HNF3β (hepatic nuclear factor-3β) and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol. Endocrinol. 13, 604–618 [DOI] [PubMed] [Google Scholar]
- 51. Wang X. L., Herzog B., Waltner-Law M., Hall R. K., Shiota M., Granner D. K. (2004) The synergistic effect of dexamethasone and all-trans-retinoic acid on hepatic phosphoenolpyruvate carboxykinase gene expression involves the coactivator p300. J. Biol. Chem. 279, 34191–34200 [DOI] [PubMed] [Google Scholar]
- 52. Wang J.-C., Stafford J. M., Granner D. K. (1998) SRC-1 and GRIP1 coactivate transcription with hepatocyte nuclear factor 4. J. Biol. Chem. 273, 30847–30850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 54. Yamada K., Duong D. T., Scott D. K., Wang J.-C., Granner D. K. (1999) CCAAT/enhancer-binding protein β is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 274, 5880–5887 [DOI] [PubMed] [Google Scholar]
- 55. Stafford J. M., Wilkinson J. C., Beechem J. M., Granner D. K. (2001) Accessory factors facilitate the binding of glucocorticoid receptor to the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 276, 39885–39891 [DOI] [PubMed] [Google Scholar]
- 56. Hall R. K., Wang X. L., George L., Koch S. R., Granner D. K. (2007) Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Mol. Endocrinol. 21, 550–563 [DOI] [PubMed] [Google Scholar]
- 57. Lucas P. C., O'Brien R. M., Mitchell J. A., Davis C. M., Imai E., Forman B. M., Samuels H. H., Granner D. K. (1991) A retinoic acid response element is part of a pleiotropic domain in the phosphoenolpyruvate carboxykinase gene. Proc. Natl. Acad. Sci. U.S.A. 88, 2184–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hall R. K., Scott D. K., Noisin E. L., Lucas P. C., Granner D. K. (1992) Activation of the phosphoenolpyruvate carboxykinase retinoic acid response element is dependent on a retinoic acid receptor/coregulator complex. Mol. Cell. Biol. 12, 5527–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scott D. K., Mitchell J. A., Granner D. K. (1996) Identification and characterization of a second retinoic acid response element in the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 271, 6260–6264 [DOI] [PubMed] [Google Scholar]
- 60. Sugiyama T., Scott D. K., Wang J.-C., Granner D. K. (1998) Structural requirements of the glucocorticoid and retinoic acid response units in the phosphoenolpyruvate carboxykinase gene promoter. Mol. Endocrinol. 12, 1487–1498 [DOI] [PubMed] [Google Scholar]
- 61. Lucas P. C., Forman B. M., Samuels H. H., Granner D. K. (1991) Specificity of a retinoic acid response element in the phosphoenolpyruvate carboxykinase gene promoter: consequences of both retinoic acid and thyroid hormone receptor binding. Mol. Cell. Biol. 11, 5164–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wolf G., Lane M. D., Johnson B. C. (1957) Studies on the function of vitamin a in metabolism. J. Biol. Chem. 225, 995–1008 [PubMed] [Google Scholar]
- 63. O'Brien R. M., Lucas P. C., Forest C. D., Magnuson M. A., Granner D. K. (1990) Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science 249, 533–537 [DOI] [PubMed] [Google Scholar]
- 64. Forest C. D., O'Brien R. M., Lucas P. C., Magnuson M. A., Granner D. K. (1990) Regulation of phosphoenolpyruvate carboxykinase gene expression by insulin: use of the stable transfection approach to locate an insulin responsive sequence (IRS). Mol. Endocrinol. 4, 1302–1310 [DOI] [PubMed] [Google Scholar]
- 65. Onuma H., Vander Kooi B. T., Boustead J. N., Oeser J. K., O'Brien R. M. (2006) Correlation between FOXO1a (FKHR) and FOXO3a (FKHRL1) binding and the inhibition of basal glucose-6-phosphatase catalytic subunit gene transcription by insulin. Mol. Endocrinol. 20, 2831–2847 [DOI] [PubMed] [Google Scholar]
- 66. Matsumoto M., Pocai A., Rossetti L., DePinho R. A., Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 67. Sutherland C., O'Brien R. M., Granner D. K. (1995) Phosphatidylinositol 3-kinase, but not p70/p85 ribosomal S6 protein kinase, is required for the regulation of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by insulin. J. Biol. Chem. 270, 15501–15506 [DOI] [PubMed] [Google Scholar]
- 68. Kuo T., McQueen A., Chen T.-C., Wang J.-C. (2015) Regulation of glucose homeostasis by glucocorticoids. in Glucocorticoid Signaling: From Molecules to Mice to Man (Wang J.-C., Harris C., eds) pp. 99–126, Springer-Verlag, New York: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Granner D. K., Pilkis S. (1990) The genes of hepatic glucose metabolism. J. Biol. Chem. 265, 10173–10176 [PubMed] [Google Scholar]
- 70. Andreone T. L., Printz R. L., Pilkis S. J., Magnuson M. A., Granner D. K. (1989) The amino acid sequence of rat liver glucokinase, deduced from cloned cDNA. J. Biol. Chem. 264, 363–369 [PubMed] [Google Scholar]
- 71. Magnuson M. A., Andreone T. L., Printz R. L., Koch S., Granner D. K. (1989) The rat glucokinase gene: structure and regulation by insulin. Proc. Natl. Acad. Sci. U.S.A. 86, 4838–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Iynedjian P. B., Gjinovci A., Renold A. E. (1988) Stimulation by insulin of glucokinase gene transcription in liver of diabetic rats. J. Biol. Chem. 263, 740–744 [PubMed] [Google Scholar]
- 73. Magnuson M. A. (1990) Glucokinase gene structure: functional implications of molecular genetic studies. Diabetes 39, 523–527 [DOI] [PubMed] [Google Scholar]
- 74. Froguel P., Vaxillaire M., Sun F., Velho G., Zouali H., Butel M. O., Lesage S., Vionnet N., Clément K., Fougerousse F., Tanizawa Y., Weissenbach J., Beckmann J. S., Lathrop G. M., Passa P., Permutt M. A., Cohen D. (1992) Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356, 162–164 [DOI] [PubMed] [Google Scholar]
- 75. Printz R. L., Koch S., Potter L. R., O'Doherty R. M., Tiesinga J. J., Moritz S., Granner D. K. (1993) Hexokinase II mRNA and gene structure, regulation by insulin and evolution. J. Biol. Chem. 268, 5209–5219 [PubMed] [Google Scholar]
- 76. Ardehali H., Yano Y., Printz R. L., Koch S., Whitesell R. R., May J. M., Granner D. K. (1996) Functional organization of mammalian hexokinase. II. Retention of catalytic and regulatory functions in both the NH2- and COOH-terminal halves. J. Biol. Chem. 271, 1849–1852 [DOI] [PubMed] [Google Scholar]
- 77. Printz R. L., Ardehali H., Koch S., Granner D. K. (1995) Human hexokinase II mRNA and gene structure. Diabetes 44, 290–294 [DOI] [PubMed] [Google Scholar]
- 78. Osawa H., Printz R. L., Whitesell R. R., Granner D. K. (1995) Regulation of hexokinase II gene transcription and glucose phosphorylation by catecholamines, cyclic AMP and insulin in muscle and fat cells. Diabetes 44, 1426–1432 [DOI] [PubMed] [Google Scholar]
- 79. Osawa H., Robey R. B., Printz R. L., Granner D. K. (1996) Identification and characterization of basal and cyclic AMP response elements in the promoter of the rat hexokinase II gene. J. Biol. Chem. 271, 17296–17303 [DOI] [PubMed] [Google Scholar]
- 80. Osawa H., Sutherland C., Robey R. B., Printz R. L., Granner D. K. (1996) Analysis of the signaling pathway involved in the regulation of hexokinase II gene transcription by insulin. J. Biol. Chem. 271, 16690–16694 [DOI] [PubMed] [Google Scholar]
- 81. O'Doherty R. M., Bracy D. P., Osawa H., Wasserman D. H., Granner D. K. (1994) Rat skeletal muscle hexokinase II mRNA and activity are increased by a single bout of acute exercise. Am. J. Physiol. 266, E171–E178 [DOI] [PubMed] [Google Scholar]
- 82. O'Doherty R. M., Bracy D. P., Granner D. K., Wasserman D. H. (1996) Transcription of the rat skeletal muscle hexokinase II gene is increased by acute exercise. J. Appl. Physiol. 81, 789–793 [DOI] [PubMed] [Google Scholar]